

Abstract
An important control step in the regulation of cytoplasmic mRNA turnover is the removal of the m7G cap structure at the 5′ end of the message. Here, we describe the functional characterization of Dhh1, a highly conserved member of the family of DEAD box-containing proteins, as a regulator of mRNA decapping in Saccharomyces cerevisiae. Dhh1 is a cytoplasmic protein and is shown to be in a complex with the mRNA degradation factor Pat1/Mtr1 and with the 5′–3′ exoribonuclease Xrn1. Dhh1 specifically affects mRNA turnover in the deadenylation-dependent decay pathway, but does not act on the degradation of nonsense-containing mRNAs. Cells that lack dhh1 accumulate degradation intermediates that have lost their poly(A) tail but contain an intact 5′ cap structure, suggesting that Dhh1 is required for efficient decapping in vivo. Furthermore, recombinant Dhh1 is able to stimulate the activity of the purified decapping enzyme Dcp1 in an in vitro decapping assay. We propose that the DEAD box protein Dhh1 regulates the access of the decapping enzyme to the m7G cap by modulating the structure at the 5′ end of mRNAs.
Keywords: Dcp1/DEAD box helicase/Dhh1/mRNA decapping/mRNA turnover
Introduction
The regulation of mRNA turnover is a powerful way to control gene expression. Work in different model systems has led to the identification of several conserved mRNA degradation pathways in eukaryotes. In the yeast Saccharomyces cerevisiae, at least four distinct mRNA turnover pathways have been described (Jacobson and Peltz, 1996; Tucker and Parker, 2000). The predominant pathway of mRNA degradation in yeast is initiated by the shortening of the poly(A) tail. In most instances, deadenylation is followed by removal of the m7G-cap structure at the 5′ end of the message in a reaction catalyzed by the decapping enzyme Dcp1 (Beelman et al., 1996; LaGrandeur and Parker, 1998). Once the protective cap is removed, Xrn1, the major yeast 5′–3′ exonuclease in the cytoplasm, degrades the remaining message very rapidly (Larimer and Stevens, 1990). An alternate way of degrading deadenylated mRNAs is catalyzed by a conserved complex of multiple 3′–5′ exonucleases, termed the exosome (Mitchell et al., 1997). The exosome functions in both the cytoplasm and the nucleus, and is involved in a variety of RNA maturation events (van Hoof and Parker, 1999). Eukaryotic messages can also be degraded by endonucleolytic cleavage in the body of the mRNA. However, this pathway seems to play only a minor role in the overall mRNA decay in yeast (Jacobson and Peltz, 1996; Tucker and Parker, 2000). In addition to these general mRNA turnover mechanisms, eukaryotic cells have also developed a surveillance machinery that recognizes aberrant messages, e.g. mRNAs containing premature stop codons (Maquat and Carmichael, 2001). In yeast, this nonsense-mediated decay (NMD) pathway does not require deadenylation, but depends on Dcp1-mediated decapping and 5′–3′ exonucleolytic degradation of the message by Xrn1 (Czapalinski et al., 1999; Hilleren and Parker, 1999).
Much emphasis has been placed on the identification and characterization of individual components of the various mRNA turnover pathways. However, the cytoplasmic deadenylase responsible for the initial poly(A) shortening in yeast has only recently been discovered. Both CCR4 and CAF1 (also called POP2) are required for normal deadenylation in vivo, and it has been proposed that they are components of the major yeast mRNA deadenylase (Tucker et al., 2001). CCR4 and CAF1 had previously been characterized as members of the CCR4–NOT transcriptional complex (Liu et al., 1998; Chang et al., 1999); however, both proteins reside in the cytoplasm and Caf1 co-purifies with a poly(A)-specific exonuclease activity (Tucker et al., 2001). In addition, it was recently shown that recombinant Caf1/Pop2 functions as a poly(A) nuclease in vitro (Daugeron et al., 2001). Since Caf1/Pop2 shares homology with the mammalian deadenylase PARN (Korner et al., 1998; Daugeron et al., 2001), these data strongly support a model in which Ccr4 and Caf1 are part of a conserved cytoplasmic deadenylase complex.
The yeast decapping enzyme, Dcp1, was identified by a combination of genetic and biochemical approaches (Stevens, 1988; Beelman et al., 1996; LaGrandeur and Parker, 1998). Strains lacking a functional DCP1 gene accumulate deadenylated messages, which contain an intact m7G cap structure (reviewed in Tucker and Parker, 2000). In addition, Dcp1 purified from yeast is able to catalyze the cleavage of the m7GpppX cap structure in vitro (LaGrandeur and Parker, 1998). Beside Dcp1, several proteins were shown to be required for full decapping activity in vivo or in yeast extracts. Dcp2 was identified as a multicopy suppressor of a temperature-sensitive allele of dcp1, and its activity is required to produce active Dcp1 (Dunckley and Parker, 1999). Two enhancers of decapping, EDC1 and EDC2, were recently characterized through their genetic interactions with Dcp1 and/or Dcp2 (Dunckley et al., 2001). Mutations in VPS16 and PAT1/MRT1 were isolated in a biochemical screen and shown to affect the activity of the decapping enzyme in extracts (Zhang et al., 1999a). Deletion of PAT1 leads to a decrease in mRNA turnover and to the appearance of deadenylated and partially capped messages in vivo (Bonnerot et al., 2000; Bouveret et al., 2000). Interest ingly, Pat1 can be found in a complex with the 5′–3′ exonuclease Xrn1 and with a group of seven Sm-like proteins, termed Lsm1–Lsm7 (Bonnerot et al., 2000; Bouveret et al., 2000; Tharun et al., 2000). This group of cytoplasmic Lsm proteins also participates in mRNA degradation and is thought to promote the decapping step by interaction with the deadenylated mRNA, thereby enhancing the interaction of the decapping complex with the message (Tharun et al., 2001). In addition to Dcp1, another decapping activity, DcpS, was recently identified in yeast and mammals. DcpS is linked to the 3′–5′ exonucleolytic activity and, in contrast to Dcp1, releases m7GMP (Wang et al., 2001).
While much has been learned about individual components of the multiple mRNA turnover pathways, little is known about how these pathways are co-ordinated or regulated in vivo. Our understanding of mRNA decay is further complicated by a multitude of interactions between the 5′ and 3′ ends of the message, and by a complex interplay between the translation and degradation machinery (reviewed in Hentze and Kulozik, 1999; Tucker and Parker, 2000). However, a key regulatory step in mRNA turnover is the decapping reaction. In this report, we describe the functional characterization of the DEAD box protein Dhh1 (Strahl-Bolsinger and Tanner, 1993) as a mRNA degradation factor involved in the regulation of decapping. DHH1 had been previously characterized as a multicopy suppressor of a caf1/pop2 deletion and was shown to associate with the Caf1–Ccr4 complex (Hata et al., 1998). However, its function had not been identified. Here it is demonstrated that loss of dhh1 specifically affects decapping in the deadenylation-dependent mRNA decay pathway. Furthermore, recombinant Dhh1 stimulates the activity of purified Dcp1 in an in vitro decapping reaction. We propose that Dhh1 functions as a regulator of mRNA decapping in yeast.
Results
Dhh1 associates with the mRNA degradation factors Pat1 and Xrn1
We have carried out a synthetic lethal screen with xpo1-1, a temperature-sensitive allele of the export factor exportin1/CRM1 (Stade et al., 1997). Seven temperature-sensitive mutants were isolated that were synthetically lethal with xpo1-1 (for details see Materials and methods). One mutant (sl9904) could be complemented by the gene coding for Dhh1, a member of the DEAD box family of proteins of putative RNA helicases (Strahl-Bolsinger and Tanner, 1993). As reported, DHH1 is not essential for viability, but its loss confers a temperature-sensitive growth phenotype (Hata et al., 1998; Moriya and Isono, 1999; data not shown). Dhh1 has previously been shown to associate with the CCR4–NOT complex (Hata et al., 1998) and it was proposed to play a role in transcriptional control (Hata et al., 1998; Moriya and Isono, 1999). To examine its cellular localization, indirect immunofluorescence was performed using a monospecific anti-Dhh1 serum. Dhh1 localized to the cytoplasm and was excluded from nuclei (Figure 1A). Identical localization was observed with a functional Dhh1–green fluorescent protein (GFP) fusion protein (data not shown). No change in Dhh1 localization was seen in xpo1-1 cells at either the permissive or non-permissive temperature (data not shown). Because of its genetic interaction with the transport factor XPO1, we also investigated whether the DEAD box protein Dhh1 plays a role in mRNA export from the nucleus. Using an oligo(dT) in situ hybridization assay, no nuclear accumulation of poly(A) RNA was detected in cells deleted for DHH1 (dhh1Δ cells) at all temperatures tested (data not shown).

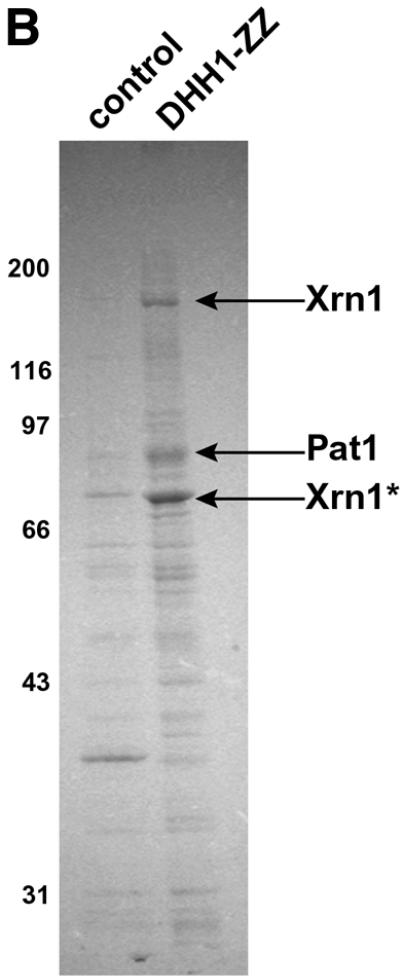
Fig. 1. (A) Dhh1 is a cytoplasmic protein. The subcellular localization of the Dhh1 protein was analyzed by indirect immunofluorescence using a polyclonal anti-Dhh1 serum. Cells were stained either with anti-Dhh1 antiserum and anti-rabbit (FITC) antibody (top panel) or only with anti-rabbit (FITC) antibody (lower panel). The right panel shows the corresponding DNA (DAPI) staining. (B) Purification of Dhh1-interacting proteins. Dhh1-interacting proteins were purified from Dhh1–ZZ-expressing cells by IgG affinity chromatography. Proteins were eluted using 1 M MgCl2, separated on an 8% SDS gel and stained with Coomassie Blue. Proteins indicated by an arrow were identified by MALDI-TOF mass spectrometry. Thirty-six peptides out of 69 were matching peptides for Xrn1 (21% sequence coverage), 24 peptides out of 43 matched Pat1 (24% sequence coverage). Molecular weight markers are indicated on the left. The names of the proteins identified by MALDI are listed on the right. Xrn1* indicates a degradation product of Xrn1. Bands that are not specific for Dhh1p interaction are not labeled. As a control strain the isogenic wild-type background was used.
To identify the function of Dhh1 and to characterize Dhh1-interacting proteins, a functional Dhh1–ZZ fusion protein containing two in-frame copies of the IgG binding domain of Staphylococcus aureus was expressed in yeast under the control of its own promoter. Following affinity purification by IgG–Sepharose chromatography, Dhh1-interacting proteins were eluted using 1 M MgCl2 and analyzed by SDS–PAGE (Figure 1B). Three polypeptides with apparent mol. wts of 180, 95 and 80 kDa specifically co-purified with Dhh1 but were not detectable in control purifications using untagged strains. These polypeptides were analyzed using MALDI mass spectrometry and unambiguously identified as the major yeast 5′–3′ exonuclease Xrn1 (Larimer and Stevens, 1990; Hsu and Stevens, 1993) involved in mRNA turnover (calculated mol. wt 175 kDa) and the mRNA degradation factor Pat1 (Wang et al., 1996; calculated mol. wt 88 kDa). The band with an apparent mol. wt of 80 kDa was identified as a degradation product of Xrn1. The interaction between Dhh1 and Pat1 or Xrn1 was not dependent on the presence of RNA, since RNase treatment did not abolish the interactions between these proteins (data not shown).
Multiple reporter mRNAs are stabilized in dhh1Δ cells
The interaction with two proteins involved in mRNA degradation suggested a function of Dhh1 in mRNA turnover. To test this, we analyzed the decay of STE2 and CYH2 mRNAs in dhh1Δ cells. Northern blot analysis was performed on RNA samples isolated from dhh1Δ and wild-type cells treated for the indicated time points with the transcriptional inhibitor 1,10-phenanthroline (Guthrie and Fink, 1991) (Figure 2; Table I). A significant stabiliz ation of STE2 and CYH2 messages was observed in dhh1Δ cells. PhosphorImager quantification of the data shown in Figure 2 revealed an ∼3-fold increase in the half-life of the STE2 message (from ∼20 min in wild-type cells to ∼60 min in dhh1Δ cells) and an ∼4-fold increase in the half-life of the CYH2 mRNA (from ∼10 min in wild-type cells to ∼40 min in dhh1Δ cells).

Fig. 2. Stabilization of CYH2 and STE2 mRNAs in dhh1Δ cells. Wild-type, dcp1Δ and dhh1Δ cells were grown to mid-log phase and transcription was inhibited using 1,10-phenanthroline. RNA was isolated at the indicated timepoints, separated by formaldehyde–agarose gel electrophoresis and analyzed by northern blotting. The decay of CYH2 mRNA (left panel) and STE2 mRNA (right panel) was examined. Numbers above the lanes indicate the time (in minutes) after transcriptional repression. RNA size markers are shown on the right.
Table I. mRNA half-lives (min) in wild-type and dhh1Δ strains.
| mRNA | Wild type | dhh1Δ |
|---|---|---|
| MFA2pG | 4 | 20 |
| PGK1pG | 20 | 80 |
| STE2 | 20 | 60 |
| CYH2 | 10 | 40 |
To further analyze the mRNA turnover defects in dhh1Δ cells, we introduced the two well-characterized reporter constructs MFA2pG and PGK1pG (Decker and Parker, 1993) into dhh1Δ and wild-type control cells. These reporter constructs are under the control of the inducible GAL promoter, allowing the measurement of decay kinetics after the transcriptional induction by galactose and subsequent repression by the addition of glucose. Furthermore, these reporter constructs permit the detection of degradation intermediates because a poly(G) tract inserted in the 3′ untranslated region (UTR) inhibits the normal progression of exoribonucleases from the 5′ and 3′ end (Decker and Parker, 1993). Northern blot analysis for MFA2pG and PGK1pG mRNA was performed with RNA samples obtained from dhh1Δ and wild-type cells grown for the indicated times after transcriptional repression (Figure 3; Table I). We observed that MFA2pG mRNA was stabilized >5-fold in the dhh1Δ strain compared with wild type (Figure 3; Table I). Similarly, PGK1pG mRNA was stabilized >4-fold in dhh1Δ cells (Table I; see also Figure 4). Taken together, the data shown in Figures 2 and 3 demonstrate that DHH1 is required for the normal decay of several yeast mRNAs and suggest that the Dhh1 protein functions in mRNA turnover.

Fig. 3. MFA2pG mRNA is stabilized in dhh1Δ cells. Wild-type and dhh1Δ strains carrying the GAL:MFA2pG reporter (Decker and Parker, 1993) were grown in 2% raffinose, 0.5% sucrose to an OD600 of 0.6. Transcription of the reporter was induced for 8 min by the addition of 2% galactose and then repressed with 4% glucose. At the indicated time points (after transcriptional repression) cells were harvested and RNA was analyzed by northern blotting using an oligo(dC) probe. The arrows mark the full-length transcript as well as the decay intermediate.

Fig. 4. Transcriptional pulse–chase analysis of the PGK1pG reporter. Wild-type and dhh1Δ cells carrying the GAL:PGK1pG reporter (Decker and Parker, 1993) were grown to an OD600 of 0.6. Transcription of the reporter was induced with 2% galactose for 8 min and then repressed with 4% glucose. At the indicated time points, cells were harvested and RNA was analyzed by northern blotting using an oligo(dC) probe. All samples were treated with RNase H to allow for the size resolution of the poly(A) tail of the reporter RNA on 6% polyacrylamide–urea gel electrophoresis. At time 0, the poly(A) tail of the RNA was also cleaved by RNAse H using an oligo(dT) as a marker for fully deadenylated mRNA (A0). The decay intermediate is marked with an arrow.
DHH1 is not required for the NMD pathway
In contrast to the deadenylation-dependent mRNA degradation, mRNAs containing nonsense codons are decapped and degraded prior to poly(A) shortening by the NMD pathway in yeast. To examine whether Dhh1 also functions in NMD, the stabilization of CYH2 pre-mRNA, a well-characterized NMD substrate (He et al., 1993), was analyzed in wild-type and dhh1Δ cells (Figure 2, left panel). As a positive control we also tested RNA that was isolated from dcp1Δ cells, since Dcp1 is required for NMD and hence for the efficient degradation of CYH2 pre-mRNA (Beelman et al., 1996). Whereas the CYH2 pre-mRNA was readily detected in the RNA isolated from dcp1Δ cells, no stabilization of CYH2 pre-mRNA was observed in either dhh1Δ or wild-type cells. We conclude that Dhh1 is not required for the NMD pathway and that the loss of dhh1 specifically affects the deadenylation-dependent decay of mRNAs.
Deletion of dhh1 does not affect the rates of mRNA deadenylation
DHH1 interacts genetically with CAF1 and CCR4 and the Dhh1 protein has been shown to associate with Caf1 (Hata et al., 1998). The recent finding that Ccr4 and Caf1 are components of the cytoplasmic deadenylase enzyme (Daugeron et al., 2001; Tucker et al., 2001) prompted us to analyze the kinetics of deadenylation in dhh1Δ cells. The PGK1pG reporter was introduced into dhh1Δ and wild-type cells, and its mRNA decay was followed after transcriptional induction and repression with galactose and glucose, respectively (Figure 4). RNA was isolated and detected by northern blotting with a probe specific for the poly(G) stretch in the PGK1pG mRNA. All samples were digested with RNase H in the presence of an oligonucleotide that binds upstream of the stop codon, allowing the visualization of size differences in the 3′ end. As a size reference, a poly(A)– RNA (Figure 4, A0) was prepared by digesting the RNA samples from the earliest timepoint with RNAse H in the presence of oligo(dT). Despite the clear difference in the half-life of PGK1pG mRNA between wild-type and dhh1Δ cells, very similar deadenylation rates or endpoints were detected in these strains (Figure 4). In both strains, the poly(A) tail was completely removed 20–30 min after the addition of galactose (Figure 4). These results indicate that Dhh1 is not required for efficient deadenylation in vivo.
The stabilized messages in dhh1Δ cells are capped
In the transcriptional pulse–chase experiments shown in Figures 3 and 4, it was noticeable that the short, poly(G)-containing intermediates, which are readily detectable in wild-type cells, did not accumulate in dhh1Δ cells. This phenotype can be explained either by a block in the decapping step or by an inhibition of exonucleolytic degradation. To test whether the stabilized messages detected in Figure 3 still contain a cap structure, the isolated RNA samples were treated with an excess of purified Xrn1 (a kind gift from A.Johnson) (Figure 5). Because the 5′–3′ exonuclease Xrn1 has a strong preference for uncapped mRNAs, protection against Xrn1-mediated degradation indicates the presence of an intact cap structure (Hsu and Stevens, 1993; Boeck et al., 1998). Thus, Xrn1 treatment provides us with a quantitative measure of the percentage of capped versus uncapped mRNA in each preparation. To examine the total amount of full-length capped and uncapped MFA2pG mRNA, samples were split into two, and one aliquot of the reaction was treated with EDTA to inhibit Xrn1 activity. The Xrn1-catalyzed degradation of the MFA2pG RNA isolated from wild-type, dhh1Δ and dcp1-2 cells (at the restrictive temperature) was then analyzed by northern blotting and the percentage of capped relative to total full-length message was quantified using PhosphorImager analysis. The degradation of the uncapped decay intermediate (Figure 5) served as an internal control for the efficiency of the exonucleolytic cleavage. At the start of transcriptional repression (t = 0), ∼95% of the RNA from wild-type, dcp1-2 and dhh1Δ cells is protected against exonucleolytic cleavage. Fifteen minutes after transcriptional suppression, ∼66% of the RNA is protected against degradation in wild-type cells, whereas ∼84% of the RNA is resistant to Xrn1 cleavage in dhh1Δ cells. In the control strain dcp1-2, which accumulates deadenylated capped messages at the restrictive temperature (Tharun and Parker, 1999), ∼89% of the RNA still contains the cap structure. Since rRNA (data not shown) and the uncapped, stabilized MFA2pG decay intermediate RNA (Figure 5) were efficiently degraded by Xrn1 in the absence of EDTA, we conclude that the intermediates that accumulate in dhh1Δ cells contain an intact cap structure. This result suggests that Dhh1 functions in mRNA turnover by regulating the decapping reaction.

Fig. 5. Stabilization of capped mRNA species in dhh1Δ cells. RNA from wild-type, dcp1-2 (at the restrictive temperature) and dhh1Δ cells transformed with the MFA2pG reporter was isolated 0 and 15 min after transcription repression with galactose. RNA samples were subjected to treatment with purified Xrn1p in the presence (+) or absence (–) of EDTA to inhibit the exonuclease activity. RNA was separated by 1.2% formaldehyde–agarose gel electrophoresis and analyzed by northern blotting with a specific probe directed against the poly(G)-rich region in the reporter construct. Percentages indicate the ratio of capped, full-length transcripts versus total full-length transcripts calculated from four independent experiments. The sizes of the full-length transcripts as well as the decay intermediates, which serve as an internal control for Xrn1, are indicated by arrows. The lower panel represents a darker exposure of the wild-type, dcp1-2 and dhh1Δ panels at 15 min.
Dhh1 activates the decapping enzyme Dcp1 in vitro
To test whether the effects of the deletion of dhh1 on decapping can be recapitulated in vitro, we performed decapping assays with extracts derived from wild-type or dhh1Δ cells. Consistently, ∼20% inhibition of decapping could be observed in the extracts derived from dhh1Δ cells compared with wild-type extracts (data not shown). Next we investigated whether recombinant Dhh1 affects the activity of purified Dcp1 in an in vitro decapping reaction (Figure 6). FLAG-tagged Dcp1 protein was expressed in yeast and purified by affinity chromatography (LaGrandeur et al., 1998). Thin-layer chromatography (TLC) analysis demonstrated that the purified Dcp1 was active (Figure 6A, lane 3) as it was able to catalyze the removal of m7GDP from an in vitro transcribed RNA substrate radioactively labeled at the cap (Zhang et al., 1999b) in a concentration-dependent manner. To show that the reaction product of Dcp1 is m7GDP, an aliquot of the reaction in Figure 6A, lane 3, was treated with nucleoside diphosphate kinase (NDPK) (lane 4). NDPK converts GDP to GTP but does not use GMP as a substrate. We next tested the effects of recombinant Dhh1 purified from Escherichia coli in this assay (Figure 6B). Dhh1 alone did not induce decapping of the RNA substrate (Figure 6B, lane 4). However, equimolar amounts of Dhh1 significantly stimulated the decapping activity of Dcp1 (Figure 6B, compare lanes 1 and 2). PhosphorImager quantification of multiple independent in vitro decapping reactions demonstrated an ∼2- to 3-fold increase in Dcp1-mediated decapping by Dhh1 (data not shown). The effect of Dhh1 on Dcp1 was concentration dependent, and maximum stimulation was observed at equimolar concentration (Figure 6B; data not shown). In addition, the activation by Dhh1 was specific as the related DEAD box protein Dbp5 involved in mRNA transport (Snay-Hodge et al., 1998; Tseng et al., 1998) did not stimulate decapping (Figure 6B, lane 3). We therefore conclude that Dhh1 specifically stimulates the decapping enzyme Dcp1.

Fig. 6. Analysis of decapping activity of FLAG-Dcp1 in the presence of Dhh1. RNA was transcribed in vitro, capped and labeled using recombinant vaccinia capping enzyme in the presence of [α-32P]GTP and S-adenosylmethionine. [α-32P]GTP cap-labeled mRNA substrate (0.1 pmol) was used in the decapping reaction performed for 30 min at 37°C in the presence of FLAG-Dcp1. Reactions were terminated by addition of 0.5 M EDTA and analyzed by TLC using PEI cellulose plates. The results were visualized and quantified using a Phosphor Imager (Molecular Dynamics). Standards (Sigma) were developed on the TLC plates simultaneously; their migration is indicated on the right. (A) Lane 1 represents the input RNA. Resolution of nuclease P1 (Sigma)-treated substrate RNA is shown in lane 2. Migration of m7GDP released by 12.6 nM Dcp1 was followed in lane 3. An aliquot of the reaction in lane 3 was treated with NDPK (Sigma) (lane 4). (B) Decapping activity of FLAG-Dcp1 was titrated from 12.6 to 1.26 nM [lane 3 in (A) and lane 1 in (B), respectively]. Lane 2 shows decapping reactions performed with 1.26 nM FLAG-Dcp1 in the presence of equimolar recombinant Dhh1 purified from E.coli. The addition of 1.26 nM Dbp5 does not stimulate the release of m7GDP by Dcp1 (compare lanes 1 and 3). Dhh1p alone (lane 4) does not induce the release of m7GDP.
Discussion
The removal of the 5′ cap structure is a decisive step in the fate of eukaryotic mRNAs. In yeast, decapping is subject to tight regulation as it almost invariably induces rapid degradation of the message (Tucker and Parker, 2000). In this study, we provide several lines of evidence that the cytoplasmic DEAD box protein Dhh1 functions in mRNA turnover by regulating the decapping step in the deadenylation-dependent mRNA decay pathway. First, the loss of dhh1 function leads to the inhibition of mRNA degradation. Secondly, messages that accumulate in dhh1Δ cells contain an intact cap structure but have lost their poly(A) tails. Thirdly, Dhh1 is in a cytoplasmic complex with the mRNA degradation factors Pat1 and Xrn1. Finally, recombinant Dhh1 is able to stimulate the purified decapping enzyme Dcp1 in in vitro decapping reactions.
A model for the function of Dhh1
Dhh1 is a member of the highly conserved family of DEAD box proteins and contains all the sequence elements characteristic of this family of proteins. Proteins of this family have been shown to play a role in multiple aspects of RNA metabolism and they are generally thought to function as RNA helicases (reviewed in de la Cruz et al., 1999). Based on this homology, we propose that Dhh1 acts as an activator of decapping by remodeling the 5′ structure of the mRNA, facilitating the access of the decapping enzyme to the message.
In the cytoplasm, the cap structure is predicted to be complexed with the cap-binding complex eIF4F, which mediates initiation of protein translation (reviewed in Gallie, 1998). One role of Dhh1 could therefore be the removal of translation initiation factors like the eIF4F complex from the mRNA. Such a model is supported by results that indicate competition between the decapping enzyme and the translation initiation machinery (Schwartz and Parker, 1999; Vilela et al., 2000). However, Dhh1 is able to stimulate decapping in vitro in the absence of translation factors. This suggests that Dhh1 either helps to recruit Dcp1 (and potentially additional decapping activators) to the 5′ end of the message or, alternatively, that Dhh1 directly activates the enzymatic activity of Dcp1.
In addition to DHH1, several other genes have been identified, the mutation of which induces stabilization of capped mRNA intermediates in vivo. These include DCP2, PAT1/MRT1, MRT3, VPS16 and components of the cytoplasmic LSM complex (Hatfield et al., 1996; Dunckley and Parker, 1999; Zhang et al., 1999a; Bonnerot et al., 2000; Bouveret et al., 2000). Dhh1 is found here in a complex with Pat1, which in turn associates with the cytoplasmic Lsm complex containing Lsm1–Lsm7 (Bonnerot et al., 2000; Bouveret et al., 2000). High-throughput two-hybrid screens have revealed interactions between Dhh1 and Dcp1 or Lsm2 (Uetz et al., 2000). We also detected by western analysis the presence of Lsm1 in the Dhh1 complex, albeit not as a major component (data not shown). Similarly to Dhh1, both Pat1 and the cytoplasmic Lsm complex act specifically in the decapping reaction of deadenylated mRNAs but do not appear to affect the decay of nonsense-containing messages (Figure 2; Bonnerot et al., 2000; Bouveret et al., 2000; Tharun et al., 2000). This supports the hypothesis that a putative Dhh1–Pat1–Lsm complex recognizes a specific feature of deadenylated messages and targets these mRNAs for destruction by recruiting or activating the decapping complex containing Dcp1 (Tharun et al., 2001). Biochemical analysis of the Dcp1 decapping activity provided evidence that Dcp1 interacts with both the cap and the body of the mRNA. It will be now of interest to analyze the substrate specificity of Dcp1 together with Dhh1 in the presence or absence of other regulators of decapping and/or translation initiation factors.
During the course of this study, another group independently reported that Dhh1 functions in mRNA decapping and interacts with proteins of the decapping and deadenylase complex (Coller et al., 2001).
Interactions of Dhh1 with Xrn1 and the deadenylase complex
Dhh1 is shown here to interact with the 5′–3′ exonuclease Xrn1 and with Pat1 (Figure 1B). Xrn1 was recently reported to also be present in the Pat1–Lsm complex (Bouveret et al., 2000; Tharun et al., 2000). Although it is not clear whether the interaction between Dhh1 and Xrn1 is direct or mediated through Pat1, these findings raise the question about the role of Xrn1 in a complex proposed to activate decapping. Xrn1 itself is not required for the decapping reaction, since a xrn1 null mutant accumulates decapped full-length messages lacking a poly(A) tail (Hsu and Stevens, 1993). In vivo data suggest that removal of the cap is almost immediately followed by 5′–3′ degradation through Xrn1 (Muhlrad et al., 1994). It is therefore conceivable that Xrn1 is recruited to the Dhh1–Pat1 complex in a standby position, ready to cleave the message once the protective cap has been removed by Dcp1. Alternatively, Dhh1 or Pat1 could also play an additional role in the exoribonucleolytic cleavage step of mRNA turnover, and could stimulate the activity of Xrn1. It is of interest that dhh1Δ cells do not accumulate the poly(G)-containing degradation intermediates of the MFA2pG or PGK1pG reporter constructs that are readily detected in wild-type or lsmΔ cells (see Figures 3 and 4; Bouveret et al., 2000; Tharun et al., 2000). In dhh1Δ cells, a ladder-like degradation pattern of the reporter RNAs was observed. A similar phenotype has also been described in pat1Δ cells (Bonnerot et al., 2000). One explanation of this could be an impaired 5′–3′ exonucleolytic degradation in dhh1Δ and pat1Δ cells; however, alternative explanations cannot be excluded. Further experiments will be required to analyze the role of the Dhh1–Pat1–Xrn1 complex.
Dhh1 has also been reported to interact physically and genetically with Caf1/Pop2 (Hata et al., 1998), a component of the recently identified cytoplasmic deadenylase complex (Daugeron et al., 2001; Tucker et al., 2001). We did not observe any significant changes in the deadenylation rates in dhh1Δ cells (Figure 4), suggesting that Dhh1 is not required for the efficient removal of the poly(A) tail in vivo. However, the connection between Dhh1 and the 3′ end processing machinery, together with previously reported interactions between the decapping machinery, the poly(A)-binding protein (Pab1) and other translation factors strongly suggests a complex regulatory network. It will be critical to determine the significance of the interactions between the 3′ and 5′ end of the message, and to understand the connection between mRNA turnover and translation.
Genetic interactions of DHH1
We isolated DHH1 in a synthetic lethal screen with the export factor XPO1/CRM. Dhh1 does not appear to function in nuclear export and is not a transport substrate of Xpo1. At present, it can only be speculated why a loss of DHH1 causes xpo1-1 cells to die. However, we have observed significant changes in the overall gene expression in dhh1Δ cells. For example, the levels of both ribosomal subunits are significantly lower in dhh1Δ cells compared with wild-type cells (data not shown). Since Xpo1 appears to play a role in the export of pre-ribosomal subunits (Ho et al., 2000; Stage-Zimmermann et al., 2000), an additional reduction in the amount of functional ribosomes could account for the synthetic lethal interaction between XPO1 and DHH1. In addition to the defects in ribosomal biosynthesis, we also noticed that the abundance of several mRNAs was either negatively (e.g. CYH2 mRNA) or positively (e.g. STE2 mRNA) affected in dhh1Δ cells (data not shown). This is consistent with earlier observations that mutations in other mRNA degradation factors can affect gene expression in either a negative or positive fashion (Liu et al., 1998; Chang et al., 1999). However, the changes in RNA levels in dhh1Δ cells do not appear to be directly regulated at the level of transcription. First, Dhh1 localizes to the cytoplasm (Figure 1A). Secondly, transcriptional pulse–chase experiments revealed no significant changes in the transcriptional induction upon deletion of dhh1 (data not shown). Further experiments are required to test whether these alterations in gene expression only occur at the level of mRNA degradation or whether Dhh1 also affects, either directly or indirectly, other transcriptional or post- transcriptional regulatory events. Of note, the loss of dhh1 confers a temperature-sensitive growth phenotype. However, as expected for a null allele, all the defects in mRNA turnover in dhh1Δ cells are independent of the temperature (data not shown). Death of dhh1Δ cells at elevated temperatures seems to occur by cell lysis, and the temperature-sensitive phenotype can be suppressed by osmotic stabilization through sorbitol or by the over-expression of PKC1 (Hata et al., 1998; Moriya and Isono, 1999; data not shown). This indicates that Dhh1 and/or the regulation of mRNA turnover may be required for cell wall biosynthesis and growth of yeast cells at elevated temperatures.
The DEAD box protein Dhh1 is highly conserved
Dhh1 is a highly conserved DEAD box protein, and putative orthologs have been identified in Schizosaccharo myces pombe (Maekawa et al., 1994), Caenorhabditis elegans, Drosophila (de Valoir et al., 1991), Xenopus (Ladomery et al., 1997), mouse (Akao et al., 1995) and humans (Lu and Yunis, 1992). The Xenopus homolog p54 was found in oocytes in cytoplasmic mRNA storage particles and was proposed to function as a translational regulator (Ladomery et al., 1997). In humans and mouse, the Dhh1 homolog DDX6 was identified as a proto-oncogene (Lu and Yunis, 1992; Akao et al., 1995; Seto et al., 1995), whereas mutations in the Drosophila homolog ME31B were shown to affect germline development (de Valoir et al., 1991). A putative function of all these proteins in mRNA turnover may be able to account for the complexity of the observed phenotypes.
In addition to Dhh1, several other members of the DEAD/DEAH box family were shown to function in mRNA turnover in prokaryotes and eukaryotes (reviewed in de la Cruz et al., 1999). For example in yeast, Ski2 is involved in 3′–5′ degradation and is thought to modulate the function of the exosome (Mitchell et al., 1997; Jacobs et al., 1998), whereas Nam7/Upf1plays a role in the NMD and probably also in the deadenylation-dependent mRNA decay pathway (Lee and Culbertson, 1995; Weng et al., 1996; He and Jacobson, 2001). Although the exact role of these proteins is not currently understood, Nam7/Upf1 is involved in decapping of nonsense-containing mRNAs, and may play a very similar mechanistic role in NMD to that of Dhh1 in deadenylation-dependent mRNA turnover.
It is now evident that RNA helicases play critical roles in all steps of RNA metabolism, including maturation, splicing, transport, translation and degradation. Despite their high degree of homology, many helicases appear to act very specifically and show little redundancy in their function (de la Cruz et al., 1999). In only very few cases have specific substrates for the helicases been identified, but potential targets include RNA structures, RNA–protein complexes or even protein–protein complexes in ribonucleoprotein particles. The challenge for the future therefore remains to identify additional protein interaction partners of Dhh1 and to characterize its in vivo RNA and RNP substrates.
Materials and methods
Plasmids and yeast strains
The plasmid pKW854 encoding DHH1 as a His6 tag fusion was constructed by PCR amplification (PFU; Stratagene) of the DHH1 gene from genomic DNA using oligos UC328, UC208 (sequences available upon request) and cloned via BamHI and SacI into pRSETA (Invitrogen).
Yeast strain KWY281 expressing a Dhh1 fusion protein containing a C-terminal GFP tag was generated using a PCR-based gene integration strategy as described (Longtine et al., 1998). The oligos used to amplify from plasmid pAF6-GFP-Kanmx were UC383 and UC214.
Yeast strain KWY407 (expressing a C-terminal protein A fusion of Dhh1) was designed using the identical strategy and identical oligos were used for amplification from plasmid pKW803 (containing S-tag-TEV protease cleavage site-ZZ-tag; HIS3 selection marker).
Isolation of synthetic lethal mutants using the xpo1-1 allele
The screening strain KWY168 was constructed for the red/white colony sectoring assay (Bender and Pringle, 1991) containing the plasmid pKW 600-URA3-ADE3-XPO1 in a xpo1::TRP1; ade2Δ; ade3Δ; his3::xpo1-1, HIS3 background, and mutagenized with UV. Details about this screen will be published elsewhere. The mutant sl9904 was transformed with a yeast genomic library inserted into YEP13-LEU2, and the complementing plasmid was recovered from transformants that regained growth at 37°C, showed a red/white colony sectoring phenotype and were able to grow on 5-FOA.
Indirect immunofluorescence microscopy of Dhh1p
Indirect immunofluorescence microscopy was performed as described (Ayscough and Drubin, 1998). The anti-Dhh1 antibody (polyclonal rabbit antiserum) and the fluorescein-conjugated secondary antibody (Jackson ImmunoResearch Laboratories) were used at 1:500. Light microscopy was performed using a Nikon eclipse E600 microscope equipped with a 100× PL Fluotar oil immersion objective. Images were acquired with a Hamamatsu digital camera CA742-98 using the Metamorph Software program (Universal Imaging). Panels were assembled with Adobe Photoshop software.
Yeast extracts
Yeast extracts were obtained by growing yeast cells in YPD medium to mid-log phase. Cells were harvested, frozen in liquid nitrogen and ground in the presence of dry ice. The yeast powder was stored at –80°C. For the affinity purification of Dhh1 from yeast strain KWY407, cell extracts corresponding to 1 l of culture were mixed with the same volume of 2× extraction buffer [60 mM HEPES pH 7.3, 200 mM KOAc, 2 mM dithiothreitol (DTT), 10 mM MgCl2, 20% glycerol, protease inhibitor mix without EDTA (Roche)], and spun down for 30 min at 10 000 g. The supernatant was incubated with 120 µl of IgG–Sepharose beads for 3 h at 4°C. Beads were washed with 40× column volume of wash buffer (30 mM HEPES pH 7.3, 100 mM KOAc, 150 mM NaCl, 2 mM DTT, 10 mM MgCl2, 10% glycerol). Proteins were eluted with 1 M MgCl2 and subsequently analyzed by SDS–PAGE. Mass spectrometry was performed using the Perseptive Voyager-DE RP Biospectrometry instrument (MALDI-TOF). Proteins were identified by searching a comprehensive non-redundant protein database using the program ProFound, Version 4.10.5, Rockefeller University Edition.
Protein expression and purification
FLAG-Dcp1p was purified from yeast as described (LaGrandeur and Parker, 1998).
Bacterial expression of His6-Dhh1p was performed following standard expression and purification protocols using Ni-NTA beads (Qiagen). Purified protein was dialyzed against reaction buffer (30 mM HEPES pH 7.6, 150 mM KOAc, 2 mM MgAc, 2 mM BME, 7% glycerol).
Bacterial expression of the His-tagged vaccinia virus capping enzyme, pET-His-D1/D12, was performed as described (Luo et al., 1995).
Generation of anti-Dhh1 antibody
The antibody was generated by injecting rabbits with the His6-Dhh1 purified protein using standard protocols. The antiserum was monospecific on western blots (data not shown).
In vivo assay for mRNA degradation
Plasmid pRP602 encoding the MFA2pG reporter as well as plasmid pRP485 encoding the PGK1pG were transformed into wild-type and dhh1Δ cells and transcriptional pulse–chase experiments were performed as described (Decker and Parker, 1993). Briefly, transcription was induced for 8 min with 2% galactose and subsequently repressed by the addition of 4% dextrose. RNA samples were analyzed on a 6% polyacrylamide gel containing 8.3 M urea followed by northern blot analysis. An oligonucleotide directed against the poly(G)-rich region was radioactively labeled and used as a probe; signals were detected using a PhosphorImager (Molecular Dynamics). mRNA amounts were normalized against the 18S rRNA. RNase H treatments were performed as described (Muhlrad and Parker, 1992).
For northern blot analysis of CYH2 and STE2 RNAs, cells were grown to mid-log phase and 1,10-phenanthroline (Sigma) was added to a final concentration of 100 µg/ml. Aliquots of the cells were removed at time points 0, 5, 10, 15, 30 and 60 min. After a quick centrifugation step, cells were frozen in liquid nitrogen. RNA was isolated as described (Parker et al., 1991) and separated by agarose gel electrophoresis. Gene-specific probes were constructed by PCR amplification using the following oligonucleotides: CYH2 (UC253 and UC254); STE2 (UC255 and UC256). Random prime labeling of the PCR product was performed using RedyprimeII™ Kit (Amersham Pharmacia). Hybridization was carried out at 65°C in Church buffer; wash steps were performed at 65°C in Church wash buffer.
Xrn1 assay and in vitro decapping assays
The Xrn1 assay was performed as described (Boeck et al., 1998). Briefly, 10 µg of total RNA were incubated with 600 ng of purified Xrn1p (kind gift of Arlen Johnson, University of Texas at Austin) in a final volume of 20 µl of 33 mM Tris–HCl pH 8, 50 mM NaCl, 2.5 mM MgCl2 and 0.2 mM DTT in the presence or absence of 5 mM EDTA. Reactions were incubated for 30 min at 37°C and stopped by the addition of 1 vol. of RNA loading buffer.
RNA labeling and decapping assays were performed in decapping buffer as described (Zhang et al., 1999b). All RNAs were gel purified prior to use as described by Wang et al. (2001). The RNA substrate was obtained by in vitro transcription of the plasmid pAS575 (Wells et al., 1998).
Acknowledgments
Acknowledgements
We wish to thank the following for generously providing reagents: A.Johnson for purified Xrn1; M.Kiledjian for plasmids, recombinant protein and helpful advice regarding Dcp1 purification; R.Parker for plasmids pRP485, pRP602 and for the yeast strains yRP1070 and yRP1340; A.Sachs for the anti-Lsm1 antibody; S.Shuman for plasmid pET-His-D1/D12; and P.Preker for purified Dbp5. We are grateful to R.Jacob and D.King for help with mass spectrometry and to A.Sachs and all members of the Weis laboratory for stimulating discussions and/or comments on the manuscript. This work was supported by a Deutsche Forschungsgemeinschaft postdoctoral fellowship to N.F., by NIH grant GM58065 (to K.W.) and by the Searle Scholars Program (to K.W.).
References
- Akao Y., Marukawa,O., Morikawa,H., Nakao,K., Kamei,M., Hachiya,T. and Tsujimoto,Y. (1995) The rck/p54 candidate proto-oncogene product is a 54-kilodalton D-E-A-D box protein differentially expressed in human and mouse tissues. Cancer Res., 55, 3444–3449. [PubMed] [Google Scholar]
- Ayscough K.R. and Drubin,D.G. (1998) A role for the yeast cytoskeleton in pheromone receptor clustering and signalling. Curr. Biol., 10, 1061–1075. [DOI] [PubMed] [Google Scholar]
- Beelman C.A., Stevens,A., Caponigro,G., LaGrandeur,T.E., Hatfield,L., Fortner,D.M. and Parker,R. (1996) An essential component of the decapping enzyme required for normal rates of mRNA turnover. Nature, 382, 642–646. [DOI] [PubMed] [Google Scholar]
- Bender A. and Pringle,J.R. (1991) Use of a screen for synthetic lethal and multicopy suppressor mutants to identify two new genes involved in morphogenesis in Saccharomyces cerevisiae. Mol. Cell. Biol., 11, 1295–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeck R., Lapeyre,B., Brown,C.E. and Sachs,A.B. (1998) Capped mRNA degradation intermediates accumulate in the yeast spb8-2 mutant. Mol. Cell. Biol., 18, 5062–5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnerot C., Boeck,R. and Lapeyre,B. (2000) The two proteins Pat1p (Mrt1p) and Spb8p interact in vivo, are required for mRNA decay and are functionally linked to Pab1p. Mol. Cell. Biol., 20, 5939–5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouveret E., Rigaut,G., Shevchenko,A., Wilm,M. and Seraphin,B. (2000) An Sm-like protein complex that participates in mRNA degradation. EMBO J., 19, 1661–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang M., French-Cornay,D., Fan,H.Y., Klein,H., Denis,C.L. and Jaehning,J.A. (1999) A complex containing RNA polymerase II, Paf1p, Cdc73p, Hpr1p and Ccr4p plays a role in protein kinase C signaling. Mol. Cell. Biol., 19, 1056–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coller J.M., Tucker,M., Sheth,U., Valencia-Sanchez,M.A. and Parker,R. (2001) The DEAD box helicase, Dhh1p, functions in mRNA decapping and interacts with both the decapping and deadenylase complexes. RNA, 7, 1717–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czaplinski K., Ruiz-Echevarria,M.J., Gonzalez,C.I. and Peltz,S.W. (1999) Should we kill the messenger? The role of the surveillance complex in translation termination and mRNA turnover. BioEssays, 21, 685–696. [DOI] [PubMed] [Google Scholar]
- Daugeron M.C., Mauxion,F. and Seraphin,B. (2001) The yeast POP2 gene encodes a nuclease involved in mRNA deadenylation. Nucleic Acids Res., 29, 2448–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker C.J. and Parker,R. (1993) A turnover pathway for both stable and unstable mRNAs in yeast: evidence for a requirement for deadenylation. Genes Dev., 7, 1632–1643. [DOI] [PubMed] [Google Scholar]
- de la Cruz J., Kressler,D. and Linder,P. (1999) Unwinding RNA in Saccharomyces cerevisiae: DEAD-box proteins and related families. Trends Biochem. Sci., 24, 192–198. [DOI] [PubMed] [Google Scholar]
- de Valoir T., Tucker,M.A., Belikoff,E.J., Camp,L.A., Bolduc,C. and Beckingham,K. (1991) A second maternally expressed Drosophila gene encodes a putative RNA helicase of the ‘DEAD box’ family. Proc. Natl Acad. Sci. USA, 88, 2113–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunckley T. and Parker,R. (1999) The DCP2 protein is required for mRNA decapping in Saccharomyces cerevisiae and contains a functional MutT motif. EMBO J., 18, 5411–5422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunckley T., Tucker,M. and Parker,R. (2001) Two related proteins, Edc1p and Edc2p, stimulate mRNA decapping in Saccharomyces cerevisiae. Genetics, 157, 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallie D.R. (1998) A tale of two termini: a functional interaction between the termini of an mRNA is a prerequisite for efficient translation initiation. Gene, 216, 1–11. [DOI] [PubMed] [Google Scholar]
- Guthrie C. and Fink,G.R. (1991) Guide to yeast genetics and molecular biology. Methods Enzymol., 194, 418. [PubMed] [Google Scholar]
- Hata H., Mitsui,H., Liu,H., Bai,Y., Denis,C.L., Shimizu,Y. and Sakai,A. (1998) Dhh1p, a putative RNA helicase, associates with the general transcription factors Pop2p and Ccr4p from Saccharomyces cerevisiae. Genetics, 148, 571–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatfield L., Beelman,C.A., Stevens,A. and Parker,R. (1996) Mutations in trans-acting factors affecting mRNA decapping in Saccharomyces cerevisiae. Mol. Cell. Biol., 16, 5830–5838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W. and Parker,R. (2001) The yeast cytoplasmic LsmI/Pat1p complex protects mRNA 3′ termini from partial degradation. Genetics, 158, 1445–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F., Peltz,S.W., Donahue,J.L., Rosbash,M. and Jacobson,A. (1993) Stabilization and ribosome association of unspliced pre-mRNAs in a yeast upf1– mutant. Proc. Natl Acad. Sci. USA, 90, 7034–7038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentze M.W. and Kulozik,A.E. (1999) A perfect message: RNA surveillance and nonsense-mediated decay. Cell, 96, 307–310. [DOI] [PubMed] [Google Scholar]
- Hilleren P. and Parker,R. (1999) Mechanisms of mRNA surveillance in eukaryotes. Annu. Rev. Genet., 33, 229–260. [DOI] [PubMed] [Google Scholar]
- Ho J.H., Kallstrom,G. and Johnson,A.W. (2000) Nmd3p is a Crm1p-dependent adapter protein for nuclear export of the large ribosomal subunit. J. Cell Biol., 151, 1057–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu C.L. and Stevens,A. (1993) Yeast cells lacking 5′→3′ exoribo nuclease 1 contain mRNA species that are poly(A) deficient and partially lack the 5′ cap structure. Mol. Cell. Biol., 13, 4826–4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs J.S., Anderson,A.R. and Parker,R.P. (1998) The 3′ to 5′ degrad ation of yeast mRNAs is a general mechanism for mRNA turnover that requires the SKI2 DEVH box protein and 3′ to 5′ exonucleases of the exosome complex. EMBO J., 17, 1497–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson A. and Peltz,S.W. (1996) Interrelationships of the pathways of mRNA decay and translation in eukaryotic cells. Annu. Rev. Biochem., 65, 693–739. [DOI] [PubMed] [Google Scholar]
- Korner C.G., Wormington,M., Muckenthaler,M., Schneider,S., Dehlin,E. and Wahle,E. (1998) The deadenylating nuclease (DAN) is involved in poly(A) tail removal during the meiotic maturation of Xenopus oocytes. EMBO J., 17, 5427–5437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladomery M., Wade,E. and Sommerville,J. (1997) Xp54, the Xenopus homologue of human RNA helicase p54, is an integral component of stored mRNP particles in oocytes. Nucleic Acids Res., 25, 965–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaGrandeur T.E. and Parker,R. (1998) Isolation and characterization of Dcp1p, the yeast mRNA decapping enzyme. EMBO J., 17, 1487–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larimer F.W. and Stevens,A. (1990) Disruption of the gene XRN1, coding for a 5′–3′ exoribonuclease, restricts yeast cell growth. Gene, 95, 85–90. [DOI] [PubMed] [Google Scholar]
- Lee B.S. and Culbertson,M.R. (1995) Identification of an additional gene required for eukaryotic nonsense mRNA turnover. Proc. Natl Acad. Sci. USA, 92, 10354–10358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.Y., Badarinarayana,V., Audino,D.C., Rappsilber,J., Mann,M. and Denis,C.L. (1998) The NOT proteins are part of the CCR4 transcriptional complex and affect gene expression both positively and negatively. EMBO J., 17, 1096–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine M.S., McKenzie,A.,III, Demarini,D.J., Shah,N.G., Wach,A., Brachat,A., Philippsen,P. and Pringle,J.R. (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast, 14, 953–961. [DOI] [PubMed] [Google Scholar]
- Lu D. and Yunis,J.J. (1992) Cloning, expression and localization of an RNA helicase gene from a human lymphoid cell line with chromosomal breakpoint 11q23.3. Nucleic Acids Res., 20, 1967–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y., Mao,X., Deng,L., Cong,P. and Shuman S. (1995) The D1 and D12 subunits are both essential for the transcription termination factor activity of vaccinia virus capping enzyme. J. Virol., 69, 3852–3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa H., Nakagawa,T., Uno,Y., Kitamura,K. and Shimoda,C. (1994) The ste13+ gene encoding a putative RNA helicase is essential for nitrogen starvation-induced G1 arrest and initiation of sexual development in the fission yeast Schizosaccharomyces pombe. Mol. Gen. Genet., 244, 456–464. [DOI] [PubMed] [Google Scholar]
- Maquat L.E. and Carmichael,G.G. (2001) Quality control of mRNA function. Cell, 104, 173–176. [DOI] [PubMed] [Google Scholar]
- Mitchell P., Petfalski,E., Shevchenko,A., Mann,M. and Tollervey,D. (1997) The exosome: a conserved eukaryotic RNA processing complex containing multiple 3′→5′ exoribonucleases. Cell, 91, 457–466. [DOI] [PubMed] [Google Scholar]
- Moriya H. and Isono,K. (1999) Analysis of genetic interactions between DHH1, SSD1 and ELM1 indicates their involvement in cellular morphology determination in Saccharomyces cerevisiae. Yeast, 15, 481–496. [DOI] [PubMed] [Google Scholar]
- Muhlrad D. and Parker,R. (1992) Mutations affecting stability and deadenylation of the yeast MFA2 transcript. Genes Dev., 6, 2100–2111. [DOI] [PubMed] [Google Scholar]
- Muhlrad D., Decker,C.J. and Parker,R. (1994) Deadenylation of the unstable mRNA encoded by the yeast MFA2 gene leads to decapping followed by 5′→3′ digestion of the transcript. Genes Dev., 8, 855–866. [DOI] [PubMed] [Google Scholar]
- Parker R., Herrick,D., Peltz,S.W. and Jacobson,A. (1991) Measurement of mRNA decay rates in Saccharomyces cerevisiae. Methods Enzymol., 194, 415–423. [DOI] [PubMed] [Google Scholar]
- Schwartz D.C. and Parker,R. (1999) Mutations in translation initiation factors lead to increased rates of deadenylation and decapping of mRNAs in Saccharomyces cerevisiae. Mol. Cell. Biol., 19, 5247–5256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seto M., Yamamoto,K., Takahashi,T. and Ueda,R. (1995) Cloning and expression of a murine cDNA homologous to the human RCK/P54, a lymphoma-linked chromosomal translocation junction gene on 11q23. Gene, 166, 293–296. [DOI] [PubMed] [Google Scholar]
- Snay-Hodge C.A., Colot,H.V., Goldstein,A.L. and Cole,C.N. (1998) Dbp5p/Rat8p is a yeast nuclear pore-associated DEAD-box protein essential for RNA export. EMBO J., 17, 2663–2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stade K., Ford,C.S., Guthrie,C. and Weis,K. (1997) Exportin 1 (Crm1p) is an essential nuclear export factor. Cell, 90, 1041–1050. [DOI] [PubMed] [Google Scholar]
- Stage-Zimmermann T., Schmidt,U. and Silver,P.A. (2000) Factors affecting nuclear export of the 60S ribosomal subunit in vivo. Mol. Biol. Cell, 11, 3777–3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens A. (1988) mRNA-decapping enzyme from Saccharomyces cerevisiae: purification and unique specificity for long RNA chains. Mol. Cell. Biol., 8, 2005–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl-Bolsinger S. and Tanner,W. (1993) A yeast gene encoding a putative RNA helicase of the ‘DEAD’-box family. Yeast, 9, 429–432. [DOI] [PubMed] [Google Scholar]
- Tharun S. and Parker,R. (1999) Analysis of mutations in the yeast mRNA decapping enzyme. Genetics, 151, 1273–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tharun S. and Parker,R. (2001) Targeting an mRNA for decapping: displacement of translation factors and association of the Lsm1p–7p complex on deadenylated yeast mRNAs. Mol. Cell, 8, 1075–1083. [DOI] [PubMed] [Google Scholar]
- Tharun S., He,W., Mayes,A.E., Lennertz,P., Beggs,J.D. and Parker,R. (2000) Yeast Sm-like proteins function in mRNA decapping and decay. Nature, 404, 515–518. [DOI] [PubMed] [Google Scholar]
- Tseng S.S., Weaver,P.L., Liu,Y., Hitomi,M., Tartakoff,A.M. and Chang,T.H. (1998) Dbp5p, a cytosolic RNA helicase, is required for poly(A)+ RNA export. EMBO J., 17, 2651–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker M. and Parker,R. (2000) Mechanisms and control of mRNA decapping in Saccharomyces cerevisiae. Annu. Rev. Biochem., 69, 571–595. [DOI] [PubMed] [Google Scholar]
- Tucker M., Valencia-Sanchez,M.A., Staples,R.R., Chen,J., Denis,C.L. and Parker,R. (2001) The transcription factor associated Ccr4 and Caf1 proteins are components of the major cytoplasmic mRNA deadenylase in Saccharomyces cerevisiae. Cell, 104, 377–386. [DOI] [PubMed] [Google Scholar]
- Uetz P. et al. (2000) A comprehensive analysis of protein–protein interactions in Saccharomyces cerevisiae. Nature, 403, 623–627. [DOI] [PubMed] [Google Scholar]
- van Hoof A. and Parker,R. (1999) The exosome: a proteasome for RNA? Cell, 99, 347–350. [DOI] [PubMed] [Google Scholar]
- Vilela C., Velasco,C., Ptushkina,M. and McCarthy,J.E. (2000) The eukaryotic mRNA decapping protein Dcp1 interacts physically and functionally with the eIF4F translation initiation complex. EMBO J., 19, 4372–4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Watt,P.M., Louis,E.J., Borts,R.H. and Hickson,I.D. (1996) Pat1: a topoisomerase II-associated protein required for faithful chromosome transmission in Saccharomyces cerevisiae. Nucleic Acids Res., 24, 4791–4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z. and Kiledjian,M. (2001) Functional link between the mammalian exosome and mRNA decapping. Cell, 107, 751–762. [DOI] [PubMed] [Google Scholar]
- Wells S.E., Hillner,P.E., Vale,R.D. and Sachs,A.B. (1998) Circulariza tion of mRNA by eukaryotic translation initiation factors. Mol. Cell, 2, 135–140. [DOI] [PubMed] [Google Scholar]
- Weng Y., Czaplinski,K. and Peltz,S.W. (1996) Identification and characterization of mutations in the UPF1 gene that affect nonsense suppression and the formation of the Upf protein complex but not mRNA turnover. Mol. Cell. Biol., 16, 5491–5506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Williams,C.J., Hagan,K. and Peltz,S.W. (1999a) Mutations in VPS16 and MRT1 stabilize mRNAs by activating an inhibitor of the decapping enzyme. Mol. Cell. Biol., 19, 7568–7576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Williams,C.J., Wormington,M., Stevens,A. and Peltz,S.W. (1999b) Monitoring mRNA decapping activity. Methods, 17, 46–51. [DOI] [PubMed] [Google Scholar]