

Abstract
We report the characterization of tadA, the first prokaryotic RNA editing enzyme to be identified. Escherichia coli tadA displays sequence similarity to the yeast tRNA deaminase subunit Tad2p. Recom binant tadA protein forms homodimers and is sufficient for site-specific inosine formation at the wobble position (position 34) of tRNAArg2, the only tRNA having this modification in prokaryotes. With the exception of yeast tRNAArg, no other eukaryotic tRNA substrates were found to be modified by tadA. How ever, an artificial yeast tRNAAsp, which carries the anticodon loop of yeast tRNAArg, is bound and modified by tadA. Moreover, a tRNAArg2 minisubstrate containing the anticodon stem and loop is sufficient for specific deamination by tadA. We show that nucleotides at positions 33–36 are sufficient for inosine formation in mutant Arg2 minisubstrates. The anticodon is thus a major determinant for tadA substrate specificity. Finally, we show that tadA is an essential gene in E.coli, underscoring the critical function of inosine at the wobble position in prokaryotes.
Keywords: adenosine deaminase/Escherichia coli/inosine/RNA editing/tRNAArg2
Introduction
The nucleotide inosine (I) has been observed in viral transcripts and eukaryotic mRNAs. In all known cases, I results from the deamination of adenosine (A), a process termed RNA editing. Because I is read as guanosine (G) by the translational machinery (Basilio et al., 1962), RNA editing can change codon specificity and therefore the amino acid sequence of the encoded protein, resulting in multiple protein products with different biological function from a single mRNA precursor. In mammals, for example, the mRNA precursors (pre-mRNAs) coding for subunits of glutamate-gated ion channel receptors (GluRs) and the serotonin receptor subunit 5-HT2C are edited (reviewed in Seeburg et al., 1998; Gott and Emeson, 2000; Maas and Rich, 2000; Gerber and Keller, 2001; Keegan et al., 2001). Editing was also detected in Drosophila melanogaster, Caenorhabditis elegans and in hepatitis delta virus (Polson et al., 1996; Smith et al., 1996; Morse and Bass, 1999; Semenov and Pak, 1999; Hanrahan et al., 2000). RNA editing therefore represents an important mechanism for increasing the genetic diversity in eukaryotes. In all these cases, pre-mRNA editing requires double-stranded RNA (dsRNA) structures, which are formed between exonic sequences encompassing the editing site and downstream intronic sequences.
RNA editing of pre-mRNAs is catalyzed by adenosine deaminases acting on RNA (ADARs; Bass et al., 1997). ADARs have a common modular organization consisting of two or three dsRNA-binding domains (dsRBD) and a catalytic deaminase domain containing three Zn2+-chelating residues and a proton-shuffling glutamate (reviewed in Bass, 1997; Maas and Rich, 2000; Gerber and Keller, 2001).
Inosine is not only present in mRNAs, but also in tRNAs. It was first found in tRNAAla from yeast (Holley et al., 1965). Eukaryotic tRNAAla contains I at two positions: at the wobble position of the anticodon (position 34) and as the derivative N1-methylinosine (m1I) at position 37, 3′ adjacent to the anticodon (Holley et al., 1965; reviewed in Grosjean et al., 1996). In eukaryotes, seven to eight tRNAs contain I at position 34, whereas in prokaryotes and plant chloroplasts only tRNAArg2 contains this modification. As in pre-mRNAs, I in tRNAs is the product of hydrolytic deamination of genomically encoded A (Auxilien et al., 1996). m1I at position 37 is formed in a two-step reaction. First, A is deaminated to I, which is further methylated by a methyltransferase (Grosjean et al., 1996; Björk et al., 2001). The genomes of Saccharomyces cerevisiae and prokaryotes do not encode classical ADAR proteins, but, based on sequence homology to ADARs, a yeast protein was identified that contains a deaminase domain but lacks a known RNA-binding motif. This protein catalyzes the deamination of A at position 37 in yeast tRNAAla and was therefore named adenosine deaminase acting on tRNA 1 and its gene tRNA-specific adenosine deaminase 1 (scADAT1/TAD1; Gerber et al., 1998). Tad1p specifically targets A37 in tRNAAla. Mutations affecting the three-dimensional structure of the tRNA or the length of the anticodon loop abolish conversion of A to I in vitro (Gerber et al., 1998). Whereas A37 is unmodified in a Δtad1 strain, modification of A34 was unaffected in all tRNAs tested, suggesting that I formation at these two positions is catalyzed by different enzymes. Whereas I in the wobble position (I34) is crucial to allow the decoding of three codons by a single tRNA (Crick, 1966), the function of m1I37 is less clear. It was postulated that this modification may prevent translational frameshifts and improve translational fidelity (Björk et al., 1989). ADAT1 proteins have also been cloned from human (Maas et al., 1999), mouse (Maas et al., 2000) and D.melanogaster (Keegan et al., 2000).
The tRNA adenosine deaminase that specifically deaminates A34 has been partially purified from yeast extracts (Auxilien et al., 1996) and has been identified by homology to Tad1p (Gerber and Keller, 1999). Activity depended on the correct tRNA structure as well as on the length and the nucleotide sequence of the anticodon loop. The tRNA:A34 deaminase is a heterodimer of sequence-related subunits named scADAT2/Tad2p and scADAT3/Tad3p. Both polypeptides contain a deaminase domain that resembles that of the cytidine deaminase (CDA) superfamily (Gerber and Keller, 1999), although Tad2p/Tad3p deaminates A. Most cytidine/deoxycytidylate deaminases catalyze deaminations of mononucleotides, but there are also a few known cytidine deaminases that act on RNA (CDARs). One of the best studied examples is APOBEC1, which catalyzes the deamination of a cytidine to uridine in the apoB mRNA, resulting in a change of a glutamine codon to a stop codon (reviewed by Chester et al., 2000). Interestingly, the deaminase domain of Tad3p lacks the glutamate that, analogous to CDAs and CDARs, is thought to be part of the active center. The lack of this glutamate suggests that Tad3p is catalytically inactive and that Tad2p is the catalytic subunit of the complex.
Deamination of tRNA is not limited to eukaryotes, but has also been detected in Escherichia coli extracts (Auxilien et al., 1996). In E.coli, the only known A to I conversion in RNA is the deamination of tRNAArg2 at position 34. Based on sequence homology, we identified an E.coli protein homologous to Tad2p. Here, we report the identification and characterization of this enzyme, which is necessary and sufficient to catalyze this editing reaction. tadA/ecADAT2 shows sequence homology to yeast Tad2p and Tad3p and is encoded by an essential gene, thus underlining the vital function of I34. The identification of the first prokaryotic tRNA-specific adenosine deaminase also provides further insight into the evolution of the deaminase family of enzymes.
Results
tadA acts specifically on tRNAArg2
Database searches with the S.cerevisiae Tad2p sequence and the FASTA program (Pearson and Lipman, 1988) revealed that the E.coli open reading frame (ORF) yfhC is 34% identical to Tad2p (Figure 1). To determine whether yfhC indeed encodes an adenosine deaminase, the protein was overexpressed in E.coli and purified to apparent homogeneity (Materials and methods; Figure 2A and B).

Fig. 1. Bacterial genomes encode a protein (tadA) related to Tad2p of S.cerevisiae. Multiple sequence alignment of E.coli tadA, yeast Tad2p and putative tadA sequences from different bacteria. Residues conserved in >83% of the proteins are shown in black, similar amino acids in gray. The three putative Zn2+-chelating residues (diamond) and the glutamate thought to mediate proton transfer (bullet) are marked. The position of the point mutation (D64E) in the tadA mutant NWL37 is indicated by an asterisk. The alignment was generated with the Clustal_W software at the European Bioinformatics Institute (Thompson et al., 1994).

Fig. 2. tadA specifically deaminates A34 in tRNAArg2. SDS– polyacrylamide gel stained with silver (A), western blot (B) and tRNA editing assay (C) of the final gel filtration column of the purification of recombinant tadA. A 20 µl aliquot of each fraction was separated on a 12% SDS–polyacrylamide gel (A) and transferred to a nitrocellulose membrane for detection with antibodies (B). The western blot was probed with a mouse α-GST monoclonal antibody. Fraction numbers are indicated at the top, the molecular masses of the size standards in kDa on the left. (C) Two microliters of the fractions indicated at the top were incubated with [33P]ATP-labeled tRNAArg2 for 30 min at 37°C. Reactions were treated with P1 nuclease and the products separated by one-dimensional TLC. The positions of AMP, IMP and the origin are indicated on the right. The position of IMP was verified with unlabeled 5′ IMP. In lane ‘–’, tRNAArg2 was incubated with buffer only. (D) Sequence analysis of in vitro modified tRNAArg2. The nucleotide sequence surrounding the anticodon is shown. tRNA from reactions shown in (C) was amplified by RT–PCR and sequenced. The nucleotide at position 34 is shaded gray, the anticodon is underlined. Control: tRNAArg2 incubated with buffer only.
Peak fractions of the final gel filtration column were tested for adenosine deaminase activity on in vitro transcribed and uniformally [α-33P]ATP-labeled tRNAArg2. The recombinant protein indeed converted A to I (Figure 2C). This activity stemmed from the recombinant protein and not from an endogenous enzyme, since glutathione S-transferase (GST) purified from E.coli or eluates from Ni2+–agarose incubated with extract from empty-vector-transformed cells had no activity (results not shown). A 2 ng aliquot of recombinant yfhC protein formed up to 0.5 mol I/mol tRNA (Figure 2C, fraction 12) and no other protein was needed for activity (Figure 2C, fraction 12). This is in contrast to the yeast enzyme, which acts as a heterodimer composed of Tad2p and Tad3p (Gerber and Keller, 1999).
To determine the site in tRNAArg2 that is modified by recombinant yfhC, in vitro modified tRNA was amplified by RT–PCR and sequenced. Since I base-pairs with C during reverse transcription, A to I deamination changes the sequence from A to G. tRNAArg2 incubated with recombinant yfhC contained a G at position 34, demonstrating that the template-encoded A at this position was deaminated to I. Furthermore, sequencing of tRNAArg2 revealed that ∼90% of A34 was deaminated to I, as can be seen from the ratio of G to A at position 34 (Figure 2D). tRNAArg2 incubated in the absence of protein carried A at position 34 and therefore was not modified (Figure 2D). No other A in tRNAArg2 was modified by the recombinant protein. These results showed that the yfhC gene encodes a protein that is sufficient to reconstitute A34:tRNA deaminase activity in vitro. Therefore, we renamed yfhC as tadA (for tRNA adenosine deaminase A).
The specificity of recombinant tadA was tested by comparing its activity on various tRNA substrates (Figure 3A). In E.coli, only one tRNA substrate is known that is deaminated at position 34 (tRNAArg2), in contrast to S.cerevisiae where seven different tRNAs are substrates for Tad2p/Tad3p (Auxilien et al., 1996; Gerber and Keller, 1999). Auxilien et al. (1996) showed that not only tRNAArg2, but also a variant of tRNAAsp from S.cerevisiae carrying the anticodon loop of yeast tRNAArg, are deaminated in vitro upon incubation with extracts from E.coli. The latter observation could be reproduced with recombinant tadA, which forms up to 0.8 mol of I in this artificial tRNA, suggesting that the anticodon loop carries important recognition signals for the enzyme (Figure 3A, lane 4). This hypothesis is supported by the result that tadA deaminated yeast tRNAArg with the same efficiency as E.coli tRNAArg2 (Figure 3A, lanes 10 and 11). These two tRNAs have a conserved anticodon loop sequence (Figure 3B). However, parts of the anticodon stem and the acceptor stem are also conserved between yeast and E.coli tRNAArg, and these nucleotides might also contribute to the recognition of the tRNA by tadA (Figure 3B).

Fig. 3. Substrate specificity of tadA. (A) tRNA-editing assay with tadA and different tRNAs. tRNA substrates were incubated with either 2 ng of recombinant His6-tadA (lanes 2, 4, 6, 8, 10 and 11), 20 ng of recombinant scTad2p/scTad3p (lane 7) or 40 µg of S.cerevisiae total protein (lanes 1, 3 and 5). One hundred femtomoles of tRNAAla from S.cerevisiae (lanes 1 and 2), a tRNAAsp mutant from S.cerevisiae (lanes 3 and 4), tRNAAla from B.mori (lanes 5 and 6), human tRNAAla (lanes 7 and 8), S.cerevisiae tRNAArg (lanes 9 and 10) and tRNAArg2 from E.coli (lane 11) were used. Mock incubation with WT yeast tRNAAsp did not result in I formation (result not shown). All reactions were incubated for 1 h at 30°C and processed as described in the legend to Figure 2 (see also Materials and methods). (B) Cloverleaf structure of E.coli tRNAArg2, S.cerevisiae tRNAArg and S.cerevisiae tRNAAsp with the anticodon loop of tRNAArg. Completely conserved nucleotides and nucleotides conserved as purines or pyrimidines between tRNAs from different species are shown in red (Klingler and Brutlag, 1993). Nucleotides that are conserved in addition between the three tRNAs are depicted in blue. (C) UV-cross-linking experiments of recombinant tadA and different tRNAs. A 400 ng aliquot of GST–tadA (lanes 2–5) and 100 fmol of [33P]ATP-labeled tRNA were irradiated and samples treated with RNaseA. Proteins were separated on a 12% SDS–polyacrylamide gel and gels exposed to a phosphoimager screen. E.c., E.coli; S.c., S.cerevisiae; B.m., B.mori; Arg2, tRNAArg2; hAla, human tRNAAla; B.m. Ala, B.mori tRNAAla; S.c. Ala, S.cerevisiae tRNAAla.
tRNAAla from S.cerevisiae, Bombyx mori and Homo sapiens are not deaminated by tadA, although they are known to contain I34 in vivo (Figure 3A, lanes 2, 6 and 8). Notably, all these tRNAs are substrates for yeast Tad2p/Tad3p (Figure 3A, lanes 1, 5 and 7; Auxilien et al., 1996; Gerber and Keller, 1999). In yeast and B.mori, tRNAAla positions 34 and 37 are deaminated and 2 mol I/mol tRNA are generated upon incubation with yeast extract (Figure 3A, lanes 1 and 5). The substrate specificity of tadA could also be shown with UV-cross-linking experiments that indicate binding of the protein to the tRNA substrate. Recombinant GST–tadA could be UV cross-linked to its natural substrate tRNAArg2 (Figure 3C, lane 2), but not to tRNAAla from H.sapiens, B.mori or S.cerevisiae (Figure 3C, lanes 4–6). GST alone could not be cross-linked to any of these substrates (results not shown). In summary, we showed that recombinant tadA specifically deaminates tRNAArg2 at position 34.
To determine whether the anticodon stem and loop is sufficient for deamination by tadA, a tRNAArg2 minisubstrate containing only the anticodon stem and loop was tested in vitro (Figure 4A). This minisubstrate was deaminated as efficiently as full-length tRNAArg2 (Figure 4B, lane 1; Table I). In the majority of the experiments, deamination of wild-type (WT) Arg2 minisubstrate yielded between 1.2 and 1.4 mol I/mol tRNA, which is slightly higher than expected. Interestingly, when A at the wobble position was mutated to G, the minisubstrate was not deaminated, indicating that this nucleotide is essential for tadA recognition (Figure 4B, lane 2). It is possible that a second A was deaminated in a fraction of the WT minisubstrate. However, we consider this possibility unlikely because no I was formed when A34 was mutated and deamination of the second A would thus depend on the presence of A34. Although the anticodon arm of Arg2 serves as a substrate for tadA, there seem to be differences in site specificity between the full-length tRNAArg2 and mini Arg2. Sequences and structures outside the anticodon arm might be important for deamination at the correct position. In order to determine nucleotides that are essential to specify these short RNAs as substrates for tadA, several mutant minisubstrates were generated and tested in vitro. No I was formed with minisubstrates that have mutations at position 35 or 36 in the anticodon (Figure 4B, lanes 3 and 4). This showed that nucleotides in the anticodon were key determinants for tadA activity. The importance of the anticodon loop size was investigated with a minisubstrate that has an 8 nucleotide (nt) loop. This substrate was still deaminated, but with a lower efficiency compared with the WT RNA (Figure 4B, lane 5; Table I), whereas a 3 nt loop was not sufficient for tadA activity (Figure 4B, lane 6). A minisubstrate that has a random loop sequence but the correct anticodon was not deaminated, showing that the anticodon is not the only determinant for tadA activity (Figure 4B, lane 7). When position 37 in mutant 6 was changed to G, I formation was not restored (Figure 4B, lane 8). G at position 37 in the WT Arg2 minisubstrate did not affect tadA activity (results not shown). However, reverting position 33 in mutant 6 to the WT U residue restored I formation, which was, however, not as efficient as with WT minisubstrate (Figure 4B, lane 9; Table I). This suggests that U33 is an important determinant for tadA activity, but additional nucleotides probably contribute to efficient deamination. To determine whether a stem–loop structure in addition to the sequence is required for tadA activity, a linear minisubstrate was analyzed (mutant 9, Figure 4A). Although mutant 9 contains the same essential nucleotides for activity as mutant 7, the linear RNA was not deaminated (Figure 4B, lane 10), indicating that a stem–loop structure is essential for tadA activity. As expected, the anticodon arm of E.coli tRNAArg3 was not a substrate for tadA (Figure 4B, lane 11) because this RNA does not contain A at the wobble position (Figure 4A). However, I formation was detected in Arg3 when C34 was mutated to A (Figure 4B, lane 12). This result provides further information about key nucleotides for tadA activity. The anticodon stem of Arg3 differs from that of Arg2, indicating that recognition of the stem by tadA is not sequence specific (Figure 4A). In summary, a minisubstrate with a stem–loop structure is deaminated at the wobble position of the anticodon if the RNA has the sequence UACG within the loop. However, it cannot be excluded that other regions of the tRNA are needed for efficient deamination. The results with the mutant minisubstrates suggest that, in addition to the correct sequence in the loop, the structure and the loop size, but not the stem sequence, are important for deamination by tadA.

Fig. 4. RNA minisubstrates derived from E.coli tRNAArg2 and tRNAArg3 are sufficient for deamination by tadA in vitro. (A) Schematic drawing of Arg minisubstrates that were tested in vitro. Nucleotides shown in gray are mutated compared with WT Arg2. A at the wobble position of the anticodon is shown in bold. Mutations in each minisubstrate are indicated by arrows. (B) Editing assay with minisubstrates and tadA. A 25–100 fmol aliquot of minisubstrate, 100 ng of recombinant Flag-tadA-His6 and 500 ng of BSA were incubated in a total volume of 25 µl. WT Arg2 (lane 1), mutant 1 (lane 2), mutant 2 (lane 3), mutant 3 (lane 4), mutant 4 (lane 5), mutant 5 (lane 6), mutant 6 (lane 7), mutant 7 (lane 8), mutant 8 (lane 9), mutant 9 (lane 10), WT Arg3 (lane 11) and mutant 10 (lane 12) were used. All reactions were incubated for 45 min at room temperature and processed as described in the legend to Figure 2 (see also Materials and methods).
Table I. Quantification of tadA activity obtained with tRNA-derived minisubstrate RNAs.
| Substrate | Mutations compared with WT | mol I/mol RNA |
|---|---|---|
| Arg2 WT | 1.2–1.4 | |
| mutant 1 | A34G | 0 |
| mutant 2 | C35G | 0 |
| mutant 3 | G36C | 0 |
| mutant 4 | 8 nt loop, U28G, A42C | 0.5 |
| mutant 5 | 3 nt loop, 7 bp G-C stem | 0 |
| mutant 6 | U28G, C32U, U33C, A37C, A38U, A42C | 0 |
| mutant 7 | U28G, C32U, U33C, A37G, A38U, A42C | 0 |
| mutant 8 | U28G, C32U, A37C, A38U, A42C | 0.26 |
| mutant 9 | Linear | 0 |
| Arg3 WT | 0 | |
| mutant 10 | C34A | 1.39 |
A point mutation in tadA leads to an inactive enzyme in vitro
Next we asked whether tadA is responsible for I formation at position 34 of tRNAArg2 in vivo and took advantage of a non-lethal point mutant described previously (Poulsen et al., 1992). The mutant form of tadA carries a glutamic acid instead of an aspartic acid at position 64 (D64E; Figure 1). This amino acid is highly conserved in bacterial, but not in eukaryotic, Tad2 proteins.
To determine whether deamination of A34 in tRNAArg2 is affected by this mutation in vivo, total RNA was isolated from the mutant (NWL37) and the corresponding parental strain (NWL32). After amplification of tRNAArg2 by RT–PCR with specific primers, the cDNAs were subcloned and multiple clones of each strain were sequenced. As expected, all clones derived from the WT strain contained G at position 34 (results not shown). Interestingly, 16 out of 17 clones derived from NWL37 also had G at position 34, and only one clone contained A at this position (results not shown). Therefore, this point mutant of tadA is almost fully active in vivo, which is not suprising given that the mutant strain showed no growth defect even at 42°C (results not shown), even though tadA is an essential gene.
Although the mutant protein was functional in cells, its activity might be reduced in an in vitro assay system. We therefore prepared extracts from mutant and parental strains, and tested equal amounts in a deamination assay. Whereas extracts from WT cells showed activity (NWL32; Figure 5, lane 2), no I formation could be detected with extracts from the mutant strain (NWL37; Figure 5, lane 3). However, activity could be restored by transforming the mutant cells with a tadA expression construct (NWL37R; Figure 5, lane 4). Thus, the mutation D64E abolishes the enzymatic activity of tadA in vitro, although it does not detectably affect its activity in cells.
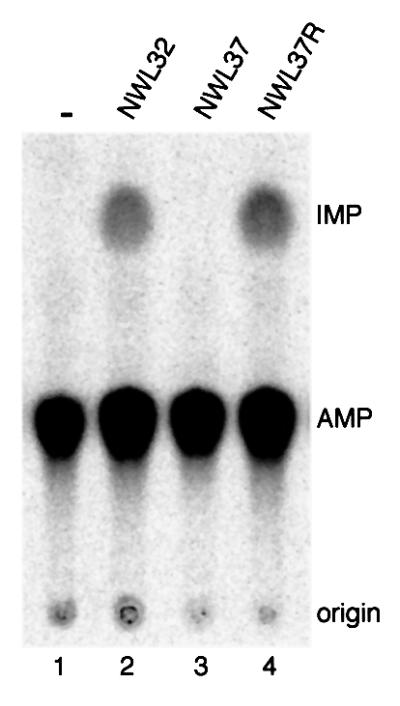
Fig. 5. The point mutation D64E abolishes tadA activity in vitro. Lane 1, no protein; lanes 2–4, 40 µg of total protein of E.coli extract. All reactions were incubated for 2 h at 37°C and processed as described in the legend to Figure 2 (see also Materials and methods).
tadA cannot substitute for yeast Tad2p in vivo and in vitro
Because tadA is 34% identical to yeast Tad2p and is functionally conserved, we attempted to replace Tad2p with tadA in a ΔTAD2 yeast strain (Gerber and Keller, 1999). tadA was cloned into the pGALΔTrp-FLIS6 vector (Gerber et al., 1998) and transformed into a Δtad2 yeast strain carrying a copy of TAD2 on a plasmid with the URA3 marker (pFL38; Gerber and Keller, 1999). Cells were then transferred to plates containing 5-fluoro-orotic acid (5-FOA) and galactose to force the loss of the URA3-marked TAD2 plasmid and induce the expression of tadA, respectively. No cells grew, showing that tadA cannot substitute for Tad2p in vivo.
Next we tested whether tadA could functionally replace Tad2p in an in vitro deaminase assay. No activity was detected when tadA pre-incubated with yeast Tad3p was incubated with B.mori tRNAAla, which is a substrate for the Tad2p/Tad3p heterodimer (Figure 6A, lanes 4 and 2). Thus, tadA cannot replace yeast Tad2p either in vivo or in vitro.

Fig. 6. tadA cannot replace yeast Tad2p in vitro and forms homodimers. (A) Recombinant yeast Tad2p, Tad3p and E.coli tadA were pre-incubated as indicated at the top and tested for activity in vitro with tRNAAla from B.mori. As a control, the yeast Tad2p/Tad3p complex was used (lane 2). (B) Flag pull-down assay with in vitro translated tadA. 35S-labeled tadA was incubated with Flag-tagged tadA (lane 2) or buffer (lane 1) and subsequently bound to Flag–agarose and eluted. Ten percent of the input is shown in lane 3. Proteins were separated on a 12% SDS–polyacrylamide gel. Molecular masses of the protein standards are indicated in kDa on the left. (C) Chemical cross-linking of recombinant tadA. A 300 ng aliquot of tadA (lanes 2–4) was incubated in a reaction mixture containing tris(2,2′-bipyridyl) dichlororuthenium(II)hexahydrate, APS and BSA (90 ng, 0.9 µg and 9 µg, respectively). tadA incubated with buffer only is shown in lane 1. Reaction products were separated on a 12% SDS–polyacrylamide gel and blotted onto a nitrocellulose membrane. tadA was detected with a mouse anti-Flag antibody. Molecular masses of protein standards are indicated in kDa on the left.
tadA forms homodimers
Because yeast Tad2p functions as a heterodimer with Tad3p, and E.coli does not encode a Tad3-like protein, we wondered whether tadA might form homodimers instead. This hypothesis was tested with three different approaches. We investigated the interaction of Flag-tadA and in vitro translated [35S]methionine-labeled tadA in a Flag pull-down assay. The tagged tadA and labeled translated tadA were pre-incubated, bound to anti-Flag– agarose, washed and eluted by boiling the matrix in SDS–gel loading buffer. The result is shown in Figure 6B (lane 2). Approximately 10% of the tadA input was pulled down by Flag-tadA. In a second experiment, recombinant Flag-tadA was chromatographed on a gel filtration column, which separates proteins according to size, and fractions were tested for tadA activity. Fifty percent of tadA was eluted at a position corresponding to a dimer and ∼50% of tadA was eluted as a monomer (results not shown). This result suggests that tadA may act as a homodimer. However, the presence of tadA monomers indicates that tadA does not interact as strongly with itself as does yeast Tad2p with Tad3p (Gerber and Keller, 1999). Formation of tadA monomers could occur by separation of tadA dimers on the gel filtration column due to the experimental conditions. To confirm dimerization, we carried out chemical cross-linking experiments and, indeed, observed a complex corresponding in size to tadA homodimers (Figure 6C, lanes 2–4). Complex formation was strongly stimulated by the addition of BSA, although a weak cross-link was also detectable in some experiments in the absence of BSA (results not shown). The reason for this stimulation is unclear; however, BSA might have a stabilizing effect on the interaction of the highly purified tadA subunits. tadA complex formation was specific and only involved tadA subunits but not BSA. Complexes with BSA would have a higher molecular mass and could therefore easily be distinguished from tadA homodimers by size. Dimeriza tion was not complete and could be observed for ∼30% of tadA. This might be due to the experimental procedure in that tadA might have been cross-linked under suboptimal dimerization conditions. The addition of E.coli total tRNA did not influence dimer formation in these assays, indicating that the observed dimerization did not require substrate tRNA (results not shown). These experiments indicate that tadA can form a homodimer in vitro; however, it remains to be shown whether it also acts as a homodimer in vivo.
tadA is essential for cell viability
tadA has previously been reported to be a non-essential gene in E.coli (Poulsen et al., 1992). tadA was deleted by inserting a chloramphenicol acetyltransferase (CAT) gene into the SphI restriction site of the ORF, which removed the last 17 amino acids of the protein. We re-investigated whether tadA is essential for viability because recombinant tadAΔC, corresponding to the knock-out gene described by Poulsen et al. (1992), was active in a tRNA editing assay (results not shown). The tadA gene was inactivated with the two-step sacB counterselection technique (Reece and Phillips, 1995). For this purpose, a plasmid-borne tadA gene was produced, which was disrupted by an interposon carrying a kanamycin-resistance cassette (pJW30; Figure 7A). The disrupted tadA cassette was introduced at the chromosomal tadA locus by homologous recombination. Thus, the first recombination event resulted in the introduction of a kanamycin-disrupted tadA gene and plasmid sequences into the genome next to WT tadA (Figure 7A). Subsequent growth of these strains on 5% sucrose selected for excision of the sacB gene, which confers sucrose sensitivity, by a second recombination event. Sucrose-resistant clones were then analyzed for kanamycin and plasmid-encoded chloramphenicol resistance. If the WT copy of tadA can be lost because the gene is not essential, one would expect to find similar numbers of WT tadA and ΔtadA clones (Figure 7A). However, all of the 400 clones analyzed were KanS/CamS and, therefore, wild type, indicating that the disrupted form of tadA was lost selectively and that tadA is an essential gene (Figure 7B). To corroborate this, we repeated the tadA deletion experiment in the presence of a functional copy of tadA under the control of the arabinose promoter (PBAD). In this case, approximately equal numbers of KanR/CamS (e.g. ΔtadA, 47%) and KanS/CamS (e.g. WT, 49%) clones were obtained (Figure 7B). The deletion of tadA was confirmed by PCR analysis of seven of the KanR/CamS clones (Figure 7C). PCR analysis of genomic DNA from WT cells led to the accumulation of a fragment of 1000 bp (Figure 7C, lane 1), corresponding to tadA + 500 bp upstream sequence. This product was not formed with genomic DNA from a ΔtadA strain. Instead, a faint band was observed at ∼2500 bp, which corresponds to tadA disrupted by the kanamycin cassette and 500 bp upstream sequence (Figure 7C, lane 2). Note that the primers used are specific for the amplification of the genomic tadA locus, whereas the rescue copy of tadA is not amplified. In addition, the deletion was confirmed by Southern blot analysis of three individual clones (results not shown). These results indicate that disruption of the WT chromosomal copy of tadA resulted in viable E.coli only when an additional plasmid-borne tadA gene was also present. The same results were obtained when these gene disruption experiments were performed with Caulobacter crescentus (results not shown). Thus, we conclude that tadA is essential for viability of eubacterial organisms.

Fig. 7. tadA is an essential gene. (A) The sacB counterselection procedure. The wild-type chromosomal copy of tadA was exchanged via a two-step homologous recombination reaction with a gene copy disrupted by the insertion of a kanamycin-resistance cassette. After the second recombination step, colonies were analyzed by their antibiotic-resistance profile for the presence or absence of the Kan cassette in the tadA locus and for mutational inactivation of the sacB gene. The mutation in sacB is indicated by an asterisk. (B) Summary of the clones obtained by the strategy described in (A). The total number of sucrose-resistant colonies analyzed is indicated in the first column (Total clones). The number of isolated colonies with a disrupted tadA (ΔtadA) gene in the chromosome is shown in the second column and the number of colonies with a restored WT tadA locus (WT) after the second recombination step is shown in the third column. Colonies that had acquired sucrose resistance through sacB inactivation are listed in the fourth column (sacB–). Plasmids pJW30 and pJW117 carry disrupted and WT copies of the tadA ORF, respectively. (C) PCR analysis of WT (lane 1) and ΔtadA (lane 2) genomic E.coli DNA. Primers are indicated by arrows and anneal 500 bp upstream of the tadA ORF and at the 3′ end of the ORF, respectively. Sizes of standards (lane M) are indicated in bp on the left.
Discussion
We describe the cloning and functional characterization of the E.coli enzyme tadA/ecADAT2. Based on the following observations, we conclude that tadA is an adenosine deaminase that specifically converts A34 to I34 in bacterial tRNAArg2. (i) Recombinant tadA, purified to homogeneity, specifically deaminates tRNAArg2 at the wobble position of the anticodon and does not need a cofactor. Highly purified tadA formed 0.5 mol I/mol tRNA. Auxilien et al. (1996) showed that 1 mol I/mol tRNAArg2 was generated upon incubation with E.coli extract. tRNAArg2 was incubated for 3 h with 130 µg of total protein in these experiments. It is, therefore, possible that pure tadA is less stable and that the presence of other proteins (e.g. tRNA modification enzymes) stabilized the enzyme. Furthermore, the structure of the in vitro transcribed tRNA may also be stabilized by additional proteins in the S100 extract and this may have contributed to the efficiency of I formation. (ii) The recombinant protein is highly specific for the wobble position of tRNAArg2. However, eukaryotic tRNAs are deaminated by tadA if they contain the anticodon loop of tRNAArg from yeast or E.coli. (iii) Recombinant tadA forms homodimers. Dimerization of tadA was not complete and tadA monomers could still be observed in all the experiments. However, this might be due to the experimental procedure and does not necessarily reflect the situation in vivo. A to I deamination at the wobble position of the anticodon is conserved from bacteria to mammals, and the enzymes catalyzing the reaction are also conserved not only in their sequence, but probably also in their property of acting as dimers. Thus, the tadA homodimers may represent the ancestor of the eukaryotic Tad2p/Tad3p heterodimer. (iv) tadA is encoded by an essential gene. The fact that E.coli cells are not viable without tadA underscores the important role of I34 in tRNA. However, it cannot be excluded that the absence of the tadA protein rather than the absence of I34 is the cause of lethality. This has been shown for other tRNA modification enzymes that were, therefore, suggested to have two different functions (Persson et al., 1992; Gutgsell et al., 2000). There are two major differences between tRNA deamination in E.coli and S.cerevisiae. First, tRNAArg2 is the only E.coli tRNA that has A at position 34, and is therefore the only tRNA target for tadA, whereas yeast has seven tRNAs containing A34. Secondly, tadA is sufficient to catalyze the reaction in E.coli, whereas in yeast the Tad2p/Tad3p heterodimer is needed to modifiy A34 to I34 in tRNAs (Gerber and Keller, 1999).
The sequences of tadA and the yeast tRNA deaminase subunit Tad2p were compared with predicted Tad2 proteins from other prokaryotic organisms (Figure 1). Comparing these sequences with those of cytidine deaminases revealed that they share conserved motifs that are typical for cytidine nucleotide deaminases rather than adenosine deaminases (Gerber and Keller, 1999). On the basis of these observations, we propose that tadA is the ancestor of the eukaryotic Tad2/Tad3 proteins. Gerber and Keller (1999) suggested that Tad2 and Tad3 are paralogs that appeared by gene duplication after the divergence of prokaryotes and eukaryotes. This hypothesis is now further supported by the identification of tadA, which acts as a single polypeptide. Prokaryotic organisms encode only Tad2-like proteins, thus the appearance of Tad3 is a later event, which most probably occurred after the divergence of prokaryotes and eukaryotes.
I-containing tRNAs are predicted to recognize codons ending in C, U and A. I at the wobble position allows a single tRNA to read three different codons (NNU, NNC, NNA), which, therefore, must encode the same amino acid (Crick, 1966; Jukes, 1973). tRNAs with I at the wobble position are present in all bacteria and eukarya for which representative sets of tRNAs have been characterized (Sprinzl et al., 1998). However, Percudani (2001) suggested that I-containing tRNAs might not contribute significantly to the decoding of NNA codons in higher eukaryotes because there is always a synonymous U-starting anticodon that can read codons ending in A without wobbling. Because prokaryotes have fewer tRNA species, I is also used to decode A. tRNAArg2 is the only tRNA that decodes the CGA arginine codon. CGA is a very rare codon with a frequency of only 0.36% in E.coli (http://www.kazusa.or.jp/codon/) and might be specifically avoided because of its poor translational properties (Curran, 1995). The possibilities of codon–anticodon base pairing are greatly improved with I at the wobble position in tRNAs. This advantage might have been important for the survival of cells under selective conditions, and might have promoted the spreading of the A34 editing mechanism because the total number of tRNA genes in an organism can be reduced with tRNAs containing I34.
An E.coli strain has been described that has a point mutation in the tadA protein (formerly designated yfhC; Poulsen et al., 1992). This mutation resulted in transient resistance to the overexpression of the membrane protein gef, which has a cell-killing function (reviewed in Gerdes et al., 1990). It is not known how the cells are killed by the proteins of the gef gene family or why a mutation in tadA leads to resistance against the gef proteins. We found that extracts from strain NWL32 showed I formation, whereas extracts from the tadA mutant strain NWL37 had no deamination activity. The absence of activity in NWL37 extracts is probably due to instability of the mutated tadA protein, which might have been inactivated during extract preparation. tadA might also be stabilized by interactions with other tRNA-modifying enzymes and these interactions could be impaired in the mutant protein. This hypothesis is supported by the observation that the mutated amino acid in NWL37 is highly conserved in bacterial tadA sequences and therefore seems to have an important function (Figure 1). NWL37 cells showed no growth defect at 42°C, indicating that the cells with the mutation did not have a major disadvantage. In agreement with this proposal, we could not detect a deficiency in tRNA deamination in strain NWL37 in vivo. This result explains the wild-type growth phenotype of the mutant strain, since a tadA mutant with significantly reduced activity in vivo would be expected to be lethal. It is not known why a mutation in the tadA gene makes the cells resistant to gef, but Poulsen et al. (1992) suggest that there is a second mutation in the genome of NWL37 because some E.coli strains were only transiently resistant to gef when the mutated tadA was introduced by P1 transduction. This other mutation has not been identified, but could be responsible for the gef resistance.
In yeast, seven different tRNAs contain I34. The Tad2p/Tad3p complex probably deaminates all these tRNAs; however, not all of them have been tested in vitro with recombinant proteins. tadA is unable to replace yeast Tad2p in vitro or in vivo. However, the yeast Tad2p/Tad3p proteins are capable of deaminating tRNAArg2 from E.coli in vitro (Auxilien et al., 1996). The importance of the anticodon loop for recognition by tadA was shown with tRNAArg minisubstrates. Mutations in the anticodon of these minisubstrates completely abolished tadA activity, whereas mutations in the stem or at position 37 (3′ adjacent to the anticodon) had no effect on I formation. In summary, the results suggest that the stem–loop structure of the anticodon arm and nucleotides in the anticodon and at position 33 are key determinants for tadA activity. It has been shown previously that other tRNA modification or processing enzymes need only a few determinants in the tRNA sequence or structure to specifically catalyze a reaction. Work with tRNA synthetases has shown that a variety of minisubstrates can be used for aminoacyl ation (reviewed in Musier-Forsyth and Schimmel, 1993; Schimmel and Alexander, 1998). Synthetic substrates for specific aminoacylation can be as short as a tetraloop with a 4 bp stem and a CCA end. For alanyl-tRNA synthetase, the critical determinant for aminoacylation of tRNAAla was shown to be the G3:U70 base pair and this was also true for specific aminoacylation of the minihelix (Francklyn and Schimmel, 1989). Furthermore, it has been shown that RNase P, the enzyme that forms the mature 5′ ends of tRNAs by cleaving off the leader sequence, can act on minisubstrates that consist only of a 3 bp stem with very short extensions at the 5′ and 3′ ends (reviewed in Altman and Kirsebom, 1999). Another example is provided by the spliceosomal 15.5 kDa protein. This protein specifically interacts with the 5′ stem–loop of U4 snRNA and plays an important role in the late stage of spliceosome assembly (Nottrott et al., 1999). The crystal structure of the 15.5 kDa protein revealed that it forms hydrogen bonds with only five bases of the U4 snRNA loop (Vidovic et al., 2000).
It is not known whether the three-dimensional architecture of the tRNA is important for efficient I formation by tadA. We cannot exclude the possibility that other nucleotides, in addition to the anticodon loop of the tRNA, are important for tadA binding. Further experiments will be required to show where tadA binds to the tRNA and which nucleotides are contacted by the enzyme. It also remains to be shown which sequences in Tad proteins are needed for tRNA binding, since none of the enzymes described so far contains a known RNA-binding motif. The high substrate specificity of tadA offers a potential explanation for the fact that only one polypeptide is required. A single protein might suffice to modify tRNAArg2 in bacteria, whereas the recognition of seven different tRNAs in yeast might require a more elaborate mechanism.
Materials and methods
Plasmids and strains
See Supplementary data, available at The EMBO Journal Online.
Cloning of the tadA gene
The ORF yfhC was amplified by PCR from 25 ng of E.coli genomic DNA with primer pairs yfhC.1/yfhC.2, yfhC.3/yfhC.4 and yfhC.5/yfhC.6. The primers anneal at the 5′ and the 3′ end of the tadA ORF, respectively. The ORF was cloned into pCR2.1 (Invitrogen) and several independent clones were sequenced. tadA was then subcloned in the appropriate restriction sites of either pQE9, pTrc-FLIS6 or pGDV1.
Expression of tadA in E.coli and purification of the recombinant protein
The procedure is described for the tadA-pGDV1 construct. The tadA-pGDV1 construct was transformed into BL21 cells (Invitrogen). Two liters of 2× YT medium, supplemented with 100 µg/l ampicillin and 10 µM ZnCl2, were inoculated with 28 ml of an overnight preculture. Cultures were grown at 37°C to an OD600 of 0.94 and then induced by adding isopropyl-β-d-galactopyranoside (IPTG) to a final concentration of 0.4 mM. Incubation was continued at 24°C for 5 h. Cells were harvested by centrifugation and the pellet was resuspended in 40 ml of lysis buffer (50 mM Tris–HCl pH 8.0, 200 mM KCl, 10% glycerol, 0.02% NP-40, 2 mM β-mercaptoethanol, 0.5 mM phenylmethylsulfonyl fluoride, 0.5 µg/ml leupeptin, 0.7 µg/l pepstatin). Cells were lysed by sonication (3 × 3 min) on ice. After centrifugation, the cleared lysate was mixed with Ni2+-NTA–agarose previously equilibrated with lysis buffer. The mixture was stirred at 4°C for 45 min and then loaded on the column. The Ni2+–agarose was washed with 100 ml of wash buffer (lysis buffer + 10 mM imidazole) and bound proteins were eluted with 250 mM imidazole. Fractions 3–5 were applied to glutathione–Sepharose (Amersham Pharmacia) equilibrated with buffer A (Tris–HCl pH 8.0, 130 mM KCl, 10% glycerol, 0.02% NP-40, 1 mM DTT). Proteins were eluted with buffer B (buffer A + 10 mM reduced glutathione). A 50 µl aliquot of fraction 1 was loaded onto a gel filtration column (Superdex 200 PC3.2/30; Smart System, Pharmacia), which had been equilibrated with buffer A.
Construction of an E.coli tadA null mutant
See Supplementary data.
In vitro transcription of tRNAs and enzyme assays
In vitro transcription of tRNAs was performed essentially as described previously by Gerber et al. (1998). For transcription of minisubstrates, T7 primer and substrate primer were mixed in a 1.2:1 ratio in 10 mM Tris pH 8 with a concentration of 20 pmol/µl substrate primer and 24 pmol/µl T7 primer. Primers were incubated for 3 min at 95°C and then quickly chilled on ice. A 1 µl aliquot of primer mix was used for each reaction, which was carried out as described previously (Gerber et al., 1998). Minisubstrates were purified on a 20% urea gel. tadA activity was assayed in a reaction mixture containing 50 mM Tris–HCl pH 8.0, 25 mM KCl, 2.5 mM MgSO4, 0.1 mM EDTA, 10% glycerol, 2 mM DTT and 100– 200 fmol of 33P-labeled tRNA substrate in a 50 µl reaction as described previously (Gerber et al., 1998). When purified tadA was used, the reaction was supplemented with 1 µg of BSA. Reactions were incubated at 37°C for 15–90 min. Unlabeled 5′ IMP (Sigma) was added to the reaction mixture as an internal standard.
Sequence analysis of tRNAs
See Supplementary data.
UV cross-linking
Ten microliter reactions were carried out with 400 ng of recombinant tadA and 100 fmol of labeled tRNA in 25 mM Tris–HCl pH 8.0, 2.5 mM MgSO4, 0.05 mM EDTA. The reactions were incubated at room temperature for 6 min and then irradiated on ice in a UV Stratalinker at 500 mJ. tRNAs were digested with 500 ng of RNaseA for 25 min at 37°C. Proteins were separated on a denaturing 12% SDS–polyacrylamide gel and exposed on a phosphoimager screen.
Chemical cross-linking
Cross-linking was carried out according to a modified version of the procedure described by Fancy and Kodadek (1999). Reactions were performed in microtiter plates in a total volume of 20 µl. Reactions contained 1× cross-linking buffer (0.1× PBS, 2.5 mM MgSO4, 0.05 mM EDTA), 300 ng of purified recombinant tadA and BSA from 90 ng to 9 µg. Proteins were dialyzed against 1× PBS since Tris buffer inhibits the cross-linking reaction. Reaction buffer [15 mM sodium phosphate pH 7.5, 150 mM NaCl, 0.125 mM tris(2,2′-bipyridyl)dichlororuthenium(II) hexahydrate] (1×) and APS to a final concentration of 2.5 mM were added in the dark just before irradiation. Reactions were irradiated with a flashlight for 30 s and immediately afterwards quenched with 5 µl of 4× Laemmli loading buffer. Proteins were denatured at 95°C for 3 min, separated on a 15% SDS–polyacrylamide gel and blotted onto a nitrocellulose membrane.
Flag pull-down assay
tadA was in vitro transcribed/translated from construct pJW106 with the TnT® Coupled Reticulocyte Lysate System (Promega) according to the manufacturer’s instructions. A 500 µl aliquot of Flag-matrix slurry was washed twice with buffer TKG25 (50 mM Tris–HCl pH 8, 25 mM KCl, 10% glycerol, 0.1 mM EDTA, 0.01% NP-40). The Flag-matrix was then blocked with 10 µg of BSA for 30 min at 4°C and afterwards washed four times with buffer TKG25. A 1 µg aliquot of Flag-tagged recombinant tadA that had been purified on Ni2+-NTA–agarose, 3 µl of in vitro translated tadA and 100 ng of BSA in buffer TKG25 in a total volume of 15 µl were incubated at room temperature for 1 h. Buffer TKG25 (2× 250 µl) was added to the protein mixture and then transferred to a tube containing the Flag-matrix and incubated at 4°C for 45 min. The Flag-matrix was washed with 3× 1 ml of buffer TKG25. A 20 µl aliquot of Laemmli loading buffer was added and the Flag-matrix boiled at 95°C for 3 min. The supernatant was loaded onto a 12% SDS–polyacrylamide gel.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We thank Dr G.Church (Harvard Medical School, Boston, MA) for providing plasmid pKO3, Dr H.Loferer (Genome Pharmaceuticals Corporation, Munich, Germany) for plasmid pRDC15 and Dr S.Molin (Technical University of Denmark, Lyngby, Denmark) for E.coli strains NWL32, NWL37 and NWL43. We are grateful to U.Jenal and members of his group for Caulobacter strains and helpful discussions. We thank B.Dichtl, U.Rüegsegger and M.Schaub for critically reading the manuscript. This work was supported by the University of Basel, the Swiss National Science Foundation and the Louis-Jeantet-Foundation for Medicine.
References
- Altman S. and Kirsebom,L. (1999) Ribonuclease P. In Gesteland,R.F., Cech,T.R. and Atkins,J.F. (eds), The RNA World. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Auxilien S., Crain,P.F., Trewyn,R.W. and Grosjean,H. (1996) Mechanism, specificity and general properties of the yeast enzyme catalysing the formation of inosine 34 in the anticodon of transfer RNA. J. Mol. Biol., 262, 437–458. [DOI] [PubMed] [Google Scholar]
- Basilio C., Wahba,A.J., Lengyel,P., Speyer,J.F. and Ochoa,S. (1962) Synthetic polynucleotides and the amino acid code. Proc. Natl Acad. Sci. USA, 48, 613–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass B.L. (1997) RNA editing and hypermutation by adenosine deamination. Trends Biochem. Sci., 22, 157–162. [DOI] [PubMed] [Google Scholar]
- Bass B.L., Nishikura,K., Keller,W., Seeburg,P.H., Emeson,R.B., O’Connell,M.A., Samuel,C.E. and Herbert,A. (1997) A standardized nomenclature for adenosine deaminases that act on RNA. RNA, 3, 947–949. [PMC free article] [PubMed] [Google Scholar]
- Björk G.R., Wikström,P.M. and Byström,A.S. (1989) Prevention of translational frameshifting by the modified nucleoside 1-methyl guanosine. Science, 244, 986–990. [DOI] [PubMed] [Google Scholar]
- Björk G.R., Jacobsson,K., Nilsson,K., Johansson,M.J.O., Byström,A.S. and Persson,O.P. (2001) A primordial tRNA modifiaction required for the evolution of life? EMBO J., 20, 231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chester A., Scott,J., Anant,S. and Navaratnam,N. (2000) RNA editing: cytidine to uridine conversion in apolipoprotein B mRNA. Biochim. Biophys. Acta, 1494, 1–13. [DOI] [PubMed] [Google Scholar]
- Crick F.H.C. (1966) Codon–anticodon pairing: the wobble hypothesis. J. Mol. Biol., 19, 548–555. [DOI] [PubMed] [Google Scholar]
- Curran J.F. (1995) Decoding with the A:I wobble pair is inefficient. Nucleic Acids Res., 23, 683–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancy D.A. and Kodadek,T. (1999) Chemistry for the analysis of protein–protein interactions: rapid and efficient cross-linking triggered by long wavelength light. Proc. Natl Acad. Sci. USA, 96, 6020–6024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francklyn C. and Schimmel,P. (1989) Aminoacylation of RNA minihelices with alanine. Nature, 337, 478–481. [DOI] [PubMed] [Google Scholar]
- Gerber A.P. and Keller,W. (1999) An adenosine deaminase that generates inosine at the wobble position of tRNAs. Science, 286, 1146–1149. [DOI] [PubMed] [Google Scholar]
- Gerber A. and Keller,W. (2001) RNA editing by base deamination: more enzymes, more targets, new mysteries. Trends Biochem. Sci., 26, 376–384. [DOI] [PubMed] [Google Scholar]
- Gerber A., Grosjean,H., Melcher,T. and Keller,W. (1998) Tad1p, a yeast tRNA-specific adenosine deaminase, is related to the mammalian pre-mRNA editing enzymes ADAR1 and ADAR2. EMBO J., 17, 4780–4789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes K., Poulsen,L.K., Thisted,T., Nielsen,A.K., Martinussen,J. and Andreasen,P.H. (1990) The hok killer gene family in gram-negative bacteria. New Biol., 2, 946–956. [PubMed] [Google Scholar]
- Gott J.M. and Emeson,R.B. (2000) Functions and mechanisms of RNA editing. Annu. Rev. Genet., 34, 499–531. [DOI] [PubMed] [Google Scholar]
- Grosjean H. et al. (1996) Enzymatic formation of adenosine to inosine and to N1-methylinosine in transfer RNAs: a review. Biochimie, 78, 488–501. [DOI] [PubMed] [Google Scholar]
- Gutgsell N., Englund,N., Niu,L., Kaya,Y., Lane,B.G. and Ofengand,J. (2000) Deletion of the Escherichia coli pseudouridine synthase gene truB blocks formation of pseudouridine 55 in tRNA in vivo, does not affect exponential growth, but confers a strong selective disadvantage in competition with wild-type cells. RNA, 6, 1870–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanrahan C.J., Palladino,M.J., Ganetzky,B. and Reenan,R.A. (2000) RNA editing of the Drosophila para Na+ channel transcript. Evolutionary conservation and developmental regulation. Genetics, 155, 1149–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holley R.W., Everett,G.A., Madison,J.T. and Zamir,A. (1965) Nucleotide sequences in yeast alanine transfer RNA. J. Biol. Chem., 240, 2122–2127. [PubMed] [Google Scholar]
- Jukes T.H. (1973) Possibilities for the evolution of the genetic code from a preceding form. Nature, 246, 22–26. [DOI] [PubMed] [Google Scholar]
- Keegan L.P., Gerber,A.P., Brindle,J., Leemans,R., Gallo,A., Keller,W. and O’Connell,M.A. (2000) The properties of a tRNA-specific adenosine deaminase from Drosophila melanogaster support an evolutionary link between pre-mRNA editing and tRNA modification. Mol. Cell. Biol., 20, 825–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keegan L.P., Gallo,A. and O’Connell,M.A. (2001) The many roles of an RNA editor. Nat. Rev. Genet., 2, 869–878. [DOI] [PubMed] [Google Scholar]
- Klingler T.M. and Brutlag,D.L. (1993) Detection of correlations in tRNA sequences with structural implications. Proc. Int. Conf. Intell. Syst. Mol. Biol., 1, 225–233. [PubMed] [Google Scholar]
- Maas S. and Rich,A. (2000) Changing genetic information through RNA editing. BioEssays, 22, 790–802. [DOI] [PubMed] [Google Scholar]
- Maas S., Gerber,A.P. and Rich,A. (1999) Identification and characterization of a human tRNA-specific adenosine deaminase related to the ADAR family of pre-mRNA editing enzymes. Proc. Natl Acad. Sci. USA, 96, 8895–8900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas S., Kim,Y.G. and Rich,A. (2000) Sequence, genomic organization and functional expression of the murine tRNA-specific adenosine deaminase ADAT1. Gene, 243, 59–66. [DOI] [PubMed] [Google Scholar]
- Morse D.P. and Bass,B.L. (1999) Long RNA hairpins that contain inosine are present in Caenorhabditis elegans poly(A)+ RNA. Proc. Natl Acad. Sci. USA, 96, 6048–6053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musier-Forsyth K. and Schimmel,P. (1993) Aminoacylation of RNA oligonucleotides: minimalist structures and origin of specificity. FASEB J., 7, 282–289. [DOI] [PubMed] [Google Scholar]
- Nottrott S., Hartmuth,K., Fabrizio,P., Urlaub,H., Vidovic,I., Ficner,R. and Lührmann,R. (1999) Functional interaction of a novel 15.5kD [U4/U6.U5] tri-snRNP protein with the 5′ stem–loop of U4 snRNA. EMBO J., 18, 6119–6133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson W.R. and Lipman,D.J. (1988) Improved tools for biological sequence comparison. Proc. Natl Acad. Sci. USA, 85, 2444–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percudani R. (2001) Restricted wobble rules for eukaryotic genomes. Trends Genet., 17, 133–135. [DOI] [PubMed] [Google Scholar]
- Persson B.C., Gustafsson,C., Berg,D.E. and Björk,G.R. (1992) The gene for a tRNA modifying enzyme, m5U54-methyltransferase, is essential for viability in Escherichia coli. Proc. Natl Acad. Sci. USA, 89, 3995–3998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polson A.G., Bass,B.L. and Casey,J.L. (1996) RNA editing of hepatitis delta virus antigenome by dsRNA-adenosine deaminase. Nature, 380, 454–456. [DOI] [PubMed] [Google Scholar]
- Poulsen L.K., Larsen,N.W., Molin,S. and Andersson,P. (1992) Analysis of an Escherichia coli mutant strain resistant to the cell-killing function encoded by the gef gene family. Mol. Microbiol., 6, 895–905. [DOI] [PubMed] [Google Scholar]
- Reece K.S. and Phillips,G.J. (1995) New plasmids carrying antibiotic-resistance cassettes. Gene, 165, 141–142. [DOI] [PubMed] [Google Scholar]
- Schimmel P. and Alexander,R. (1998) Diverse RNA substrates for aminoacylation: clues to origins? Proc. Natl Acad. Sci. USA, 95, 10351–10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeburg P.H., Higuchi,M. and Sprengel,R. (1998) RNA editing of brain glutamate receptor channels: mechanism and physiology. Brain Res. Rev., 26, 217–219. [DOI] [PubMed] [Google Scholar]
- Semenov E.P. and Pak,W.L. (1999) Diversification of Drosophila chloride channel gene by multiple posttranscriptional mRNA modifications. J. Neurochem., 72, 66–72. [DOI] [PubMed] [Google Scholar]
- Smith L.A., Wang,X., Peixoto,A.A., Neumann,E.K., Hall,L.M. and Hall,J.C. (1996) A Drosophila calcium channel α1 subunit gene maps to a genetic locus associated with behavioral and visual defects. J. Neurosci., 16, 7868–7879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprinzl M., Horn,C., Brown,M., Ioudovitch,A. and Steinberg,S. (1998) Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res., 26, 148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J.D., Higgins,D.G. and Gibson,T.J. (1994) Clustal_W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res., 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidovic I., Nottrott,S., Hartmuth,K., Lührmann,R. and Ficner,R. (2000) Crystal structure of the spliceosomal 15.5 kD protein bound to a U4 snRNA fragment. Mol. Cell, 6, 1331–1342. [DOI] [PubMed] [Google Scholar]