

Abstract
Type II DNA topoisomerases actively reduce the fractions of knotted and catenated circular DNA below thermodynamic equilibrium values. To explain this surprising finding, we designed a model in which topoisomerases introduce a sharp bend in DNA. Because the enzymes have a specific orientation relative to the bend, they act like Maxwell's demon, providing unidirectional strand passage. Quantitative analysis of the model by computer simulations proved that it can explain much of the experimental data. The required sharp DNA bend was demonstrated by a greatly increased cyclization of short DNA fragments from topoisomerase binding and by direct visualization with electron microscopy.
Type II topoisomerases are essential enzymes that pass one DNA through another and thereby remove DNA entanglements. They make a transient double-stranded break in a gate segment (G segment) that allows passage by another segment (T segment) of the same or another DNA molecule (reviewed in refs. 1 and 2). Thus, these enzymes have the potential to convert real DNA molecules into phantom chains that freely pass through themselves to generate an equilibrium distribution of knots, catenanes, and supercoils.
The actual picture is more complex and more interesting. The observed steady-state fractions of knotted, catenated, and supercoiled DNAs produced by type II topoisomerases are up to two orders of magnitude lower than at equilibrium (3). Thermodynamically, there is no contradiction in this finding because the enzymes use the energy of ATP hydrolysis. Active topology simplification by topoisomerases has an important biological consequence. It helps explain how topoisomerases can remove all DNA entanglements under the crowded cellular conditions which favor the opposite outcome. The challenge, though, is to understand how type II topoisomerases actively simplify DNA topology. Topology is a global property of circular DNA molecules, and yet it is determined by the much smaller topoisomerases, which can act only locally.
Two models have been suggested to explain active simplification of DNA topology. First, if type II topoisomerases corral the T segment within a small loop of DNA containing the G segment, active disentanglement would result (3). However, it was pointed out when this model was suggested (3) that to account for the large effects observed, the loop trapping would need substantial energy input from ATP hydrolysis for the transport of the DNA along the enzymes, and these enzymes are energetically efficient (4). Moreover, no direct experimental data supporting the model have been presented.
Second, a kinetic proofreading model proposed that two successive bindings of T segments are required for strand passage (5). The first binding event converts the enzyme bound with a G segment to an activated state. An assumption of the model is that segment collision in the knotted state occurs about 1/Pk times more often than in the unknotted state, where Pk is the equilibrium probability of knotting. Our computer simulations below show that this assumption is incorrect.
Here we suggest a model simpler than either of these two for the action of type II topoisomerases (6). Using computer simulations, we show that our model can explain much of the experimental data. We also describe experiments which demonstrate that a eukaryotic and a prokaryotic topoisomerase have a key feature of this model: they bend DNA sharply upon binding. We conclude that the mechanism we describe is used by type II topoisomerases to actively disentangle DNA.
Materials and Methods
Computer Analysis of the Enzyme Action.
Circular DNA was modeled as a discrete worm-like chain consisting of N rigid cylinders of equal length, as described in ref. 7. The values of the model parameters were for DNA in a 0.2 M NaCl solution. Hairpin and straight G segments were modeled by four and two adjacent cylinders, respectively, which maintained their geometry during the simulations, as shown in Fig. 1 Left. The Metropolis Monte Carlo procedure was used to simulate the equilibrium set of DNA conformations (7). A particular topology was preserved during a simulation run by rejecting all trial conformations that changed chain topology. The topology of each conformation was determined by calculating the Alexander polynomial Δ(t), for t = −1 (8). The fraction of the chains that have a segment juxtaposed with the G segment was estimated by direct inspection of the constructed sets. A segment was considered to be juxtaposed with a G segment if all distances between the ends of the testing segment and the ends of the G segment were less than a distance ρo, and the angle between the tested segment and the plane of the hairpin (or between the tested segment and the straight G segment) was larger than φo. Although the probabilities of juxtaposition depend on the values chosen for ρo and φo, for sufficiently small ρo the ratio of the probabilities for knotted and unknotted chains does not depend on this choice. In most of the calculations, we used ρo = 14 nm and φo = 35°. Local deformations of the chain, as diagramed in Fig. 1, were used to determine whether strand passage would change the topology of a particular conformation. Visual inspection of numerous examples showed that the deformations do provide passage of the T segment through the G segment. After the deformation, the topology of the new conformation was determined by calculating Δ(−1). Sets of up to 109 DNA conformations were simulated to obtain statistically reliable estimations of the required parameters.
Figure 1.

Test of topology change by strand passage. For each case where a hairpin G segment (red) was juxtaposed with a potential T segment, the topology of the new conformation resulting from strand passage was calculated. To perform the test, we replaced the actual G segment by a bypass (pink) of the T segment. (A) For a hairpin G segment, the bypass was obtained by a 180° rotation of the hairpin. (B) For a straight G segment, the bypass was a four-segment loop directed toward the potential T segment.
Cyclization Frequency of DNA.
Eight DNA fragments ranging in length from 190 bp to 204 bp and containing a common 190-bp sequence were prepared by using the PCR and HpaII digestion. The digested DNA was purified by gel electrophoresis and end-labeled with 32P by using T4 polynucleotide kinase. Ligation was conducted at 16°C in 20 mM Tris⋅HCl, pH 7.0/20 nM DNA/30 mM KCl/100 μg/ml BSA/1.8 mM ATP/10 mM MgCl2, and 8 units/μl T4 DNA ligase. In half of the reactions, 100 nM purified topoisomerase IV (9) was added 5 min before the addition of ligase. At measured times thereafter, samples were removed, the DNA was purified and analyzed by gel electrophoresis, and the extent of the reaction was quantified with a Fuji phosphorimager. The measured relative amounts of circular monomers, C(t), and linear dimers, D(t), were used to calculate j factors, a measure of cyclization efficiency, as
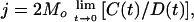 |
1 |
where t is the reaction time and Mo is the initial concentration of the DNA fragment (10).
Electron Microscopy (EM).
pBR322 DNA (200 ng) was incubated with either 10 ng of yeast topoisomerase II or 60 ng of Escherichia coli topoisomerase IV in 20 mM potassium phosphate (pH 7.5)/80 mM potassium acetate/2 mM 2-mercaptoethanol/5 mM MgCl2/1 mM ATP/20 μg/ml BSA for 5 min at room temperature. The purified yeast enzyme was provided by J. Lindsley (University of Utah, Salt Lake City, UT). After incubation, the samples were fixed by treatment with 0.6% glutaraldehyde for 5 min, adsorbed to thin carbon foils, washed with water and ethanol, air dried, and rotary shadowcast with tungsten (11). Samples were examined in a Philips CM12 electron microscope at 40 kV. Images on sheet film were scanned with a Nikon LS4500 film scanner and processed by using Adobe PHOTOSHOP software.
Results
The Model for Topoisomerase Action.
Pulleyblank suggested (12) that in simplifying DNA topology, type II topoisomerases work like Maxwell's famous demon, who sits near a door that separates a box into two parts, each part containing a gas. The demon opens the door only for molecules moving in one direction and thereby shifts the system away from equilibrium. The demon must consume energy to perform its work. The type II topoisomerases can harness the energy of ATP hydrolysis and promote passage of DNA in one direction relative to themselves (13–15). Thus, these enzymes share a basic feature with the demon of converting chemical energy into unidirectional DNA movement. However, simplification of DNA topology would not result if the enzymes bind DNA without distortion. DNA is locally straight, and all axial orientations of the enzyme bound to the G segment, and thus relative to the whole DNA, would have equal probability. Instead, we suggest that type II topoisomerases create a local asymmetry in DNA upon binding, so that the unidirectionality of strand passage could shift the system away from equilibrium.
We suggest that the DNA asymmetry results because type II topoisomerases bend the G segment sharply upon binding, forming a hairpin as diagramed in Fig. 2. The enzyme would then have a specific orientation relative to the hairpin, and we suggest further that the entrance gate for the T segment is located inside the hairpin. Thus, during topoisomerization, the enzyme would transfer the T segment from inside to outside the hairpin (Fig. 2). This directionality of strand passage is only local, because the hairpin can have any orientation relative to the DNA chain. Surprisingly though, our quantitative analysis of the model shows that it leads to a large decrease of the steady-state fraction of knots and catenanes compared with the equilibrium levels.
Figure 2.

Model of type II topoisomerase action. The enzyme (green) bends a G segment of DNA (red) into a hairpin. The entrance gate for the T segment of DNA (yellow) is inside the hairpin. Thus, the T segment can pass through the G segment only from inside to outside the hairpin.
Computational Analysis.
We present our method of analysis of the effect of type II topoisomerases on the fraction of knots in circular DNA in detail and just outline below the comparable results for catenanes. The calculation used a circular DNA 7 kb in length. We considered unknotted chains and the simplest knot, the trefoil, which is the only one detected in the experiments that showed active simplification of DNA topology (3). The steady-state concentrations of knotted and unknotted molecules reached in the presence of the topoisomerases, Ck and Cu, respectively, are related by the equation defining a steady state:
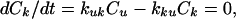 |
2 |
where kuk is the rate constant for conversion of unknotted molecules into knotted ones, and kku is the reverse rate constant. We assume that the reaction is not diffusion limited, so that the probability of finding a potential T segment inside the hairpin formed by the G segment, Pjux, is equal to the equilibrium probability of this localization in the absence of strand passage. In this case,
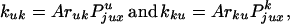 |
3 |
where ruk and
rku are the fractions of strand passages
that change topology,
P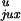 and
P
and
P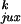 are the
respective probabilities of juxtaposition of a potential T segment near
the enzyme entrance for unknotted and knotted conformations, and
A is a coefficient. The terms
ruk and rku
need to be introduced because many strand passages do not change the
topology of a nicked DNA molecule. Combining Eqs. 2 and
3, we obtain
are the
respective probabilities of juxtaposition of a potential T segment near
the enzyme entrance for unknotted and knotted conformations, and
A is a coefficient. The terms
ruk and rku
need to be introduced because many strand passages do not change the
topology of a nicked DNA molecule. Combining Eqs. 2 and
3, we obtain
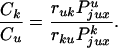 |
4 |
Because all values on the right side of Eq. (4) can be estimated well by computer simulation, we can calculate the steady-state fraction of knotted molecules. We performed such simulations for chains with both hairpin and straight G segments. For straight G segments, Eq. 4 should give the well established equilibrium fraction of knots (3). This calculation provided some insight into the computational results and served as an internal control for the validity of the computational approach.
We modeled DNA molecules as discrete worm-like chains and used the
Monte Carlo procedure to generate equilibrium sets of chain
conformations. Previous studies showed that the DNA model and
simulation method give reliable quantitative descriptions of
large-scale conformational properties of DNA (see ref. 16 and citations
therein). The G segment was modeled as a short region whose geometry
did not change during the simulation (Fig.
3). Using the algorithm described in the
previous section, we selected conformations from the constructed set in
which a second segment was properly juxtaposed with the G segment to
become the T segment, thus yielding Pjux.
These conformations were further analyzed to determine whether strand
passage would change their topology. The values of
P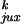 ,
P
,
P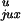 ,
rku, and ruk
for a 7-kb DNA are shown in Table 1.
,
rku, and ruk
for a 7-kb DNA are shown in Table 1.
Figure 3.

Typical simulated conformation of a knotted DNA with a hairpin G segment (red). Another segment of the 7-kb model chain is inside the hairpin in this conformation, which was selected from the set generated by a Metropolis Monte Carlo procedure.
Table 1.
Calculated chain conformation values that specify the steady-state fraction of knots for 7-kb circular DNA
| G segment conformation |
P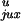
|
ruk |
P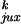
|
rku | Ck/Cu |
P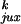 /P /P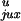
|
rku/ruk | Pk |
|---|---|---|---|---|---|---|---|---|
| Hairpin | 0.00064 | 0.018 | 0.0085 | 0.95 | 0.0014 | 13 | 53 | 0.014 |
| Straight | 0.0028 | 0.050 | 0.0093 | 0.76 | 0.020 | 3.3 | 15 | 0.014 |
P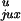 and
P
and
P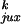 are the probabilities of juxtaposition
of a potential T segment and G segment; ruk and
rku are coefficients which specify the fractions
of strand passages that change topology; Pk is
the equilibrium probability of knotting.
are the probabilities of juxtaposition
of a potential T segment and G segment; ruk and
rku are coefficients which specify the fractions
of strand passages that change topology; Pk is
the equilibrium probability of knotting.
The simulations showed that for a DNA chain with a hairpin G segment, the steady-state fraction of knots, Ck/Cu, is reduced by a factor of 14 compared with the results for DNA with a straight G segment (Table 1). This is a surprisingly large effect because the hairpin occupies such a small portion of the DNA chain (see Fig. 3). The decrease in the fraction of knots from the experimentally observed equilibrium value for five different topoisomerases varied from 5 to 90 (3); the reason for the variation in the efficiency of topology simplification is unknown. Although the calculated 14-fold decrease in knots is smaller than the maximum experimental value, we believe that this result nevertheless supports our model. Because there are approximations in the computational analysis, we cannot expect quantitative agreement with the experimental data. The reasonable agreement between the calculated steady-state fraction of knots for a model chain with a straight G segment and the directly simulated equilibrium fraction (Table 1) confirms the validity of our analysis.
Both
P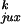 /P
/P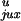 and rku/ruk
contribute to the reduction of the steady-state fraction of knots in
our model (Table 1). It was expected that
P
and rku/ruk
contribute to the reduction of the steady-state fraction of knots in
our model (Table 1). It was expected that
P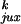 would be greater
than P
would be greater
than P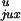 even when
the G segment is straight, because knotted conformations are more
compact, on average, than unknotted ones, and thus knotting promotes
segment collision. It was not obvious, however, that
P
even when
the G segment is straight, because knotted conformations are more
compact, on average, than unknotted ones, and thus knotting promotes
segment collision. It was not obvious, however, that
P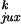 /P
/P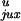 would be larger for a molecule with a hairpin G segment than for a
molecule with a straight G segment. The chain conformation shown in
Fig. 3 helps to understand this important result. One can see that two
of three crossings that define a trefoil are made by the juxtaposed T
and hairpin G segments. As a result, the rest of the chain has much
more conformational freedom. Thus, the hairpin promotes juxtaposition
of T and G segments in a knotted molecule by making such conformations
more entropically favorable.
would be larger for a molecule with a hairpin G segment than for a
molecule with a straight G segment. The chain conformation shown in
Fig. 3 helps to understand this important result. One can see that two
of three crossings that define a trefoil are made by the juxtaposed T
and hairpin G segments. As a result, the rest of the chain has much
more conformational freedom. Thus, the hairpin promotes juxtaposition
of T and G segments in a knotted molecule by making such conformations
more entropically favorable.
A similar argument explains why rku/ruk is greater for a chain containing a hairpin, a result that also was not predicted. The value of ruk, which is small for a chain with a straight G segment, is even smaller when a strand passes through a hairpin. This is because a knotted conformation generated by the passage of a T segment through a hairpin G segment has a lower probability of appearance in the equilibrium set of conformations than one made by the passage through a straight G segment. Indeed, juxtaposition of a straight segment to the outside of a hairpin (see Fig. 2, Right) typically makes no contribution to the crossings of a knot.
Type II topoisomerases also actively unlink catenanes, an activity critically important in the segregation of chromosomes. The computed steady-state fraction of the catenanes with a hairpin G segment was 4-fold less than the equilibrium one, which is in the range of experimentally observed values (3).
If type II topoisomerases use our suggested mechanism, they must bend the G segment sharply. Because of conflicting data on this matter (17), we reinvestigated the issue by using the complementary approaches of DNA cyclization in solution and direct visualization by EM.
Effect of Topoisomerase II Binding on the Efficiency of Cyclization of Short DNA Fragments.
If the topoisomerase–DNA complex bends DNA, it must increase the efficiency of cyclization of short DNA fragments (18). Cyclization efficiency is usually specified by the j factor, which is the effective concentration of one end of a DNA molecule in the vicinity of the other (see ref. 19 for review). We measured j factors for DNA fragments in the presence and absence of topoisomerase IV. T4 DNA ligase was added to seal any joints formed. Fragments of eight different lengths, from 190 to 204 bp, were used in the experiments to account for the effect of torsional orientation of the ends of the DNA on j factor values. A representative gel is shown in Fig. 4A. The j factors are approximately 30 times higher in the presence of topoisomerase IV than in the absence of the enzyme (Fig. 4B). Therefore, the binding of the enzyme strongly bends DNA in solution. The similar periodic variation of the j factor with DNA length with and without topoisomerase IV shows that the enzyme introduces a bend into the DNA rather than a swivel. We performed Monte Carlo simulations of j factors in the presence and absence of enzyme (see ref. 20 for details of j factor calculations). A bend angle of 130° gave the best fit with the experimental results (Fig. 4B).
Figure 4.

The effect of topoisomerase IV binding on the efficiency of DNA cyclization. (A) Gel electrophoretic separation of ligation products obtained in the absence and presence of topoisomerase IV for a 190-bp DNA. The results after the indicated times of ligation are shown. The bands are circular monomers (CM) and dimers (CD), and linear monomers (M), dimers (LD), and trimers (LT). (B) j Factors for eight DNA fragments in the absence (●) and presence (○) of topoisomerase IV. Error bars (standard deviation) smaller than the symbols are not shown. Solid lines represent a theoretical fit based on a Monte Carlo simulation using a DNA helical repeat of 10.55 bp, a persistence length of 40 nm, and a DNA torsional rigidity of 2 × 10−19 erg⋅cm (1 erg = 0.1 mJ). Enzyme binding was modeled by wrapping the model chain around a cylinder 14 nm in diameter with an angle α = 130° (see Inset for the angle definition).
We showed that both subunits of topoisomerase IV have to be present for DNA bending to result (data not shown). To determine whether bending requires ATP binding or hydrolysis, we measured cyclization in the presence of ATP or AMP-PNP (adenosine 5′-[β,γ-imido]triphosphate, a nonhydrolyzable ATP analog) and in the absence of nucleotide. Because T4 DNA ligase requires ATP, these experiments were performed with E. coli DNA ligase, which is powered by NAD. The j factors obtained both in the presence of AMP-PNP and without nucleotide were identical, but were approximately 2-fold lower than the values obtained in the presence of ATP (data not shown). Thus, the large majority of the cyclization enhancement is independent of ATP binding or hydrolysis and occurs in the initial binding to DNA.
EM of the Enzyme-DNA Complexes.
We provided direct proof of sharp bending of DNA by type II topoisomerases by using EM visualization of the complexes. Both enzymes used—topoisomerase IV from E. coli and yeast topoisomerase II—caused a sharp bend in relaxed circular DNA. Representative complexes are shown in Fig. 5 A and B. The bend angle varied, but angles larger than 90° were observed for about one-half of the complexes with both enzymes. In some cases, the angle appeared to exceed 180° (Fig. 5A).
Figure 5.

Visualization of type II topoisomerase molecules bound to relaxed and (−) supercoiled DNA. pBR322 DNA was incubated with yeast topoisomerase II (A, B, D) or E. coli topoisomerase IV (C, E), as described in the text. The DNA was relaxed in A and B and supercoiled in C–E. The complex in C is an enlargement of a molecule in E. After incubation, the samples were prepared for EM by fixation and rotary shadowcasting with tungsten. Images are shown in reverse contrast. [Bar = 0.33 kb of DNA (A–C) and 1.0 kb of DNA (D, E).]
We looked next at complexes with (−) supercoiled DNA (Fig. 5, C–E). Because the double helix is strongly bent at superhelix apices, the apices should be preferential points of protein binding if DNA is sharply bent by the topoisomerase. Indeed, we found that 89% ± 4% of topoisomerase IV and 85% ± 3% of yeast topoisomerase II molecules bound to supercoiled DNA were localized at the apices. The same strong preference for apical binding was found in the presence and absence of ATP. Thus, the EM data confirm and extend the results of the cyclization analysis. We conclude that the type II topoisomerases use the suggested mechanism to simplify DNA topology.
Discussion
Our data strongly support the model of topoisomerase II action based on sharp bending of the G segment upon binding and the specific orientation of the enzyme relative to the bend. It is important to emphasize that the specific orientation of the entrance gate of the enzyme relative to the hairpin is the major element of our model. Indeed, simulation of the equilibrium knotting probability shows that the presence of hairpins in DNA does not change Pk for circular chains by more than 10% (data not shown). The steady-state fraction of knots is reduced dramatically in our model only because the enzyme catalyzes the strand passage reaction in one direction, from inside to outside the hairpin. If the reverse transport of the T segment were equally likely, the steady-state fraction of knots would equal Pk.
Applying Eq. 4 to the model with a straight G segment, we
determined whether
P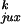 /P
/P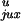 or rku/ruk
makes the equilibrium knotting probability,
Pk, so small. Table 1 shows that the major
contribution comes from the very low value of
ruk, the probability of forming a knot by
strand passage, so that
rku/ruk
equals 15. The higher probability of segment collisions in the knotted
compared with the unknotted state,
P
or rku/ruk
makes the equilibrium knotting probability,
Pk, so small. Table 1 shows that the major
contribution comes from the very low value of
ruk, the probability of forming a knot by
strand passage, so that
rku/ruk
equals 15. The higher probability of segment collisions in the knotted
compared with the unknotted state,
P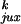 /P
/P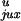 ,
makes a smaller contribution of 3.3. The kinetic proofreading model for
topology simplification by type II topoisomerases (5) can diminish the
steady-state fraction of knots only by
P
,
makes a smaller contribution of 3.3. The kinetic proofreading model for
topology simplification by type II topoisomerases (5) can diminish the
steady-state fraction of knots only by
P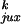 /P
/P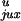 ,
which is a very small gain compared with the values observed
experimentally (3).
,
which is a very small gain compared with the values observed
experimentally (3).
Our model also helps explain the paradoxical result that the intermolecular reaction of decatenation of (−) supercoiled catenanes by type II topoisomerases is greatly favored over the intramolecular reaction of (−) supercoil relaxation (9, 21). If the enzymes are usually located at the apices of the interwound superhelix rather than at crossings, as we observed, it may be difficult for another segment of the same (−) supercoiled DNA to become a T segment. This juxtaposition can be done easily, however, by a segment of the linked molecule. Indeed, Roca and Wang (21) showed that in the unlinking of a heminicked catenane, the G segment is provided by the (−) supercoiled ring, and the T segment is provided by the nicked ring. Finally, the high resolution crystal structures of type II topoisomerases also suggest a bend of the G segment toward the T segment (ref. 22; J. Berger, personal communication).
Our model for enzyme action is a culmination of the two-gate model for type II topoisomerases of Roca and Wang (13–15). In their model, the transport of the T segment goes in one direction relative to the enzyme, which has separate entrance and exit gates for the T segment. According to the principles of statistical physics, such a unidirectional transport requires energy consumption, and this energy is provided by ATP hydrolysis. It was not clear, however, what the benefits of this unidirectional transport are. Indeed, with a straight G segment, the unidirectional transport of a T segment through the enzyme cannot shift the steady-state fraction of knots and catenanes from equilibrium. Equilibrium could be reached without the hydrolysis of ATP by some other mechanism, because the breaking and resealing of DNA does not require energy consumption. Coupling the unidirectional transport with DNA bending and a specific orientation of the enzyme relative to the bend makes the two-gate mechanism rational: now the energy of ATP hydrolysis is used to simplify DNA topology below thermodynamic equilibrium so that these enzymes can carry out their physiological roles.
Acknowledgments
This work was supported by National Institutes of Health Grants GM54215 (to A.V.), GM31657 (to N.R.C.), and GM31819 (to J.G.).
Abbreviations
- G segment
gate segment
- T segment
transported segment
- EM
electron microscopy
References
- 1.Berger J M, Wang J C. Curr Opin Struct Biol. 1996;6:84–90. doi: 10.1016/s0959-440x(96)80099-6. [DOI] [PubMed] [Google Scholar]
- 2.Wang J C. Q Rev Biophys. 1998;31:107–144. doi: 10.1017/s0033583598003424. [DOI] [PubMed] [Google Scholar]
- 3.Rybenkov V V, Ullsperger C, Vologodskii A V, Cozzarelli N R. Science. 1997;277:690–693. doi: 10.1126/science.277.5326.690. [DOI] [PubMed] [Google Scholar]
- 4.Baird C L, Harkins T T, Morris S K, Lindsley J E. Proc Natl Acad Sci USA. 1999;96:13685–13690. doi: 10.1073/pnas.96.24.13685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yan J, Magnasco M O, Marko J F. Nature (London) 1999;401:932–935. doi: 10.1038/44872. [DOI] [PubMed] [Google Scholar]
- 6.Vologodskii A V. In: RECOMB98: Proceedings of the Second Annual International Conference on Computational Molecular Biology. Istrail S, Pevsner P, Waterman M, editors. New York: Association for Computing Machinery; 1998. pp. 266–269. [Google Scholar]
- 7.Rybenkov V V, Cozzarelli N R, Vologodskii A V. Proc Natl Acad Sci USA. 1993;90:5307–5311. doi: 10.1073/pnas.90.11.5307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vologodskii A V, Lukashin A V, Frank-Kamenetskii M D, Anshelevich V V. Sov Phys, J Exp Theor Phys. 1974;39:1059–1063. [Google Scholar]
- 9.Ullsperger C, Cozzarelli N R. J Biol Chem. 1996;271:31549–31555. doi: 10.1074/jbc.271.49.31549. [DOI] [PubMed] [Google Scholar]
- 10.Taylor W H, Hagerman P J. J Mol Biol. 1990;212:363–376. doi: 10.1016/0022-2836(90)90131-5. [DOI] [PubMed] [Google Scholar]
- 11.Griffith J D, Christiansen G. Annu Rev Biophys Bioeng. 1978;7:19–35. doi: 10.1146/annurev.bb.07.060178.000315. [DOI] [PubMed] [Google Scholar]
- 12.Pulleyblank D E. Science. 1997;277:648–649. doi: 10.1126/science.277.5326.648. [DOI] [PubMed] [Google Scholar]
- 13.Roca J, Wang J C. Cell. 1992;71:833–840. doi: 10.1016/0092-8674(92)90558-t. [DOI] [PubMed] [Google Scholar]
- 14.Roca J, Wang J C. Cell. 1994;77:609–616. doi: 10.1016/0092-8674(94)90222-4. [DOI] [PubMed] [Google Scholar]
- 15.Roca J, Berger J M, Harrison S C, Wang J C. Proc Natl Acad Sci USA. 1996;93:4057–4062. doi: 10.1073/pnas.93.9.4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rybenkov V V, Vologodskii A V, Cozzarelli N R. J Mol Biol. 1997;267:312–323. doi: 10.1006/jmbi.1996.0877. [DOI] [PubMed] [Google Scholar]
- 17.Wang J C. Annu Rev Biochem. 1996;65:635–695. doi: 10.1146/annurev.bi.65.070196.003223. [DOI] [PubMed] [Google Scholar]
- 18.Ulanovsky L, Bodner M, Trifonov E N, Choder M. Proc Natl Acad Sci USA. 1986;83:862–866. doi: 10.1073/pnas.83.4.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crothers D M, Drak J, Kahn J D, Levene S D. Methods Enzymol. 1992;212:3–29. doi: 10.1016/0076-6879(92)12003-9. [DOI] [PubMed] [Google Scholar]
- 20.Podtelezhnikov A A, Mao C, Seeman N C, Vologodskii A. Biophys J. 2000;79:2692–2704. doi: 10.1016/S0006-3495(00)76507-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roca J, Wang J C. Genes Cells. 1996;1:17–27. doi: 10.1046/j.1365-2443.1996.01001.x. [DOI] [PubMed] [Google Scholar]
- 22.Morais Cabral J H, Jackson A P, Smith C V, Shikotra N, Maxwell A, Liddington R C. Nature (London) 1997;388:903–906. doi: 10.1038/42294. [DOI] [PubMed] [Google Scholar]