

Abstract
The catalytic subunit of DNA-dependent protein kinase (DNA-PKCS) is required for a non-homologous end-joining pathway that repairs DNA double-strand breaks produced by ionizing radiation or V(D)J recombination; however, its role in this pathway has remained obscure. Using a neutravidin pull-down assay, we found that DNA-PKCS mediates formation of a synaptic complex containing two DNA molecules. Furthermore, kinase activity was cooperative with respect to DNA concentration, suggesting that activation of the kinase occurs only after DNA synapsis. Electron microscopy revealed complexes of two DNA ends brought together by two DNA-PKCS molecules. Our results suggest that DNA-PKCS brings DNA ends together and then undergoes activation of its kinase, presumably to regulate subsequent steps for processing and ligation of the ends.
Keywords: DNA-dependent protein kinase/DNA repair/electron microscopy/non-homologous end-joining/V(D)J recombination
Introduction
Many environmental agents induce double-strand breaks (DSBs) in DNA. Some of these agents are used to treat cancer, including ionizing radiation from radon decay and topoisomerase II inhibitors such as adriamycin from the bacterium Streptomyces and epidophyllotoxin from the mandrake plant. Unrepaired DSBs lead to chromosomal fragmentation and cell death, while improperly repaired DSBs often generate chromosome translocations and cancer.
DSBs are repaired by either homologous recombination or non-homologous end-joining (NHEJ). This paper will focus on NHEJ, which is essential for generating antibody diversity via V(D)J recombination (Smider and Chu, 1997) and class switch recombination (Rolink et al., 1996; Casellas et al., 1998; Manis et al., 1998; Chen et al., 2000). To achieve NHEJ, several factors act together to recognize and bind the broken DNA ends, bring the ends together in a synaptic complex, process the ends into a ligatable form and ligate the ends to restore chromosomal integrity.
Six of the gene products required for NHEJ have been identified using mutant mammalian cell lines that are abnormally sensitive to ionizing radiation and defective in V(D)J recombination. Ku70 and Ku80 are subunits of the Ku heterodimer, which binds to DNA ends (Smider et al., 1994; Taccioli et al., 1994). The catalytic subunit of DNA-dependent protein kinase (DNA-PKCS) is a 465 kDa serine–threonine protein kinase that is activated by DNA ends (Gottlieb and Jackson, 1993). Xrcc4 and ligase IV form a heterodimer with DNA ligase activity (Li et al., 1995; Grawunder et al., 1997). Artemis, which is mutated in human RS-SCID (severe combined immunodeficiency with radiosensitivity), interacts with DNA-PKCS and may be involved in processing DNA ends (Moshous et al., 2001; Ma et al., 2002). Ku, DNA-PKCS, Xrcc4 and ligase IV are required in a cell-free system for NHEJ, which also requires MgCl2, ATP and inositol hexakisphosphate (IP6) (Baumann and West, 1998; Hanakahi et al., 2000).
Considerable attention has been focused on DNA-PKCS because of its striking property of remaining quiescent until activated by DNA ends (Gottlieb and Jackson, 1993). Of the 4096 amino acids that comprise DNA-PKCS, the C-terminal 380 amino acids encode a kinase domain that shares homology with members of the phosphatidylinositol 3-kinase (PI3K) superfamily of lipid and protein kinases. Many of these kinases play roles in responding to DNA damage, including the mammalian proteins DNA-PKCS, ATM and ATR (reviewed in Durocher and Jackson, 2001).
The kinase activity of DNA-PKCS is likely to be involved in NHEJ. The fungal metabolite wortmannin, which inactivates kinases in the PI3K family, inhibits NHEJ in cell-free extracts (Baumann and West, 1998). Mutation of the conserved aspartate in the kinase domain of DNA-PKCS abolishes kinase activity and prevents DNA-PKCS from restoring radiation resistance to mutant rodent cells (Kurimasa et al., 1999).
During NHEJ, proteins must bind the DNA ends and bring them together in a synaptic complex. Because of its abundance and high affinity for DNA ends, Ku is likely to be the first protein to bind the broken DNA ends. Under some conditions, Ku is the only detectable DNA end-binding activity when cell extracts are analyzed in an electrophoretic mobility-shift assay (Rathmell and Chu, 1994a,b). Upon binding to DNA, Ku recruits DNA-PKCS to the DNA ends (Hammarsten and Chu, 1998). Photocrosslinking experiments indicate that Ku then translocates into the DNA by one helical turn, leaving DNA-PKCS bound at the DNA terminus, occupying the 10 bp proximal to the free end (Yoo and Dynan, 1999). Thus, DNA-PKCS, not Ku, binds to a critical position at the DNA terminus. However, a prior study reported that Ku stimulated the synapsis of DNA molecules (Ramsden and Gellert, 1998). To address this apparent paradox, we hypothesized that initial association of two DNA ends is mediated by DNA-PKCS.
To test this hypothesis, we used biochemical methods and electron microscopic analysis to show that DNA-PKCS brings DNA ends together in a synaptic complex that contains two DNA-PKCS molecules. DNA synapsis was unaffected by wortmannin inhibition of the kinase. However, DNA synapsis led to full activation of the kinase, since kinase activity was cooperative with respect to DNA concentration. Thus, the kinase activity of DNA-PKCS is not required for synapsis, and the primary role of the kinase may be to regulate later steps in NHEJ. These results suggest a defined sequence of events during NHEJ, in which DNA-PKCS binds to the DNA ends upon recruitment by Ku, brings the ends together in a synaptic complex and then undergoes activation of its kinase. We will discuss a model in which Ku and other proteins align the DNA ends during later steps of NHEJ.
Results
DNA-PKCS mediates DNA synapsis
To determine whether purified DNA-PKCS could mediate synapsis of two DNA fragments, we exploited the strong interaction between biotin and neutravidin, a neutral derivative of streptavidin. We synthesized a blunt 42 bp double-stranded DNA fragment (f42) and derivatives that were either radiolabeled (f42*) or biotinylated (f42B) at one of the ends. DNA-PKCS was incubated at 4°C with f42* and f42B bound to neutravidin-conjugated agarose beads. The beads were then washed in low salt buffer. The radioactivity that remained attached to the beads represented the formation of a synaptic complex containing f42B and f42*.
Synapsis between f42B and f42* increased with increasing DNA-PKCS concentration, such that up to 45% of the f42* was pulled down with the agarose beads (Figure 1A). Non-specific associations between f42* and the beads were ruled out, because the signal depended on the addition of DNA-PKCS and the presence of biotin on the unlabeled DNA (Figure 1B). High salt concentrations disrupt most protein–protein and protein–DNA interactions. As expected, synapsis was lost with 530 mM NaCl (Figure 1B).

Fig. 1. DNA-PKCS mediates synapsis of DNA ends. (A) DNA synapsis versus DNA-PKCS concentration. In the pull-down assay, DNA-PKCS was incubated with neutravidin–agarose beads in binding buffer with two types of 42 bp DNA fragments: radiolabeled f42* (0.4 nM) and biotinylated f42B (4 nM). ‘Radioactivity pulled down’ is the fraction of total f42* that remained attached to the beads after washing, and signifies the amount of synapsis, which increased with increasing DNA-PKCS concentration. (B) Specificity of the pull-down assay. The pull-down assay was performed in the presence or absence of DNA-PKCS (14.4 nM), f42* (0.4 nM), f42B (4 nM) and unlabeled f42 (4 nM). The signal required formation of a complex containing DNA-PKCS, f42* and f42B. The complex was non-covalent, since it was disrupted by high salt concentration. (C) Effect of salt concentration on DNA synapsis. DNA-PKCS (14.4 nM), Ku (14.4 nM) or both were incubated with f42B (4 nM) and f42* (0.4 nM) in binding buffer at 30 or 150 mM NaCl. Synapsis occurred at both salt concentrations, but was not stimulated by Ku. (D) Effect of Ku on DNA synapsis. DNA-PKCS (14.4 nM), Ku (14.4 nM) or both were incubated with the longer DNA fragments f181B (4 nM) and f181* (0.4 nM) in a buffer of 10 mM HEPES pH 7.4, 20% glycerol and 80 mM NaCl. Under these modified conditions, Ku was able to stimulate DNA synapsis weakly.
Ku weakly stimulates DNA synapsis
Ku was tested in the pull-down assay to investigate its effect on DNA synapsis (Figure 1C). As shown in Figure 1A and B, experiments were performed with 42 bp DNA fragments in a Tris-based buffer containing 5% glycerol and 30 mM NaCl. In addition, the salt concentration was increased to the physiological level of 150 mM NaCl. Synapsis mediated by DNA-PKCS still occurred, albeit less efficiently than at 30 mM NaCl. Under both conditions, Ku failed to mediate synapsis of DNA ends either by itself or when added to DNA-PKCS (Figure 1C).
To determine whether an effect of Ku might be detected under other conditions, we varied the assay conditions. An effect was observed when we used a 10 mM HEPES pH 7.4 buffer with 20% glycerol and 80 mM NaCl, and increased the DNA length to 181 bp to promote binding of both DNA-PKCS and Ku to the same DNA fragment. Under these conditions, Ku was able to mediate DNA synapsis, although not as effectively as DNA-PKCS (Figure 1D). When both Ku and DNA-PKCS were present, synapsis was ∼1.5-fold greater than with DNA-PKCS alone, after background subtraction. Ku and DNA-PKCS could be acting independently to bring DNA ends together. Alternatively, this effect could be due to Ku stimulating DNA-PKCS-mediated synapsis. We turned to electron microscopy to address this question.
Electron microscopy reveals DNA synapsis mediated by two DNA-PKCS molecules
Because DNA-PKCS binds DNA at the ends (Hammarsten and Chu, 1998; Yoo and Dynan, 1999), we hypothesized that the pull-down assay was detecting a synaptic complex of DNA fragments bridged end-to-end by DNA-PKCS. To test this hypothesis, DNA-PKCS and/or Ku were incubated with a linear 3.5 kb DNA molecule with non-complementary 5′ overhangs. The resulting complexes were prepared for electron microscopy by incubating the DNA and proteins under conditions optimized in the pull-down assay to reveal effects of both Ku and DNA-PKCS on synapsis (10 mM HEPES pH 7.4, 20% glycerol, 80 mM NaCl). The samples were fixed with 0.6% glutaraldehyde for 5 min and free protein was eliminated by chromatography over a Bio-Gel A-5m column. Samples were then mounted on carbon-coated copper foils and rotary shadowcast with tungsten (see Materials and methods).
More than 200 DNA molecules were scored in each experiment (Table I; Figure 2). When DNA-PKCS was incubated with linear DNA at an 11:1 molar ratio of protein to DNA, 50.8% of the DNA contained DNA-PKCS bound to one end, 10.2% of the DNA contained DNA-PKCS bound to both ends (Figure 2A and E, respectively) and 17.6% of the DNA molecules were protein free (Figure 2B). Most significantly, 16.3% of the DNA molecules formed circles bound by DNA-PKCS (Figure 2C and D). At the DNA concentration used here (2 nM), intramolecular synapsis of ends on the same DNA molecule should be strongly favored over intermolecular synapsis. As predicted, two DNA molecules joined end-to-end by DNA-PKCS were observed at a very low frequency (1.6%). When the linear DNA was prepared for electron microscopy in the absence of protein, less than 1% of the DNA molecules formed circular structures spontaneously.
Table I. Electron microscopic analysis of DNA incubated with DNA-PKCS and Ku.
| DNA–protein complex | DNA-PKCS (%) | Ku (%) | DNA-PKCS + Ku (%) |
|---|---|---|---|
| DNA with one end bound by protein | 50.8 | 31 | 18.2 |
| DNA with both ends bound by protein | 10.2 | 3.7 | 41.5 |
| Circularized DNA bound by protein | 16.3 | 1.9 | 29.7 |
| DNA bound internally by protein | 1 | 12 | 0.6 |
| DNA not bound by protein | 17.6 | 47.2 | 3.5 |
| Two DNA molecules joined by protein | 1.6 | 0 | 1.3 |
| DNA–protein aggregates | 2.6 | 4.2 | 1.9 |
Linearized plasmids were incubated with protein as described (see Materials and methods). Percentages reflect the distribution of DNA configurations after incubation with DNA-PKCS, Ku or both.

Fig. 2. Electron microscopic visualization of DNA-PKCS and Ku associated with DNA. Linearized plasmid DNA (100 ng, 2 nM) terminating in 5′ non-complementary, single-stranded overhangs was incubated with either DNA-PKCS (22 nM), Ku (22 nM) or both proteins, and visualized by electron microscopy. DNA–protein complexes are displayed in order of observed abundance from top to bottom for each of the reactions. Bar = 1 kb DNA. For quantification of the distribution of DNA molecules, see Table I. (A–E) Linearized plasmid DNA incubated with DNA-PKCS. (A) DNA with DNA-PKCS bound to one end. (B) Unbound DNA. (C and D) DNA circularized by DNA-PKCS. (E) DNA with DNA-PKCS bound to both ends. (F–J) Linearized plasmid DNA incubated with Ku. (F) Unbound DNA. (G) DNA with Ku bound to one end. (H and I) DNA with Ku translocated to internal sites. (J) DNA with Ku bound to both ends. (K–O) Linearized plasmid DNA incubated with DNA-PKCS and Ku. DNA-PKCS is much larger than Ku; therefore, we are unable to determine by this technique whether the protein represents DNA-PKCS alone or DNA-PKCS plus Ku. (K) DNA with protein bound to both ends. (L and M) DNA circularized by protein. (N) DNA with protein bound to one end. (O) Unbound DNA.
When Ku was incubated with linear DNA at the same 11:1 molar ratio of protein to DNA, protein particles significantly smaller than those observed with DNA-PKCS were seen bound to the DNA. Nearly half of the linear DNA (47.2%) was not bound by Ku (Figure 2F). The remainder of the linear DNA was bound by Ku at one end (31%; Figure 2G), at both ends (3.7%; Figure 2J) or at internal sites (12%; Figure 2H and I). Some of the DNA molecules bound internally by Ku were associated with a single mass of Ku (Figure 2H), while other DNA molecules were bound by several distinct masses of Ku (Figure 2I). Such internal binding may be explained by the translocation of Ku along the DNA, a phenomenon predicted by biochemical experiments (de Vries et al., 1989; Yaneva et al., 1997). Only 1.9% of the DNA molecules were found in a circular configuration when incubated with Ku alone (Table I).
When Ku (22 nM) and DNA-PKCS (22 nM) were incubated with linear DNA (2 nM), 29.7% of the DNA molecules formed circles bound by a mass of protein (Figure 2L and M). This corresponded to a 1.8-fold increase in circularization when both Ku and DNA-PKCS were added compared with the effect of DNA-PKCS alone. Much of this increase might be due to more efficient recruitment of DNA-PKCS to the DNA ends, since there was a 4-fold increase in the percentage of DNA with both ends bound by protein. The protein complexes associated with the circularized DNA were too large to be attributed to Ku alone and were consistent with one or two particles of DNA-PKCS. The resolution of our electron microscope method was not adequate to determine whether the complex also contained Ku. These results suggest that Ku stimulates the synapsis of DNA ends by facilitating the recruitment of DNA-PKCS to DNA ends rather than by acting alone.
Quantitative analysis of electron microscopic images allowed us to assess the stoichiometry of the DNA–protein complexes. Several complexes revealed what appeared to be two individual DNA-PKCS molecules forming a bridge between two DNA ends (Figure 3). To confirm these observations, we measured the projected surface area of individual protein complexes bound either to the ends of linear DNA or to circularized DNA molecules (Figure 4). When DNA-PKCS was incubated with DNA in the absence of Ku, the average projected surface area of protein complexes bound to circularized DNA was 1.8-fold greater than that of protein complexes bound to DNA ends. When both DNA-PKCS and Ku were incubated with the DNA, the average projected surface area of the protein complexes bound to circularized DNA was 1.7-fold greater than for end-bound protein complexes. These results indicate that DNA synapsis involves two DNA-PKCS molecules. There remains the formal possibility that the complexes at the ends of the linear DNA consist of two DNA-PKCS molecules and that the circle junctions therefore contain four DNA-PKCS molecules. However, the DNA-PKCS molecules bound to DNA ends did not have a dimeric appearance in the electron microscope images. Furthermore, the appearance of the DNA-PKCS molecules at the DNA ends was consistent with a 465 kDa DNA-PKCS monomer, based on our previous visualization of similar-sized DNA binding proteins such as topoisomerase II (Vologodskii et al., 2001).

Fig. 3. Electron microscopic image of DNA synapsis mediated by DNA-PK. Linearized plasmid DNA (100 ng, 2 nM) was incubated with DNA-PKCS (22 nM) and Ku (22 nM) and visualized by electron microscopy. Each end of the DNA molecule in the upper left is bound to a single protein complex. The DNA molecule in the lower right has been circularized with what appears to be two protein complexes bound to the ends of the DNA. Bar = 1 kb DNA.

Fig. 4. Distribution of projected surface areas for protein complexes bound to DNA. (A) DNA incubated with DNA-PKCS. (B) DNA incubated with DNA-PKCS and Ku. The top histograms display the distribution of projected surface area for end-bound protein, and the bottom histograms display the distribution for circle-bound protein. Surface area measurements are in the same relative units for both panels. Note the difference in scale on the y-axis for (A) and (B). The mean projected surface areas of circle-bound protein are 1.8- and 1.7-fold larger than the projected surface areas of end-bound protein in (A) and (B), respectively, indicating that DNA synapsis involves two DNA-PKCS molecules.
The kinase activity of DNA-PKCS is not required for DNA synapsis
To determine whether DNA-PKCS kinase activity was required for DNA synapsis, we performed the pull-down assay under kinase-permissive and kinase-inhibitory conditions (Figure 5A). Wortmannin binds covalently to the active site of PI3K-like kinase domains and inhibits the kinase activity of DNA-PKCS with an IC50 of 0.25 µM (Hartley et al., 1995; Sarkaria et al., 1998; Izzard et al., 1999). Purified DNA-PKCS was incubated at 4°C with or without 10 µM wortmannin. To assess the effect of kinase activity, the pull-down assay was performed as in Figure 1, except for the addition of ATP, MgCl2 and peptide substrate, and an increase in the incubation temperature to 20°C. DNA-PKCS can also undergo autophosphorylation, but the effect of autophosphorylation is minimal with the saturating amount of peptide substrate used here (Chan and Lees-Miller, 1996). Under these conditions, synapsis was unaffected by wortmannin (Figure 5A, upper panel). Efficiency of synapsis was decreased ∼3-fold at 20°C compared with parallel reactions at 4°C (data not shown), probably due to the instability of DNA-PKCS at higher temperatures (O.Hammarsten, L.DeFazio and G.Chu, unpublished observations).

Fig. 5. Role of DNA-PKCS kinase activity in synapsis. (A) DNA-PKCS kinase activity is not required for DNA synapsis. DNA-PKCS (14.4 nM) was incubated with f42* and f42B in kinase buffer plus ATP and peptide substrate in the presence or absence of wortmannin (10 µM). The upper panel shows the pull-down assay, in which DNA synapsis was unaffected by wortmannin. The lower panel shows the gel-based kinase assay, in which phosphorylation of the peptide substrate was completely inhibited by wortmannin. (B) Activation of DNA-PKCS by DNA is cooperative. The upper panel shows the gel-based kinase assay, in which DNA-PKCS (5.8 nM) was incubated with f42 (0.04–2.05 nM, increasing in 1.3-fold increments) and synthetic peptide (285 µM) in kinase buffer. In the lower panel, kinase activation was plotted as a function of DNA concentration. The inset shows kinase activity for the 11 subsaturating DNA concentrations in a log–log plot. The arrows represent the highest DNA concentration used in the linear and log–log plots. The slope of 1.7 demonstrates significant cooperativity in the activation of DNA-PKCS by DNA.
To confirm successful inhibition of the kinase under these conditions, phosphorylation of the peptide was measured at 20°C in a parallel reaction containing [γ-32P]ATP. We devised an assay designed to detect very low levels of kinase activity. Instead of measuring peptide phosphorylation by the standard filter binding assay (described in Anderson and Lees-Miller, 1992), we separated phosphorylated peptide from excess [γ-32P] ATP by non-denaturing PAGE and quantitated activity by phosphoimager (see Materials and methods). This assay was at least 10 times more sensitive than filter binding (data not shown). In the assay, kinase activity was inhibited by 99% in the presence of 10 µM wortmannin (Figure 5A, lower panel). Thus, kinase activity is not required for DNA synapsis.
Synapsis is required for full activation of the DNA-PKCS kinase
Activation of the DNA-PKCS kinase requires binding to DNA ends, and at low salt concentrations both DNA binding and kinase activation occur in the absence of Ku (Yaneva et al., 1997; Hammarsten and Chu, 1998). To determine whether one or two DNA ends are required for kinase activation, we incubated DNA-PKCS with peptide substrate, [γ-32P]ATP and increasing concentrations of f42 DNA. To detect kinase activity at low DNA concentrations, we used the highly sensitive gel-based assay described in the previous paragraph and in Materials and methods.
Kinase activity showed evidence of cooperativity, increasing with DNA concentration along a sigmoidal curve (Figure 5B). A log–log plot of the data revealed a slope of 1.7 (Figure 5B, lower panel inset). When f42 was replaced with a 32 bp DNA fragment with five bases of single-stranded DNA on both the 3′ and 5′ ends (f32-ss3′/5′), a slope of 1.7 was again observed (data not shown). A slope of 2 implies that two DNA molecules are required for kinase activation. A slope of <2 could be observed if DNA-PKCS is weakly activated upon binding to one DNA end. Alternatively, the binding step of DNA-PKCS to a DNA end could limit the rate of formation of the final synaptic complex. In either case, this result demonstrates that full activation of DNA-PKCS involves more than one DNA molecule, and occurs only after formation of the synaptic complex.
Discussion
Roles of DNA-PKCS and Ku in the synapsis of DNA ends
After a DNA DSB is created, the broken ends must be brought together before processing and ligation can occur. Numerous studies have shown that DNA-PKCS is required for NHEJ (reviewed in Chu, 1997; Smith and Jackson, 1999). DNA-PKCS binds DNA ends, and upon binding, undergoes activation of its kinase. However, the biochemical role of DNA-PKCS in NHEJ has remained obscure.
To clarify the role of DNA-PKCS, we used a biochemical pull-down assay and electron microscopy to demonstrate that DNA-PKCS mediates synapsis of DNA ends. The synaptic activity of DNA-PKCS did not require cohesive DNA ends, since the DNA fragments used in the pull-down assay were blunt, and the plasmids used in electron microscopy were linearized to give non-complementary overhangs.
On the other hand, DNA-PKCS is capable of holding cohesive DNA ends in a ligatable configuration. DNA-PKCS preserved the ligation efficiency of Xrcc4/ligase IV acting on a 400 bp DNA fragment with cohesive ends, while altering the outcome of ligation from intramolecular to intermolecular ligation products (Chen et al., 2000). Thus, binding of DNA-PKCS prevented the DNA fragment from forming a closed circle for the favored intramolecular ligation reaction. Instead, DNA-PKCS preserved ligation efficiency despite being restricted to the less favored intermolecular ligation reaction.
We examined the stoichiometry of DNA-PKCS bound to DNA using electron microscopy. Visual inspection of the images and calculation of protein surface area projections indicated that a single DNA-PKCS molecule will bind to a DNA end. However, when two DNA ends are brought together, the synaptic complex contains two DNA-PKCS molecules (Figures 3 and 4). A previous atomic force microscope study raised the possibility that DNA-PKCS might bring two DNA ends together, although it did not resolve the presence of two DNA-PKCS molecules in the synaptic complex (Yaneva et al., 1997).
In solution, DNA-PKCS exists primarily as a monomer. DNA-PK catalytic activity in HeLa cell extracts elutes from a gel filtration column as a single peak between marker proteins of 232 and 669 kDa (Dvir et al., 1993), consistent with the monomeric molecular weight of 465 kDa. DNA-PKCS also exists as a monomer in gel filtration experiments with purified protein (O.Hammarsten and G.Chu, unpublished data). Furthermore, a cryo-electron microscopy reconstruction of the DNA-PKCS structure reveals a monomer in solution (Chiu et al., 1998). These results support the conclusion that DNA-PKCS dimerizes only when it forms a synaptic complex with two DNA ends.
Since DNA-PKCS mediates DNA synapsis, what is the role of Ku in this process? We confirmed a previous report that Ku can stimulate the synapsis of DNA molecules (Ramsden and Gellert, 1998). In the previous report, synapsis was relatively weak, since only 8% of the radiolabeled DNA was pulled down with biotinylated DNA in the presence of Ku. In our experiments, Ku was capable of mediating limited DNA synapsis, but only after DNA length was increased to 181 bp and glycerol in the binding buffer was increased to 20%. However, our electron microscope experiments failed to detect DNA ends brought together by Ku. A previous atomic force microscope study found that Ku formed clusters along linearized plasmid DNA and induced the formation of loops created by end-to-side juxtapositions of the DNA (Cary et al., 1997). These structures may have produced the signals observed in the pull-down assays.
The pull-down assay and electron microscopy showed that synapsis was greater with both DNA-PKCS and Ku than with DNA-PKCS alone. Furthermore, electron microscopy suggested that all of the synaptic complexes contained DNA-PKCS. Thus, Ku acting alone has limited activity in bringing DNA ends together, but does enhance the ability of DNA-PKCS to mediate DNA synapsis, perhaps by recruiting DNA-PKCS to the DNA ends.
Mechanism for DNA-PKCS kinase activation
Because the kinase activity of DNA-PKCS is likely to be involved in DSB repair (Baumann and West, 1998; Kurimasa et al., 1999), we examined the role of the kinase in DNA synapsis. Our results show that DNA-PKCS undergoes activation of its kinase only after synapsis occurs. In the pull-down assay, kinase activity was not required for synapsis. However, kinase activity was cooperative with DNA concentration, suggesting that synapsis is required for full kinase activation. We conclude that in the early steps of NHEJ, DNA-PKCS binds to the DNA ends upon recruitment by Ku, brings the ends together in a synaptic complex, and then undergoes activation of its kinase (Figure 6).
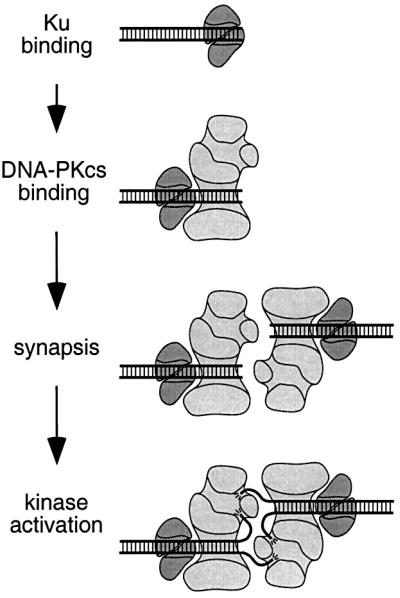
Fig. 6. Model for DNA-PKCS binding and kinase activation. Ku binding: the Ku heterodimer binds to DNA ends, forming a ring structure (depicted in cross-section) that encircles the DNA (Walker et al., 2001). DNA-PKCS binding: DNA-PKCS is recruited to the DNA end by Ku, which then translocates inward. Double-stranded DNA binds to an open channel in DNA-PKCS. Synapsis: two DNA-PKCS molecules bring two DNA ends together to form a synaptic complex. Kinase activation: one single-stranded DNA end is inserted into an opening to the enclosed cavity adjacent to the open channel in DNA-PKCS. The other single-stranded end is inserted into an opening to the enclosed cavity on the opposing DNA-PKCS molecule. The interactions between DNA-PKCS and single-stranded DNA ends stabilize the synaptic complex and fully activate the kinase.
Suggestions for how the DNA-PKCS kinase is activated by DNA have come from its structure (Leuther et al., 1999). The 22 Å electron crystallography structure contains an open channel large enough to accommodate double-stranded DNA, and an enclosed cavity just large enough to permit the insertion of single-stranded DNA. The enclosed cavity has three openings, one of which is adjacent to the open channel.
These structural features suggest that DNA-PKCS interacts with both single- and double-stranded DNA. Competition experiments demonstrate that DNA-PKCS binds to single- and double-stranded DNA via separate but interacting sites (Leuther et al., 1999). The kinase is activated most efficiently by a double-stranded DNA molecule with five bases of unpaired single strands at both 3′ and 5′ ends (Hammarsten et al., 2000). Indeed, one single-stranded DNA end is not sufficient for kinase activation (Martensson and Hammarsten, 2002). Since full kinase activation does not occur until after DNA synapsis, we propose that activation of DNA-PKCS occurs only after single-stranded DNA ends are threaded into two openings to the enclosed cavity (Figure 6). The interaction of a single-stranded end with the opposing DNA-PKCS molecule may stabilize the synaptic complex as well as stimulate the kinase.
Model for NHEJ
Our findings suggest a model for NHEJ with a defined order of events. Ku is the first protein to bind the broken DNA end. Upon recruiting DNA-PKCS to the DNA end, Ku translocates inward by one helical turn, leaving DNA-PKCS bound directly to the DNA end (Yoo and Dynan, 1999). In this conformation, DNA-PKCS is positioned to mediate synapsis, while Ku stabilizes binding of DNA-PKCS to the DNA ends. In vitro, DNA synapsis mediated by DNA-PKCS is less efficient at physiological salt concentration than at low salt concentration. In vivo, the synaptic complex may be stabilized by protein concentration effects or by additional proteins.
DNA synapsis activates the DNA-PKCS kinase, which phosphorylates proteins required for further steps in the reaction. Phosphorylation of Artemis may regulate its nuclease activity and facilitate processing of the DNA ends (Ma et al., 2002). The Rad50–Mre11–Nbs1 complex also has a nuclease activity (Paull and Gellert, 1998, 2000; Trujillo et al., 1998). During end-processing, multimers of Rad50–Mre11 may tether the two DNA ends in a complex, as suggested by scanning electron microscopy (de Jager et al., 2001). At a later step, DNA-PKCS phosphorylates Xrcc4 and stimulates the ligation activity of Xrcc4– ligase IV (Leber et al., 1998). DNA-PKCS may then undergo autophosphorylation and dissociate from the DNA (Chan and Lees-Miller, 1996).
After dissociation of DNA-PKCS, Ku may align DNA ends to facilitate ligation by Xrcc4–ligase IV (Ramsden and Gellert, 1998; McElhinny et al., 2000; Walker et al., 2001). The Xrcc4–ligase IV complex itself can bridge DNA ends, but only when the ends are cohesive (Chen et al., 2000). In the absence of DNA-PKCS, the weak end-bridging activities of Ku and Xrcc4–ligase IV may be sufficient to provide residual NHEJ activity, consistent with the milder phenotype observed in DNA-PKCS-deficient cells.
In conclusion, our results suggest that DNA-PKCS is the protein that brings DNA ends together for NHEJ. Full kinase activity occurs only after formation of the synaptic complex, and thus appears to be important for subsequent steps in the NHEJ reaction. We have presented a model in which the DNA bridging activities of Rad50–Mre11– Nbs1, Ku and Xrcc4–ligase IV are involved in maintaining the synaptic complex during later steps in the NHEJ reaction, which involve processing, alignment and ligation of the ends. Future experiments must address how these proteins are regulated by the DNA-PKCS kinase. Only then will we understand the mechanism of NHEJ for DSB repair, V(D)J recombination and class switch recombination.
Materials and methods
Purification of DNA-PKCS and Ku
DNA-PKCS and Ku were purified from 30 l of HeLa cell pellets (National Cell Culture Center, Minneapolis, MN) using a modification of a previously described procedure (Hammarsten and Chu, 1998). Briefly, cells were extracted by mixing in a blender in buffer A (50 mM Tris–HCl pH 7.4, 5 mM EDTA, 10% glycerol, 0.02% NaN3, protease inhibitors) supplemented with 600 mM NaCl, 2.5 mM EGTA and 0.02% Tween-20. High Q resin (80 ml; Bio-Rad, Hercules, CA) equilibrated in buffer A supplemented with 600 mM NaCl was added to the extract and the resin slurry was poured through a column. Protein was concentrated by ammonium sulfate precipitation, dissolved in buffer A supplemented with 100 mM NaCl and 2.5 mM EGTA, and dialyzed against 0.5× buffer A supplemented with 250 mM NaCl. The dialyzate was applied to a 1 ml oligonucleotide column (Hammarsten and Chu, 1998). DNA-PKCS and Ku were step-eluted in buffer A supplemented with 700 mM NaCl. Relevant fractions were concentrated by ammonium sulfate precipitation, dissolved in gel filtration running buffer (buffer A supplemented with 350 mM NaCl, 20% glycerol) and applied to an 80 ml gel filtration column (Sephacryl S300; Amersham Biosciences, Piscataway, NJ). The fractions containing DNA-PKCS and Ku were then applied to a 0.5 ml f32 column (Hammarsten and Chu, 1998). DNA-PKCS was step-eluted in buffer A supplemented with 300 mM NaCl and Ku in buffer A supplemented with 1000 mM NaCl. DNA-PKCS- or Ku-containing fractions were pooled separately and concentrated on a 0.7 ml High Q resin (Bio-Rad). Protein was step-eluted with buffer A supplemented with 300 mM NaCl, 20% glycerol. Proteins were judged to be purified to homogeneity by Coomassie Blue staining. Western blotting showed that DNA-PKCS preparations contained <1% Ku and that Ku preparations contained <1% DNA-PKCS (data not shown). Prior to use, Ku and DNA-PKCS were diluted in storage buffer [buffer A supplemented with 250 mM NaCl, 50% glycerol, 0.3 mg/ml bovine serum albumin (BSA)].
DNA preparation
The 42 base oligonucleotide (5′-AACCTAGATCGTAGTTGTAC CCCATGGTGGACGCGTGCGGCC-3′) and its complement were synthesized and purified to remove truncated synthesis products (Poly-Pak™ cartridges; Glen Research, Sterling, VA). To make biotinylated f42, a biotin group was attached to the 3′ end of the 42 base oligonucleotides described above (Glen Research; Stanford Protein and Nucleic Acid Facility, Stanford, CA). Double-stranded f42 DNA was made by annealing of the complementary oligonucleotides in a 1:1 molar ratio in TE buffer (10 mM Tris–HCl pH 7.6, 0.1 mM EDTA). Double-stranded DNA was separated from single-stranded DNA by non-denaturing PAGE, ethidium bromide staining and excision of the appropriate band. The DNA was electroeluted as described previously (Leuther et al., 1999) and dialyzed against TE buffer (Spectra/Por® MicroDialyzer; Spectrum Laboratories, Rancho Dominguez, CA). To make radiolabeled f42 and to confirm its successful purification, DNA was labeled by incubation with [γ-32P]ATP and T4 polynucleotide kinase (New England BioLabs, Beverly, MA). Unincorporated [γ-32P]ATP was removed by gel filtration (Sepharose G-50; Amersham Biosciences). Samples were resolved by 20% non-denaturing PAGE and successful purification was confirmed when a single band was detected by phosphoimager analysis (data not shown).
To make f32-ss3′/5′, a 32 bp DNA fragment with five bases of T on both the 3′ and 5′ ends, the 42 base oligonucleotide (5′-TTT TTAACCTAGATCGTAGTTGTACCCCATGGTGGACTTTTT-3′) was annealed to an oligonucleotide that was complementary to the middle 32 bases of the above sequence but also contained five bases of T on the ends (5′-TTTTTGTCCACCATGGGGTACAACTACGATCTAGGTTT TTTT-3′) and purified as described for f42.
Double-stranded 181 bp DNA was made by PCR amplification of a region of plasmid pBluescript II SK+ (Stratagene, La Jolla, CA) using Pfu Turbo DNA polymerase (Stratagene). For biotinylated 181 bp DNA, one of the primers for PCR amplification contained a biotin group on the 5′ end (MWG Biotech, High Point, NC). PCR products were purified using QIAquick PCR Purification Kit (Qiagen, Valencia, CA) and analyzed by agarose gel electrophoresis and ethidium bromide staining. Double-stranded 181 bp DNA was radiolabeled and successful purification was confirmed by non-denaturing PAGE and phosphoimager analysis.
Pull-down assay
DNA synapsis was measured by a pull-down assay using neutravidin-coated agarose beads (Pierce, Rockford, IL). Neutravidin is a neutral derivative of streptavidin and forms a tight interaction with biotin. Neutravidin-conjugated agarose beads (20 µl slurry; Pierce) were added to a 500 µl Eppendorf tube and washed twice in wash buffer (100 µl, 10 mM Tris–HCl pH 7.4, 1 mM EDTA, 5% glycerol, 10 mM β-mercaptoethanol, 1 mg/ml BSA) with centrifugation for 1 min at 3300 g. Biotinylated f42 (f42B; 4 nM) was added to the beads in wash buffer (16 µl) and allowed to bind to the neutravidin on the beads. Radiolabeled f42 (f42*; 0.4 nM) and DNA-PKCS or Ku were added, so the final binding buffer was wash buffer plus 30 mM NaCl, 4% glycerol. For experiments at physiological salt concentration, binding buffer was adjusted to 150 mM NaCl. The tubes were rotated for 10–30 min at 4°C and centrifuged for 1 min at 3300 g. Supernatant was removed, and beads were washed several times in excess wash buffer. Supernatants and beads were spotted on filter paper and analyzed by phosphoimager or collected in tubes and measured by scintillation counting. Synapsis between f42B and f42* was measured as the fraction of radioactivity pulled down with the beads. BSA (1 mg/ml) was included in this assay to minimize the possibility that f42* might be pulled down by a non-specific interaction of DNA-PKCS with the beads.
For some experiments containing Ku, binding was carried out in HEPES buffer [10 mM HEPES pH 7.4, 1 mM EDTA, 5% glycerol, 0.1 mM dithiothreitol (DTT), 1 mg/ml BSA]. Storage buffer was added to increase the glycerol concentration to 20% and NaCl to 80 mM in the binding reaction. After incubation, samples were washed in HEPES buffer and analyzed by scintillation counting.
We demonstrated that the synaptic complex of DNA-PKCS and DNA was stable for the pull-down assay as follows. Beads attached to the synaptic complex were added to wash buffer (500 µl) and then rotated at 4°C. Supernatant was removed after centrifugation from parallel reactions at different times, and radioactivity in the beads and supernatant was analyzed by scintillation counting. Dissociation of the synaptic complex from the beads was measured as the transfer of radiolabeled f42* to the supernatant. Less than 10% of the activity was lost from the beads after 30 min, which is longer than the time it took to complete the washes during the pull-down assay.
Kinase assay
A highly sensitive assay was devised to measure DNA-PKCS kinase activity. DNA-PKCS (5.8–14.4 nM) was incubated with the specific peptide substrate EPPLSQEAFADLWKK (285 µM; Promega, Madison, WI) in the presence of f42 DNA (5 nM), [γ-32P]ATP (0.6 µCi), and saturating concentrations of ATP (200 µM) in kinase buffer (binding buffer supplemented with 4 mM MgCl2). Reactions were incubated at 30°C for 4 min, during which phosphorylation of the peptide increased linearly with time (data not shown), and then quenched by addition of excess (4 mM) cold ATP. Phosphorylated peptide was resolved from excess [γ-32P]ATP by electrophoresis in a non-denaturing 20% polyacrylamide gel. Peptide phosphorylation was analyzed by phosphoimager. For experiments with wortmannin, either wortmannin (10 µM) dissolved in dimethylsulfoxide (DMSO), or the same volume of DMSO was added to the reaction. For experiments examining the number of DNA molecules necessary for DNA-PKCS kinase activation, peptide phosphorylation was measured at DNA concentrations ranging from 0.04 to 2.05 nM.
Preparation of DNA–protein complexes and electron microscopy
The plasmid pBluescript II SK+ (Stratagene) was linearized with BamHI and HindIII to give non-complementary 5′ overhangs. The linearized plasmid DNA (100 ng, 2 nM) was incubated with DNA-PKCS (22 nM) or Ku (22 nM) for 10 min at 20°C in a buffer containing 10 mM HEPES pH 7.4, 0.1 mM EDTA, 5% glycerol and 1 mM DTT. Addition of protein in storage buffer raised the glycerol concentration to 20% and NaCl concentration to 80 mM. Binding reactions were terminated by adding glutaraldehyde to 0.6% for 5 min at 20°C. The samples were applied to Bio-Gel A-5m (Bio-Rad) columns equilibrated with TE buffer. Filtered samples were mixed with a spermidine-containing buffer, adsorbed to glow-charged thin carbon-coated copper foils, dehydrated through a series of water–ethanol washes and rotary shadowcast with tungsten as described previously (Griffith and Christiansen, 1978). Samples were examined in a CM12 instrument. Micrographs were scanned from negatives using a Nikon multi-format film scanner. The contrast was optimized, and panels in Figures 2 and 3 were arranged using Adobe Photoshop. Morphometric measurements were carried out using a Summagraphics digitizer with software developed by J.D.Griffith. Images of single molecules were captured from the negatives using a Sony CCD camera and the relative areas measured using NIH Image software.
Acknowledgments
Acknowledgements
We thank Joe Budman, Ola Hammarsten and Bob Lehman for helpful discussions, and Timothy Maynor for assisting with analysis of projected surface areas in the electron micrographs. L.G.D. was supported by a grant from the National Science Foundation. This work was supported by NIH grant RO1 GM 58120 to G.C. and NIH grants RO1 GM 31819 and CA 70343 to J.D.G.
References
- Anderson C.W. and Lees-Miller,S.P. (1992) The nuclear serine/threonine protein kinase DNA-PK. Crit. Rev. Eukaryot. Gene Expr., 2, 283–314. [PubMed] [Google Scholar]
- Baumann P. and West,S.C. (1998) DNA end-joining catalyzed by human cell-free extracts. Proc. Natl Acad. Sci. USA, 95, 14066–14070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cary R.B., Peterson,S.R., Wang,J., Bear,D.G., Bradbury,E.M. and Chen,D.J. (1997) DNA looping by Ku and the DNA-dependent protein kinase. Proc. Natl Acad. Sci. USA, 94, 4267–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casellas R. et al. (1998) Ku80 is required for immunoglobulin isotype switching. EMBO J., 17, 2404–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan D.W. and Lees-Miller,S.P. (1996) The DNA-dependent protein kinase is inactivated by autophosphorylation of the catalytic subunit. J. Biol. Chem., 271, 8936–8941. [DOI] [PubMed] [Google Scholar]
- Chen L., Trujillo,K., Sung,P. and Tomkinson,A.E. (2000) Interactions of the DNA ligase IV–Xrcc4 complex with DNA ends and the DNA-dependent protein kinase. J. Biol. Chem., 275, 26196–26205. [DOI] [PubMed] [Google Scholar]
- Chiu C., Cary,R., Chen,D., Peterson,S. and Stewart,P. (1998) Cryo-EM imaging of the catalytic subunit of the DNA-dependent protein kinase. J. Mol. Biol., 284, 1075–1081. [DOI] [PubMed] [Google Scholar]
- Chu G. (1997) Double-strand break repair. J. Biol. Chem., 272, 24097–24100. [DOI] [PubMed] [Google Scholar]
- de Jager M., van Noort,J., van Gent,D.C., Dekker,C., Kanaar,R. and Wyman,C. (2001) Human Rad50/Mre11 is a flexible complex that can tether DNA ends. Mol. Cell, 8, 1129–1135. [DOI] [PubMed] [Google Scholar]
- de Vries E., van Driel,W., Bergsma,W.G., Arnberg,A.C. and van der Vliet,P.C. (1989) HeLa nuclear protein recognizing DNA termini and translocating on DNA forming a regular DNA–multimeric protein complex. J. Mol. Biol., 208, 65–78. [DOI] [PubMed] [Google Scholar]
- Durocher D. and Jackson,S.P. (2001) DNA-PK, ATM and ATR as sensors of DNA damage: variations on a theme? Curr. Opin. Cell Biol., 13, 225–231. [DOI] [PubMed] [Google Scholar]
- Dvir A., Stein,L.Y., Calore,B.L. and Dynan,W.S. (1993) Purification and characterization of a template-associated protein kinase that phosphorylates RNA polymerase II. J. Biol. Chem., 268, 10440–10447. [PubMed] [Google Scholar]
- Gottlieb T.M. and Jackson,S.P. (1993) The DNA-dependent protein kinase: requirement for DNA ends and association with Ku antigen. Cell, 72, 131–142. [DOI] [PubMed] [Google Scholar]
- Grawunder U., Wilm,M., Xiantuo,W., Kulezla,P., Wilson,T.E., Mann,M. and Lieber,M.R. (1997) Activity of DNA ligase IV stimulated by complex formation with Xrcc4 protein in mammalian cells. Nature, 388, 492–494. [DOI] [PubMed] [Google Scholar]
- Griffith J.D. and Christiansen,G. (1978) Electron microscope visualization of chromatin and other DNA–protein complexes. Annu. Rev. Biophys. Bioeng., 7, 19–35. [DOI] [PubMed] [Google Scholar]
- Hammarsten O. and Chu,G. (1998) DNA-dependent protein kinase: DNA binding and activation in the absence of Ku. Proc. Natl Acad. Sci. USA, 95, 525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarsten O., DeFazio,L. and Chu,G. (2000) Activation of DNA-dependent protein kinase by single-stranded DNA ends. J. Biol. Chem., 275, 1541–1550. [DOI] [PubMed] [Google Scholar]
- Hanakahi L.A., Bartlet-Jones,M., Chappell,C., Pappin,D. and West,S.C. (2000) Binding of inositol phosphate to DNA-PK and stimulation of double-strand break repair. Cell, 102, 721–729. [DOI] [PubMed] [Google Scholar]
- Hartley K.O. et al. (1995) DNA-dependent protein kinase catalytic subunit: a relative of phosphatidylinositol 3-kinase and the ataxia telangiectasia gene product. Cell, 82, 849–856. [DOI] [PubMed] [Google Scholar]
- Izzard R.A., Jackson,S.P. and Smith,G.C.M. (1999) Competitive and noncompetitive inhibition of the DNA-dependent protein kinase. Cancer Res., 59, 2581–2586. [PubMed] [Google Scholar]
- Kurimasa A., Kumano,S., Boubnov,N., Story,M., Tung,C.S., Peterson,S. and Chen,D. (1999) Requirement for the kinase activity of human DNA-dependent protein kinase catalytic subunit in DNA strand break rejoining. Mol. Cell. Biol., 19, 3877–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leber R., Wise,T., Mizuta,R. and Meek,K. (1998) The Xrcc4 gene product is a target for and interacts with the DNA-dependent protein kinase. J. Biol. Chem., 273, 1794–1801. [DOI] [PubMed] [Google Scholar]
- Leuther K.K., Hammarsten,O., Kornberg,R.D. and Chu,G. (1999) Structure of DNA-dependent protein kinase: implications for its regulation by DNA. EMBO J., 18, 1114–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Otevrel,T., Gao,Y., Cheng,H.-L., Seed,B., Stamato,T.D., Taccioli,G.E. and Alt,F.W. (1995) The Xrcc4 gene encodes a novel protein involved in DNA double-strand break repair and V(D)J recombination. Cell, 83, 1079–1089. [DOI] [PubMed] [Google Scholar]
- Ma Y., Pannicke,U., Schwarz,K. and Lieber,M.R. (2002) Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell, 108, 781–794. [DOI] [PubMed] [Google Scholar]
- Manis J.P., Gu,Y., Lansford,R., Sonoda,E., Ferrini,R., Davidson,L., Rajewski,K. and Alt,F.W. (1998) Ku70 is required for late B cell development and immunoglobulin heavy chain class switching. J. Exp. Med., 187, 2081–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martensson S. and Hammarsten,O. (2002) DNA-dependent protein kinase catalytic subunit: structural requirements for kinase activation by DNA ends. J. Biol. Chem., 277, 3020–3029. [DOI] [PubMed] [Google Scholar]
- McElhinny S., Snowden,C., McCarville,J. and Ramsden,D. (2000) Ku recruits the Xrcc4–ligase IV complex to DNA ends. Mol. Cell. Biol., 20, 2996–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moshous D. et al. (2001) Artemis, a novel DNA double-strand break repair/V(D)J recombination protein, is mutated in human severe combined immune deficiency. Cell, 105, 177–186. [DOI] [PubMed] [Google Scholar]
- Paull T. and Gellert,M. (1998) The 3′ to 5′ exonuclease activity of Mre 11 facilitates repair of DNA double-strand breaks. Mol. Cell, 1, 969–979. [DOI] [PubMed] [Google Scholar]
- Paull T.T. and Gellert,M. (2000) A mechanistic basis for Mre11-directed DNA joining at microhomologies. Proc. Natl Acad. Sci. USA, 97, 6409–6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsden D.A. and Gellert,M. (1998) Ku protein stimulates DNA end joining by mammalian DNA ligases: a direct role for Ku in repair of DNA double-strand breaks. EMBO J., 17, 609–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathmell W.K. and Chu,G. (1994a) A DNA end-binding factor involved in double-strand break repair and V(D)J recombination. Mol. Cell. Biol., 14, 4741–4748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathmell W.K. and Chu,G. (1994b) Involvement of the Ku autoantigen in the cellular response to DNA double-strand breaks. Proc. Natl Acad. Sci. USA, 91, 7623–7627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolink A., Melchers,F. and Andersson,J. (1996) The SCID but not the RAG-2 gene product is required for S µ-S ε heavy chain class switching. Immunity, 5, 319–330. [DOI] [PubMed] [Google Scholar]
- Sarkaria J.N., Tibbetts,R.S., Busby,E.C., Kennedy,A.P., Hill,D.E. and Abraham,R.T. (1998) Inhibition of phosphoinositide 3-kinase related kinases by the radiosensitizing agent wortmannin. Cancer Res., 58, 4375–4382. [PubMed] [Google Scholar]
- Smider V. and Chu,G. (1997) The end-joining reaction in V(D)J recombination. Semin. Immunol., 9, 189–197. [DOI] [PubMed] [Google Scholar]
- Smider V., Rathmell,W.K., Lieber,M. and Chu,G. (1994) Restoration of X-ray resistance and V(D)J recombination in mutant cells by Ku cDNA. Science, 266, 288–291. [DOI] [PubMed] [Google Scholar]
- Smith G.C. and Jackson,S.P. (1999) The DNA-dependent protein kinase. Genes Dev., 13, 916–934. [DOI] [PubMed] [Google Scholar]
- Taccioli G.E. et al. (1994) Ku80: product of the Xrcc5 gene and its role in DNA repair and V(D)J recombination. Science, 265, 1442–1445. [DOI] [PubMed] [Google Scholar]
- Trujillo K.M., Yuan,S.S., Lee,E.Y. and Sung,P. (1998) Nuclease activities in a complex of human recombination and DNA repair factors Rad50, Mre11, and p95. J. Biol. Chem., 273, 21447–21450. [DOI] [PubMed] [Google Scholar]
- Vologodskii A., Zhang,W., Rybenkov,V., Podtelezhnikov,A., Subramanian,D., Griffith,J. and Cozzarelli,N. (2001) Mechanism of topology simplification by type II DNA topoisomerases. Proc. Natl Acad. Sci. USA, 98, 3045–3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker J.R., Corpina,R.A. and Goldberg,J. (2001) Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature, 412, 607–614. [DOI] [PubMed] [Google Scholar]
- Yaneva M., Kowalewski,T. and Lieber,M. (1997) Interaction of DNA-dependent protein kinase with DNA and with Ku: biochemical and atomic-force microscopy studies. EMBO J., 16, 5098–5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo S. and Dynan,W.S. (1999) Geometry of a complex formed by double strand break repair proteins at a single DNA end: recruitment of DNA-PKcs induces inward translocation of Ku protein. Nucleic Acids Res., 27, 4679–4686. [DOI] [PMC free article] [PubMed] [Google Scholar]