

Abstract
Embryonic stem (ES) cells contain a p53-dependent apoptosis mechanism to avoid the continued proliferation and differentiation of damaged cells. We show that mouse ES cells lacking Ets1 are deficient in their ability to undergo UV-induced apoptosis, similar to p53 null ES cells. In Ets1–/– ES cells, UV induction of the p53 regulated genes mdm2, perp, cyclin G and bax was decreased both at mRNA and protein levels. While p53 protein levels were unaltered in Ets1–/– cells, its ability to transactivate genes such as mdm2 and cyclin G was reduced. Furthermore, electrophoretic mobility shift assays and immunoprecipitations demonstrated that the presence of Ets1 was necessary for a CBP/p53 complex to be formed. Chromatin immunoprecipitations demonstrated that Ets1 was required for the formation of a stable p53–DNA complex under physiological conditions and activation of histone acetyltransferase activity. These data demonstrate that Ets1 is an essential component of a UV-responsive p53 transcriptional activation complex in ES cells and suggests that Ets1 may contribute to the specificity of p53-dependent gene transactivation in distinct cellular compartments.
Keywords: apoptosis/CBP/Ets1/p53/UV
Introduction
Pre-implantation embryos are hypersensitive to DNA damage and can rapidly undergo apoptosis without cell-cycle arrest (Aladjem et al., 1998; Heyer et al., 2000). Indeed, apoptosis is observed in the inner cell mass of up to 75% of pre-implantation embryos (Hardy et al., 1989). The pro-apoptotic protein p53 plays a critical role in the cellular stress response and appears to be involved in these early embryonic checkpoint controls. Significantly, the absence of p53 resulted in a high rate of embryonic malformations (Corbet et al., 1999; Heyer et al., 2000). This suggests that mechanisms exist to avoid the expansion of potentially abnormal embryonic cells, which could contribute to developmental abnormalities.
Murine embryonic stem (ES) cells, which are derived from the inner cell mass of pre-implantation embryos, have provided an important resource to investigate these early embryonic checkpoints. Exposure of these cells to DNA-damaging agents, such as UV irradiation, have demonstrated that p53 levels rapidly increase, together with associated transcriptional activity (Ko and Prives, 1996; Levine, 1997; Corbet et al., 1999). The factors which contribute to this activation of p53 could thus have significant roles in ensuring the normal development of healthy embryos.
Although the mechanisms that regulate p53 transcriptional activity are not precisely defined, this complex process is known to include post-translational events such as phosphorylation and acetylation (Meek, 1998; Sakaguchi et al., 1998). Phosphorylation of specific residues alters the stability of p53 and its ability to form complexes with DNA and/or other proteins (Kapoor and Lozano, 1998; Lu et al., 1998; Unger et al., 1999). For example, phosphorylation of N-terminal serine residues is believed to be important for the interaction of p53 with the transcriptional co-activators p300/CREB-binding protein (CBP) and p300/CBP-associated factor (PCAF; Lambert et al., 1998; Sakaguchi et al., 1998). Furthermore, p300/CBP binding and subsequent acetylation of p53 has also been shown to enhance sequence-specific p53–DNA binding (Gu and Roeder, 1997). These complex regulatory events, which are just beginning to be defined, result in p53-dependent transactivation of a number of genes involved in the apoptotic process. These include MDM2, GADD45, cyclin G, bax, p21 and PERP (El-Deiry et al., 1993; Perry et al., 1993; Ko and Prives, 1996; Matsuyama et al., 1998; Attardi et al., 2000). Production of these proteins activates a signaling cascade that results in cell death via DNA cleavage and nuclear fragmentation characteristic of apoptosis.
There is considerable circumstantial evidence to link p53 and the transcription factor Ets1. Both p53 and Ets1 induce apoptosis, are regulated by Ras pathway phosphorylation, bind to a comparable region of CBP and have similar expression patterns during development (Schmid et al., 1991; Kola et al., 1993; Maroulakou et al., 1994). Indeed, the p42 Ets1 splice variant has been shown to induce apoptosis in human colon cancer cells (Li et al., 1999) and Ets1 is involved in the cellular response to stress (Seth et al., 1999; Lelievre et al., 2001). Ets1 transcriptional activity is also enhanced following phosphorylation by the Ras pathway (Wasylyk et al., 1997; for a review, see Watson et al., 2002). Furthermore, the CH3 domain of CBP binds Ets1, is the same domain that has been shown to bind p53 (Yang et al., 1998), and is involved in the regulation of genes controlling apoptosis and the cell cycle (Avantaggiati et al., 1997). Based upon this evidence, we propose that Ets1 and p53 may compete and/or complex with CBP and other proteins to modulate transcriptional activity of genes in p53-dependent signaling pathways.
In the absence of direct data linking Ets1 and p53, we generated Ets1 null mouse ES cells by homologous recombination and determined that they have a reduced apoptotic response following exposure to UV. This was strikingly similar to that shown for p53 null ES cells (Corbet et al., 1999). Ets1 null ES cells also had significantly lower levels of mdm2, perp, cyclin G and bax mRNA, which are all defined p53 target genes. Significantly, Ets1 was found to be part of a p53 and CBP-containing complex bound to a p53 consensus binding site in electrophoretic mobility shift assays (EMSA) and was necessary for the assembly of CBP to this UV-induced p53 complex.
Results
Generation of Ets1 null mouse ES cells
Ets1-deficient ES cells were generated by homologous recombination using the loxP-CRE system. The vector detailed in Figure 1 was used to generate a targeted allele where exons 3–6 and a neomycin resistance gene were floxed. This strategy was designed such that specific recombination by CRE recombinase removed the floxed exons (and neomycin), which would result in a frameshift and premature stop codon (should mRNA still be synthesized). Heterozygous targeted ES clones were obtained from screening G418 and gancyclovir-resistant colonies (Figure 1C and D). Correctly targeted clones were identified with a 5′ external probe, which detected 5.5 kb wild-type and 3.8 kb targeted bands on EcoRV-digested ES cell DNA (Figure 1C). These clones were also confirmed using a 3′ internal probe (Figure 1D). Clones with two targeted alleles were generated by culture in 2.0 mg/ml G418 and identified by loss of the wild-type band (5.5 kb) using a larger 5′ probe on EcoRV-digested DNA (e.g. clones 2 and 17; Figure 1E). Two independent double-targeted Ets1 ES cell clones (2 and 17) were transiently transfected with a PGK-CRE recombinase vector. Both targeted clones were able to generate sub-clones with appropriately spliced Ets1 alleles (2–15 and 17–34, respectively), which gave the expected 13 kb EcoRV band on a Southern blot (Figure 1E). To verify that these ES cell clones were truly Ets1 null, RNA was extracted and the absence of Ets1 expression was confirmed by northern blot analysis (Figure 1F; data not shown). These targeted clones (2 and 17) and their CRE-recombined derivatives (2–15 and 17–34) were used in subsequent assays.


Fig. 1. Targeted mutagenesis of the Ets1 gene. (A) The wild-type Ets1 locus and targeting vector showing the targeting strategy and restriction enzyme sites used for screening. The construct was designed such that exons 3–6 and a neomycin cassette were floxed for subsequent excision by CRE recombinase. 5′ and 3′ probes, which were used to discriminate between alleles, are also shown. LoxP sites are indicated by filled triangles. (B) Schematic representation of the wild-type and modified Ets1 locus: (1) wild-type locus; (2) targeted locus with appropriate insertion of loxP sites flanking exons 3–6; (3) double-targeted Ets1 locus generated by High G418 selection; and (4) double-knockout ES cells generated by CRE-mediated excision. (C) EcoRV-digested DNA probed with the 1.5 kb genomic fragment external to the targeting construct. WT and clone 270 show the 5.5 kb WT allele only, whereas clone 269 shows both the WT and 3.8 kb targeted band. (D) Confirmation of targeting of clone 269. Genomic DNA from wild-type and Ets1-targeted clone 269 probed with the 1.7 kb 3′ internal genomic fragment. Only clone 269 shows the expected 2.5 and 5.7 kb targeted bands (HindIII and BglII digested DNA, respectively) in addition to the expected WT bands. (E) Double-targeted ES cell clones generated by High G418 resistance (17, 2) and subsequent removal of the Ets1 exons 3–6 by CRE recombinase (17–15, 2–34). The CRE spliced and unspliced targeted alleles are indicated by the 13 and 1.7/3.8 kb bands, respectively, using the 5.5 kb 5′ probe. (F) Northern blotting showing absence of Ets1 mRNA expression in Ets1-targeted and spliced ES cell clones using a murine Ets1 cDNA probe. Poly(A)+ mRNA (3 µg) of each was used and reprobed for GAPDH to demonstrate equal loading. These clones were subsequently considered Ets1–/–.
Ets1–/– ES cells have decreased p53 mRNA levels
Ets1 and Ets2 have been shown to activate p53 promoter constructs in vitro and have high affinity for an element within the promoter containing palindromic Ets-binding sites (Venanzoni et al., 1996). We therefore analysed the level of p53 mRNA in feeder cell-depleted wild-type and Ets1–/– ES cells by northern blot analysis (Figure 2A). Interestingly, Ets1–/– ES cells expressed less p53 mRNA than that observed in wild-type ES cells and our targeted cells prior to CRE-mediated Ets1 inactivation. Similar results were observed for a second independently derived Ets1–/– clone (data not shown). Thus the absence of Ets1 was associated with a reduction in p53 mRNA levels in these ES cells.
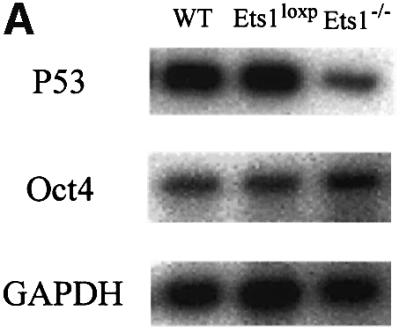

Fig. 2. Reduced expression of p53 mRNA in Ets1–/– ES cells is not due to altered morphology or differentiation status. (A) Northern blotting showing reduced expression of p53 in Ets1–/– ES cells compared with wild-type (WT), double-targeted (Ets1loxP) ES cells. The same blot was used to determine the that relative levels of Oct4 mRNA were unaltered and GAPDH was used to demonstrate equal loading. (B) Photomicrographs of wild-type (1 and 4), Ets1loxP (2 and 5) and Ets1–/– (3 and 6) ES cells. The upper panel is a phase-contrast image whereas the lower panel is labeling with anti-SSEA1–FITC, which is expressed only in undifferentiated ES, indicating that these cultures contain very few differentiated cells. Bar corresponds to 100 µm.
Morphology and differentiation status of Ets1–/– ES cells
Since p53 has been reported to be down-regulated after ES cell differentiation (Louis et al., 1988), we examined the morphology and differentiation status of the ES cell clones. The double-targeted ES cell lines (before CRE, and are Ets1+/+) had a normal phenotype and were indistinguishable from wild-type cells in all assays. Ets1–/– ES cells demonstrated morphology similar to control cells (Figure 2B). We have also determined that these ES cells have similar expression of SSEA1 [immunohistology and flow cytometry (>95%)], alkaline phosphatase (histology) and Oct4 [northern blot and flow cytometry (>95%)] (Figure 2; data not shown). Each of these markers are only expressed in undifferentiated ES cells and not in differentiated derivatives (Wobus et al., 1984; Scholer et al., 1989). These data indicate that the differentiation status of control and Ets1 null ES cells was similar.
ES cells lacking Ets1 are less susceptible to UV-induced apoptosis
Since there was a lower level of p53 mRNA in Ets1–/– ES cells and Ets1 has been reported to be proapoptotic, we examined UV-induced apoptosis in these cells by propidium iodide staining. There was no significant difference in the rates of proliferation or apoptosis between untreated wild-type, Ets1loxP and two independent Ets1–/– ES cell lines (15 and 34; Figure 3; data not shown). However, when we exposed these cells to various doses of UV irradiation, which is effective at inducing p53-mediated apoptosis in ES cells (Sabapathy et al., 1997; Corbet et al., 1999), the number of apoptotic cells was significantly reduced in the Ets1–/– ES cells (Figure 3). The proportion of cells undergoing apoptosis in wild-type ES cells (and the parental Ets1 double-targeted ES cells) increased with increasing doses of UV irradiation with ∼60% of Ets1+/+ ES cells apoptosing after 40 J/m2 UV (Figure 3A and B). In contrast, only 30% apoptosis was observed in Ets1–/– ES cells exposed to the same dose of UV (Figure 3A and B). Time-course analysis after 40 J/m2 UV revealed that there was initially no significant difference in proportion of apoptotic cells, as all genotypes had a relatively small fraction of apoptotic cells 4 h post-irradiation (Figure 3C), and by 9 h, 20–30% of cells from both genotypes were apoptotic. After this initial period of cell death, the percentage of apoptotic Ets1–/– cells did not significantly increase, whereas the fraction of wild-type and Ets1loxP cells continued to increase for up to 12 h. To confirm that this was truly apoptosis, we also examined these cells for morphological features characteristic of apoptosis by Hoechst staining (Figure 3D–I) and the characteristic fragmentation of DNA (Figure 3J). Untreated wild-type and Ets1–/– ES cells showed very similar morphological features (Figure 3D–F); however, after 40 J/m2 of UV treatment, the wild-type and targeted (Figure 3G and H) but not Ets1–/– cells (Figure 3I) demonstrated nuclear condensation and fragmentation. Similarly, agarose gel electrophoresis of DNA extracted from wild-type cells 12 h after UV irradiation (40 J/m2) showed DNA laddering, which was not observed in UV-treated Ets1–/– cells (Figure 3J).



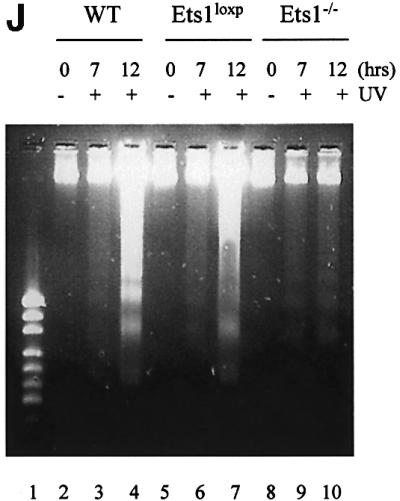
Fig. 3. Ets1–/– ES cells are resistant to UV irradiation-induced apoptosis. (A) Flow cytometric analysis of fixed wild-type, Ets1-targeted and two independent Ets1 null ES cell clones stained with propidium iodide 12 h after UV irradiation (40 J/m2). Percentage of cells with a hypodiploid (sub-2N) DNA content indicative of apoptosis are shown. (B) Graphical representation of the percentage of cells undergoing apoptosis after various doses of UV irradiation. Data is shown as mean ± SD of three independent experiments. (C) Percentage of cells undergoing apoptosis at different time points after irradiation with 40 J/m2 of UV. Data is shown as mean ± SD of three independent experiments. (D–I) Immunofluorescent images of Hoechst 33342-stained WT (D and G), Ets1loxP (E and H) and Ets1–/– (F and I) ES cells, with and without UV irradiation. Significant nuclear condensation and fragmentation is not observed in untreated cells (D–F) or UV-irradiated Ets1 null cells (I). However, a significant number of WT and Ets1-targeted ES cells showed nuclear fragmentation after UV irradiation (G and H). Bar corresponds to 20 µm. (J) Ethidium bromide staining of DNA from WT, Ets1loxP and Ets1–/– ES cells after 40 J/m2 UV irradiation analysed by agarose gel electrophoresis, demonstrating bands of DNA fragmentation in WT and targeted but not Ets1–/– cells. Lane 1 shows pUC19/HpaII marker.
Addition of exogenous Ets1, but not exogenous p53, restores sensitivity to UV irradiation-induced apoptosis in Ets1–/– ES cells
To verify that the resistance to apoptosis in Ets1 null ES cells was only due to the Ets1 status, we transfected Ets1–/– cells with FLAG-tagged Ets1 or vector alone constructs (pEFBOS 1 FLAG) as a control. Transfected cells were shown to express FLAG-Ets1 protein by αFlag western blotting (Figure 4A). These ES cell lines were exposed to 40 J/m2 UV and examined for apoptosis by propidium iodide staining and DNA fragmentation. There was no significant difference in the apoptosis rates between untreated Ets1–/– ES cells expressing FLAG-Ets1 or FLAG alone (Figure 4C), indicating these Ets1 expression alone does not activate apoptosis. However, after UV treatment, Ets1–/– cells transfected with FLAG-Ets1 showed increased sensitivity to apoptosis, ∼57% cell death at 12 h after UV irradiation, similar to that observed for wild-type cells (Figure 4C). In contrast, cells transfected with FLAG alone showed only 27% apoptosis (Figure 4C). These observations demonstrate that the inability of our ES cells lacking Ets1 to undergoing UV-induced apoptosis is due to their Ets1 status only, since exogenous Ets1 can restore the normal response.
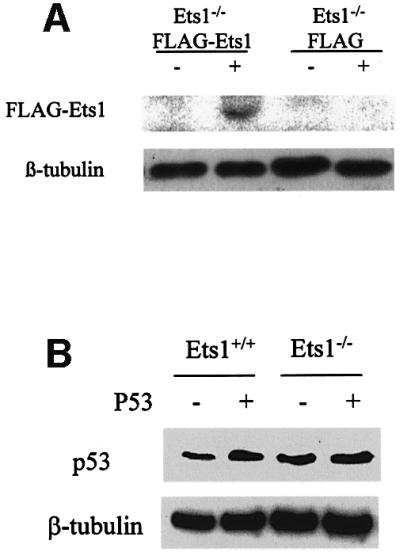

Fig. 4. Addition of exogenous Ets1, but not exogenous p53, restores sensitivity to UV irradiation-induced apoptosis in Ets1–/– ES cells. Western blots of ES cell lysates after transfection with FLAG-Ets1 (A) and exogenous p53 (B), which demonstrate expression of FLAG-Ets1 and increased expression of p53, respectively. (C) FACS analysis of propidium iodide-stained wild-type and Ets1–/– ES cells expressing exogenous CMV-p53 or EF1α-FLAG/Ets1 after UV treatment. These demonstrate characteristic apoptosis after UV treatment of ES cells, which express endogenous or exogenous Ets1, regardless of p53 status.
Since we determined that p53 mRNA levels were also decreased in Ets1–/– ES cells and a similar reduced sensitivity to UV-induced apoptosis has been observed in ES cells lacking p53 (Corbet et al., 1999), we tested whether addition of p53 would reconstitute the apoptotic response. Ets1–/– and wild-type ES cells were transfected with a CMV-p53 construct that has previously been shown to induce cell-cycle arrest and apoptosis in human and murine cells (Wills et al., 1994; Linke et al., 1996). CMV-p53 transfectants did show increased levels of p53 protein by western blotting; however, unlike p53 mRNA levels (Figure 2A), there was no significant difference in endogenous p53 protein levels between Ets1–/– and wild-type cells (Figure 4B). Furthermore, high levels of exogenous p53 did not induce apoptosis or cell-cycle arrest, and the ability of Ets1–/– ES cells to undergo apoptosis was not restored even after UV treatment, as determined by propidium iodide staining (Figure 4C) and DNA laddering analysis (data not shown). For example, in Ets1–/– cells expressing increased levels of p53, only 35% of cells were apoptotic compared with 27% with endogenous p53 only. There was also no significant difference in the proportion of Ets1+/+ cells undergoing apoptosis after increasing p53 levels by transfection of exogenous p53. Thus the decreased sensitivity of Ets1–/– ES cells to UV-induced apoptosis appears to be due to the lack of Ets1, and not related to levels of p53 protein.
The expression of p53-dependent UV-activated genes is reduced in cells lacking Ets1
Although UV-induced ES cell apoptosis has been shown to be p53 dependent (Sabapathy et al., 1997; Corbet et al., 1999), our data demonstrate that UV-induced apoptosis is Ets1 dependent and is reduced in the absence of Ets1. Furthermore, it has previously been shown that Ets1 can be proapoptotic via a p53-independent mechanism (Li et al., 1999, 2000), thus we examined the expression of a number of p53-dependent and -independent apoptosis-regulating genes, including perp, mdm2, cyclin G, bax (p53 dependent), Bcl-2 and Bcl-XL (p53 independent; Figure 5). Northern blot analysis of wild type and two Ets1–/– ES cell clones demonstrated that there was no significant difference between the non-induced levels of the p53-dependent genes perp, mdm2, cyclin G and bax (Figure 5A). How ever, these genes were strongly induced by UV irradiation in wild-type and Ets1-targeted ES cells, but were poorly induced by UV treatment in Ets1–/– ES cells (Figure 5A). These data correlated with western blot analysis, which demonstrated that mdm2 and Bax proteins were induced by UV in Ets1+/+ but not Ets1–/– ES cells (Figure 5B). In contrast, levels of the p53-independent proteins Bcl-2 and Bcl-XL were not induced by UV irradiation in wild-type or Ets1–/– cells (Figure 5B). Interestingly, western blot analysis of p53 protein indicated that p53 protein was expressed and induced by UV similarly in Ets1+/+ and Ets1–/– ES cells.

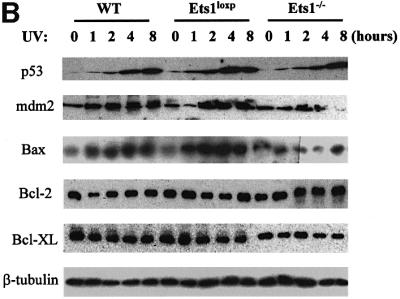
Fig. 5. The expression of p53 transactivated genes is reduced in cells lacking Ets1 following UV irradiation. (A) Results of a typical northern blot experiment measuring the expression of p53-regulated genes in wild-type (wt), Ets1-targeted (Ets1loxP) and Ets1–/– ES cells before and after UV irradiation (40 J/m2). Cells were analysed and RNA isolated at times indicated after UV treatment. Probes specific for mouse perp, mdm2, cyclin G and bax were used for northern blot analyses with GAPDH as a loading control. Data from five independent experiments of two independent Ets1–/– clones were quantified using a Fuji Image Reader VI.3E and expressed as mean ± SD, relative to a GAPDH loading control. (B) Expression of p53-regulated proteins in wild-type and Ets1–/– ES cells. Cells were isolated at the indicated time points (0, 1, 2, 4 and 8 h) post-UV irradiation (40 J/m2). Antibodies specific for mouse p53, mdm2, Bax, Bcl-2, Bcl-XL and β-tubulin (loading control) were used for western blot analyses. Analysis was performed at least three times on two independent Ets1–/– clones and data from a representative experiment is shown.
These data suggest that Ets1 may regulate perp, mdm2, cyclin G and bax, which are defined p53 target genes, either independently of p53 or via post-translational regulation of p53 transactivational activity.
p53 transactivational activity induced by UV requires Ets1
p53 transactivational activity is regulated, at least in part, by post-translational modification such as phosphorylation, acetylation and interaction with other proteins (Meek, 1998; Sakaguchi et al., 1998). In the absence of Ets1, the levels of p53-regulated genes were altered although p53 protein levels were unchanged, thus we investigated whether the transactivational activity of p53 was altered in the absence of Ets1. Mdm2 and cyclin G promoter constructs were used to determine whether the alterations observed in mRNA and protein levels were due to alterations in transcription. Consistent with our other data, when these constructs were introduced into Ets1+/+, Ets1loxP or two Ets1–/– ES cell clones, they were more efficiently activated by UV in wild-type rather than Ets1–/– cells (Figure 6). To demonstrate that this action was mediated by p53, constructs with mutations in the p53REI site of the mdm2 promoter and p53REII site of cyclin G promoter, which have been shown to remove p53 responsiveness (Zauberman et al., 1995a,b; Yardley et al., 1998), were similarly introduced into these cell lines. These mutations removed all responsiveness of these promoters to UV irradiation regardless of the Ets1 status (Figure 6). Indeed, the response of the promoters containing a p53RE site mutation were transactivated less in Ets1+/+ cells than the wild-type promoters in Ets1–/– ES cells. In contrast, the mdm2 promoter containing mutations in one of the Ets-binding elements was able to respond to UV treatment of wild-type ES cells at a reduced level (Figure 6A), similar to that observed for the wild-type mdm2 promoter in Ets1 null cells. The Ets-binding element mutation did not alter the response in Ets1–/– ES cells.

Fig. 6. UV enhances p53 transcriptional activity in wild-type but not Ets1–/– ES cells. (A) Wild-type and Ets1–/– ES cells were transfected with wild-type mdm2 promoter-luciferase constructs or those containing mutations in the defined p53- or Ets-binding sites (mdm2, mdm2Δp53I and mdm2ΔEtsA, respectively). Cells were exposed to UV irradiation 24 h after transfection and harvested at 36 h. Luciferase activity was determined relative to a β-gal control. Each assay was performed at least three times in triplicate. Data is shown as the mean of independent experiments ± SD. (B) As above, except the mouse cyclin G promoter and p53 element mutant cyclin GΔp53II promoter-reporter vectors were transfected into wild-type and Ets1–/– ES cells and exposed to UV irradiation. Mean of at least three independent experiments is shown ± SD.
These data demonstrate that the p53-binding element of these promoters is required for the UV response, but the presence of the transcription factor Ets1 and the Ets(A)-binding site of the mdm2 promoter is necessary for optimal transactivation.
The p53–CBP complex induced by UV irradiation contains and requires Ets1
Our data indicate that the reduced induction of p53-dependent genes after UV irradiation in Ets1–/– ES cells was transcriptionally mediated; however, the mechanism by which Ets1 contributed to the transactivation of these genes was unclear. Thus we determined whether the ability of p53 to bind DNA was altered in these cells. Nuclear extracts were isolated from UV-irradiated ES cells and EMSA performed with a p53-binding element consensus sequence that has previously been shown to bind p53 after UV exposure (Figure 7; El-Deiry et al., 1992; Sabapathy et al., 1997). These data demonstrated that p53-binding activity was induced in both wild-type and Ets1 null cells at 5 h after UV treatment (Figure 7A, lanes 3 and 4, and lanes 8 and 9). Specificity of the p53–DNA complex is demonstrated both by competition with unlabelled oligonucleotide (Figure 7A, lanes 5 and 10) and the ability to supershift the complex with αp53 antibody (Figure 7B, lanes 4 and 8). Importantly, the p53 complex derived from wild-type but not Ets1 null nuclear extracts was also supershifted by αEts1 antibody (Figure 7B, lane 3), indicating that Ets1 and p53 are part of a stable complex bound to the p53-binding element. Anti-FLAG and monoclonal anti-Ets1 antibodies were also able to supershift the p53 complex in Ets1–/– ES cells reconstituted with FLAG-Ets1 (Figure 7C, lanes 3–5). Anti-Ets2 or anti-Ets1 antibodies pre-absorbed with recombinant Ets1 protein were unable to supershift the complex (Figure 7B, lanes 2, 5 and 9). Since both p53 and Ets1 have been shown to interact with CBP, we also used αCBP antibody in these assays (Figure 7D). Similar to previous studies (Lill et al., 1997), this antibody was able to inhibit p53-p300/CBP binding to p53-binding element in wild-type ES cells (Figure 7D, lanes 3–5), but not Ets1–/– cells (Figure 7D, lanes 8–10). These results suggest that Ets1 is normally present in the UV-induced p53–DNA complex. Although, we did not detect variation in the size of this complex between wild-type and Ets1–/– cells, we have not yet determined whether, due to the large size of this complex, our assays were unable to resolve these differences or perhaps other protein/s bind this complex in the absence of Ets1.


Fig. 7. The UV-induced complex that binds the consensus p53–DNA-binding sequence in ES cells contains p53, Ets1 and CBP. (A) Nuclear extracts were prepared from untreated or UV-irradiated wild-type and Ets1–/– ES cells (40 J/m2) 5 h post-irradiation. The indicated volume of extracts were incubated with [γ-32P]dATP-labeled p53 consensus oligonucleotides with or without a 5-fold excess of unlabeled oligonucleotide competitor and separated on acrylamide gels. The position of the specific p53–DNA complex is indicated. (B) Nuclear extracts of ES cells treated with UV as above were incubated with or without Ets1, p53 or Ets2 antibodies as indicated and analyzed in EMSA using the [γ-32P]dATP-labeled p53 consensus oligonucleotide. Ets1Abs indicates that anti-Ets1 antibody was preabsorbed with recombinant His-tagged Ets1. (C) Nuclear extracts were prepared from UV-irradiated Ets1–/– ES cells transfected with FLAG-Ets1 or FLAG alone, incubated with 3/7 µl antibodies to p53, Ets1 (monoclonal) or FLAG as indicated, and analysed using the [γ-32P]dATP-labeled p53 consensus oligonucleotide. The complex was supershifted by p53, Ets1 and anti-FLAG antibodies only in ES cells containing FLAG-Ets1. (D) EMSA of nuclear extracts from UV-irradiated wild-type and Ets1–/– ES cells with [γ-32P]dATP-labeled p53 consensus oligonucleotides and antibodies to p53 (3 µl) or CBP (3–10 µl) as indicated. The formation of the complex was inhibited by CBP antibody in wild-type ES cells, but not in Ets1–/– ES cells.
The ability of p53 to complex with proteins necessary for transcriptional activation (e.g. CBP) is altered in UV-irradiated Ets1–/– cells. To further investigate the nature of this complex, we used the combination of immunoprecipitation and western blotting to examine p53–CBP–Ets1 interactions in untreated or UV-irradiated Ets1+/+ and Ets1–/– ES cells. We confirmed that CBP was present at a similar level in all lysates (Figure 8A) and demonstrated that p53 and Ets1 were co-precipitated with CBP from wild-type cells after UV treatment (Figure 8B). In contrast, p53 was not immunoprecipitated with CBP in Ets1–/– ES cells or in untreated wild-type cells, although similar amounts of CBP were present (Figure 8B). Similarly, CBP was co-precipitated with p53 only in the presence of Ets1 in immunoprecipitations using αp53 antibody (Figure 8C). Since we demonstrated that introduction of FLAG-Ets1 into Ets1–/– cells restored the apoptotic response, we examined whether ES cells with exogenous FLAG-Ets1 only were capable of forming the p53–CBP–FLAG-Ets1 complex. Lysates from these cells were immunoprecipitated with αp53 and precipitates probed for the presence of p53, FLAG-Ets1 and CBP by western blotting (Figure 8D). CBP was only co-precipitated in cells containing endogenous or exogenous Ets1 after UV irradiation, although p53 was present in all samples (Figure 8D). FLAG-Ets1 was also co-precipitated with p53.

Fig. 8. UV induces a complex formation between p53, Ets1 and CBP. Total cell lysates were prepared from wild-type and Ets1–/– ES cells 6 h after exposure to UV irradiation (40 J/m2). These lysates were shown to have similar levels of CBP by western blotting using β-tubulin as a loading control (A). To determine whether complexes containing CBP and p53 were formed, these lysates were immunoprecipitated with CBP (B) or p53 (C) antibodies and the bound complexes were analysed for the presence of p53 and CBP by western blotting (B and C). In each case, similar amounts of CBP (B) or p53 (C) were immunoprecipitated from each ES cell genotype; however, p53 and CBP were co-precipitated after UV irradiation in wild-type but not Ets1–/– ES cells. (D) Similar immunoprecipitations with p53 antibody of wild-type, Ets1–/– (+ FLAG) and Ets1–/– (+ FLAG-Ets1) ES cell lysates. These precipitated complexes were examined for p53, CBP and FLAG-Ets1 by western blotting demonstrating that CBP was co- precipitated with p53 in the presence of exogenous Ets1.
These data indicate that after UV irradiation of ES cells, Ets1 is necessary for the formation of a p53–CBP (and Ets1) complex, which is believed to be critical for p53 transactivation (Lill et al., 1997; Espinosa and Emerson, 2001).
Ets1 is required for p53 or acetylated histone H3 co-precipitation with the mdm2 promoter in ChIP assays
It is clear from other studies that p53 associates with a complex in vivo containing p300/CBP, which has histone acetyltransferase activity and this complex plays a role in p53-mediated activation. Since we have demonstrated that Ets1 is required for the formation of the p53–CBP (Ets1) complex and optimal transcriptional activation of promoter reporter constructs after UV irradiation, we investigated whether p53 required Ets1 to bind the mdm2 promoter under physiological conditions by ChIP.
Thus, we cross-linked chromatin from wild-type and Ets1–/– ES cells with or without UV treatment, and fragmented chromatin was immunoprecipitated with αp53 antibodies and analysed for the mdm2 promoter by PCR and Southern blotting. Phosphorimager scans of representative slot blots probed with the mdm2 probes are shown in Figure 9A. Similar semi-quantitative PCR specific for the PDH2 (testis-specific gene) and GAPDH promoters were performed as controls. This analysis demonstrated that while an increased number of mdm2 promoter fragments were precipitated from wild-type cells after UV irradiation compared with untreated and Ets1–/– ES cells, no UV-induced increase in p53–mdm2 promoter complexes were observed in the absence of Ets1.
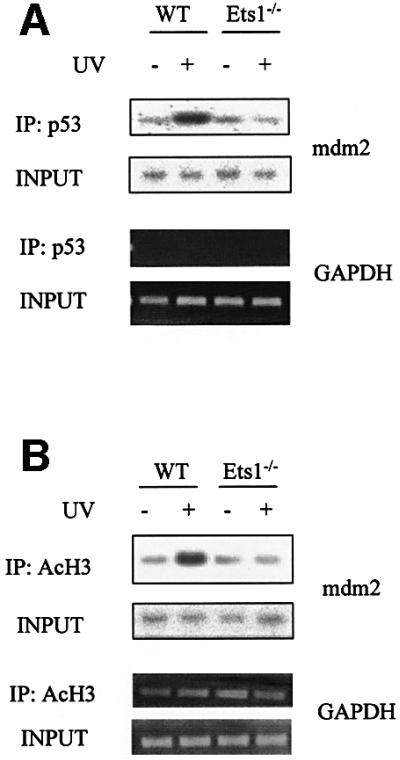
Fig. 9. p53 binds and activates the mdm2 promoter after UV irradiation under physiological conditions in wild-type but not Ets1–/– ES cells. Hybridization of a mdm2 promoter-specific probe to ChIP DNA from wild-type and Ets1–/– ES cells with and without treatment with 40 J/m2 UV irradiation. (A) The top panel shows hybridization to DNA precipitated with p53 antibody and amplified by PCR with mdm2 promoter-specific primers flanking the p53 sites. The second panel is a similar hybridization to input DNA. The specificity of p53 immunoprecipitation is shown by similar PCR amplification of input and p53-precipitated DNA for GAPDH. (B) Precipitation of DNA with antibody to acetylated H3 (IP: AcH3) and input DNA similarly amplified with oligonucleotides to mdm2 or GAPDH, demonstrating specific induction of mdm2 by UV irradiation only in wild-type cells.
Gene activation under physiological conditions has also been shown to correlate with histone acetylation. Furthermore, CBP, which we have shown to complex with Ets1 and p53, has intrinsic histone acetyltransferase activity. Thus we also assessed whether the mdm2 promoter was bound to acetylated H3 in these cell lines after UV irradiation using antibody to acetylated H3 in ChIP assays. These assays demonstrated that while the GAPDH promoter was consistently precipitated, the mdm2 promoter was only co-precipitated with acetylated histones after UV irradiation in the presence of Ets1 (Figure 9B). This indicates that Ets1 is required for p53 binding and transcriptional activation of the mdm2 promoter under physiological conditions.
Discussion
The role of p53 in the regulation of UV-induced apoptosis in ES cells is well established. Following UV exposure, ES cells show a rapid rise in p53 protein levels (Figure 5B; Graeber et al., 1994) and may undergo apoptosis (Savatier et al., 1994; Canman et al., 1995). These mechanisms have been shown to be p53 dependent. Our data indicate that Ets1 null ES cells are less sensitive to UV-induced apoptosis. This is due to the reduced transactivational activity of p53, since Ets1 is demonstrated herein to be necessary for the formation or stabilization of the p53–CBP-containing transcriptional activation complex and subsequent histone acetylation after UV irradiation. Therefore, since p53 is not as transcriptionally active in Ets1–/– cells, their apoptotic-resistant phenotype resembles that observed in p53–/– ES cells.
This is the first direct demonstration of a role for Ets1 in p53-dependent apoptosis and the mechanism by which it facilitates p53 transactivational activity. Previously, there has been contradictory data regarding the role of Ets1 in apoptosis. In Rag2–/– Ets1–/– chimeras, where T and B lymphocytes are derived from Ets1–/– cells, Ets1 was shown to be required for the normal survival and activation of T cells, and Ets1–/– T cells underwent spontaneous apoptosis, indicating a possible anti-apoptotic role (Bories et al., 1995; Muthusamy et al., 1995). Interestingly, our data herein indicate that the anti-apoptotic gene Bcl-2 is not regulated by Ets1 in ES cells; however, Bcl-2 is transactivated by Ets1 in some other cellular contexts (Li et al., 2000). In contrast, in p53 null colon carcinoma cells, interaction of Ets1 and the Fas-binding protein Daxx suppresses Bcl-2 transactivation (Torii et al., 1999; Li et al., 2000) and Ets1 has been shown to up-regulate the pro-apoptotic gene caspase-1 (Li et al., 1999). Ets1 has also been shown to have opposing roles in endothelial cells where Ets1 has recently been shown to induce pro-apoptotic genes such as bid, caspase-4 and p21 (Teruyama et al., 2001), together with genes involved in angiogenesis. Dominant-negative Ets1 was also shown to inhibit angiogenesis (Nakano et al., 2000). These data suggest that Ets1 can also have a pro-apoptotic role, perhaps when interacting with other regulators. These apparently conflicting data on the role of Ets1 in apoptosis, together with our demonstration that Ets1 is necessary for UV-induced apoptosis in ES cells, may be explained by differences in the regulation of other factors and particular cellular context. For example, in thymocytes and T lymphocytes, which can undergo apoptosis by both p53-dependent and -independent mechanisms, Ets1–/– but not p53–/– cells undergo spontaneous apoptosis (Clarke et al., 1993; Lowe et al., 1993; Bories et al., 1995; Muthusamy et al., 1995). In these cells, Ets1 appears to positively regulate Bcl-2 and act in the survival pathway (Li et al., 2000). In contrast, in the context of UV-induced apoptosis of ES cells, which has previously been shown to be p53 dependent (Corbet et al., 1999), our data demonstrate that Ets1 performs an important pro-apoptotic role.
During pre-implantation embryonic development, cells within the developing embryo become hypersensitive to DNA damage, and after exposure to a DNA-damaging agent, undergo p53-dependent apoptosis without cell-cycle arrest, presumably to avoid a high rate of embryonic malformations (Corbet et al., 1999; Heyer et al., 2000). In ES cells, which are derived from this restricted developmental window, we have demonstrated that the apoptotic response is impaired in Ets1 null cells. Our observation may also indicate a wider role for this Ets transcription factor in the modulation of apoptosis, especially since p53 and Ets1 display a similar temporal and tissue-specific expression during development (Schmid et al., 1991; Kola et al., 1993; Maroulakou et al., 1994). The decreased mRNA levels of p53 in our Ets1-deficient cells initially suggested that the alteration in the apoptotic response in these cells may be due to transcriptional regulation of p53, especially since Ets1 binds to an element in the p53 promoter and has been shown to activate p53 promoter constructs in vitro (Venanzoni et al., 1996). However, the protein levels of p53 were unaltered in Ets1–/– ES cells. It is unclear why the differences in mRNA levels are not reflected in the amount of p53 protein. One possibility is that any additional p53 produced is susceptible to degradation through an autoregulatory feedback loop (e.g. via mdm2) (Haupt et al., 1997; Kubbutat et al., 1997). Thus our data suggested that Ets1 is not essential for the maintenance of p53 protein levels, but may be acting in post-translational regulation of p53 transcriptional activity.
DNA damage in mammalian cells stabilizes the p53 protein, which then functions as a cell-cycle checkpoint leading to cell-cycle arrest or apoptosis (Dulic et al., 1994; Chao et al., 2000). This effect is mediated by p53 transcriptional activity, in which p53 regulates the expression of many genes including mdm2, cyclin G, perp, bax, gadd45 and p21. Significantly, ES cells preferentially undergo apoptosis without cell-cycle arrest (Aladjem et al., 1998; Heyer et al., 2000), and in Ets1 null ES cells the levels of many of these pro-apoptotic genes were less inducible by UV. However, a subset of p53 target genes involved in cell-cycle arrest, including p21 and gadd45, do not respond to UV irradiation (data not shown; Savatier et al., 1995; Sabapathy et al., 1997). Thus it is possible that in the cellular context of ES cells, Ets1 is required for the mechanism that preferentially activates genes involved in p53-dependent apoptosis.
We propose that additional proteins are required to form a stable DNA–p53 transcription initiation complex under physiological conditions and, at least in ES cells, Ets1 is an essential element. The p53-responsive elements in the mdm2 and cyclin G promoters have been defined (Zauberman et al., 1995a,b; Yardley et al., 1998), and we demonstrate herein that mutation of these elements inhibits their response to UV irradiation regardless of Ets1 status, indicating that there is no Ets1-dependent, p53-independent activation of these promoters. Although these promoter-reporter constructs also had significant but reduced transcriptional activity in the absence of Ets1 or when the Ets-binding site was mutated in vitro, our data also indicate that, under the more physiological conditions of ChIP assays, the endogenous mdm2 promoter was only stably bound by p53 and histone H3 acetylated in the presence of Ets1. Thus under physiological conditions in ES cells, p53 requires Ets1 to form a stable DNA–p53 transcriptional complex.
p53-dependent activation of bax, fas, mdm2 and p21 after γ-irradiation has also been shown to depend upon cellular context (Bouvard et al., 2000), most likely due to tissue specific expression of other factors. Since Ets1 is expressed in ES cells, lymphocytes and endothelial cells (Kola et al., 1993; Maroulakou et al., 1994), it could function as a p53 co-activator in these cellular contexts. Indeed Ets1 has been shown to contribute to apoptosis in both ES cells (this paper) and endothelial cells (Teruyama et al., 2001). In other cell types, other proteins could contribute to p53-dependent transactivation. For example, Sp1 (or a sp1-like factor) has been shown to be required for p53-dependent transcriptional activation of the bax promoter in osteosarcoma cells (Thornborrow and Manfredi, 2001). Thus, under physiological conditions and depending upon cellular context, Ets1 and other factors contribute to the specificity and extent of p53-dependent transactivational activity.
In ES cells, Ets1 plays a vital role in the formation of a complex between p53 and CBP/p300. CBP/p300 are closely related histone acetyltransferases that interact with p53 through its N-terminal transactivational domain and function as coactivators for p53-mediated transcription (Avantaggiati et al., 1997; Lill et al., 1997; Ito et al., 2001). Although not required for p53–DNA binding, CBP acetylation of p53-bound nucleosomes was shown to be required for gene transactivation (Espinosa and Emerson, 2001). Mutation of p53 acetylation sites has also been shown to decrease co-factor binding and transactivation activity (Barlev et al., 2001). CBP also interacts with many other proteins, including at least three domains which bind transcription factors. Ets1 physically interacts with two distinct regions of CBP (CH1 and CH3) (Yang et al., 1998; Jayaraman et al., 1999). The CH3 region of CBP is known as a binding site of various nuclear proteins involved in cell-cycle regulation and apoptosis including Ets1, p53, EIA, HTLV-1, c-Myb, cyclin E/CDK2 and AP-1 (Bannister and Kouzarides, 1995; Lundblad et al., 1995; Dai et al., 1996; Kamei et al., 1996; Lee et al., 1996; Ariumi et al., 2000). Our data indicate that in ES cells CBP forms a stable complex with p53 only in the presence of Ets1, which is associated with p53–DNA binding, histone H3 acetylation and target gene transcriptional activation. These specific interactions provide further insight into the mechanisms that control p53 function. Furthermore, our data indicate that in addition to DNA-specific binding, Ets proteins may also be important in the formation of protein complexes required for optimal transcription responses.
We thus propose a working model (Figure 10) where, in unstimulated ES cells, there is little or no transcription of UV-responsive p53 target genes. After UV irradiation, proteins including p53 are increased and activated, resulting in the formation of a complex containing DNA-bound p53, Ets1 and CBP. This promoter-bound complex is capable of histone acetylation and interaction with the basal transcription machinery, leading to strong transcriptional activation. In the absence of Ets1 (or perhaps the presence of other inhibitors) the complex is not stable and only modest transcriptional activation would be observed. Although this role of Ets1 in the regulation of p53-dependent transactivation may be restricted to certain cellular contexts, similar factors elsewhere could contribute to the specificity and extent of p53 transcriptional activity.

Fig. 10. Model for the role of Ets1 in the induction of p53 transcriptional activity after UV irradiation of ES cells. In this model, after exposure to UV, there is a formation of a complex, which includes promoter-bound p53, Ets1 and CBP (where X represents other undefined factors) that interact with the basal transcription machinery leading to strong transcriptional activation. When Ets1 is absent (or other inhibitors present?), the p53–DNA–CBP complex is not stably formed, which leads to only modest transcriptional activation.
Materials and methods
Construction of the targeting vector
A 12 kb murine Ets1 genomic clone, which spanned exons 3–6, was isolated from a 129SVJ mouse genomic library (Stratagene) by screening with a 600 bp fragment of the murine Ets1 cDNA. This genomic clone was used to construct the targeting vector by inserting one loxP site into the NdeI site in intron A. To test for correct integration of loxP into the targeted clones, an EcoRV site was introduced into the same position for diagnostic purposes. A floxed PGK-NEO cassette was then inserted into a SmaI site in intron 6 of the cloned Ets1 genomic DNA (Figure 1A). The thymidine kinase gene was inserted at one end of the Ets1 genomic DNA to allow negative selection for random integration using gancyclovir.
Generation of homozygous Ets1–/– ES cells
The targeting construct was linearized with NotI, electroporated into J1 ES cells and selected with 0.3 mg/ml G418 and 1 µM gancyclovir as described (Hwang et al., 1995). Correct targeting was confirmed by Southern blot analysis (Figure 1C and D). Heterozygous mutant ES cells were cultured in 2.0 mg/ml G418 and surviving cells were screened by Southern blot analysis (Figure 1E). Ets1 was deleted by transient transfection of targeted cells with 20 µg of PGK-CRE (with a puromycin resistance gene), selection with 2 µM puromycin for 2 days and Southern blot analysis (Figure 1E). ES cell lines with the appropriate genotype were expanded and multiple vials cryopreserved after a similar number of passages to ensure consistency throughout experiments. One representative vial was thawed and karyotyped to ensure that all cell lines displayed a normal chromosome number.
Reconstitution of Ets1–/– ES cells with FLAG-Ets1 protein
FLAG-Ets1 was generated by inserting full-length mouse Ets1 cDNA downstream of the EF-1α promoter. We co-transfected Ets1–/– ES cells (clone 2–15) using Fugene 6 with 20 µg FLAG-tagged Ets1 (or pEFBOS 1 FLAG) and a puromycin resistance gene. After selection as above, individual ES cell colonies were isolated and screened by PCR. Another Ets1–/– ES cell clone (17–34) was also transfected; however, only bulk cultures were analysed. Ets1 protein expression was confirmed by western blotting. The cell line and multiple clones displayed various levels of FLAG-Ets1 protein; however, there was no difference between their growth or responses to stimuli (data not shown) and a representative clone is shown in all experiments.
Cell culture and UV irradiation
Cells were cultured using standard ES cell protocols. Prior to analysis, cells were depleted of feeder cells by differential adherence and grown in monolayers on gelatinized 10 cm petri dishes. Before assays, samples were stained with propidium iodide and analysed by FACS to ensure similar cell-cycle status of samples. For UV irradiation, all medium was removed from plates before exposure to UV at 254 nm. Dosage was mean ± <5%.
Apoptosis and cell-cycle analysis
Cell-cycle analysis and quantitation of apoptosis were performed as described by Gong et al. (1994). Briefly, both adherent and floating cells were fixed with 80% cold ethanol, pelleted by centrifugation, stained with propidium iodide and analyzed using an EPICS 752 flow cytometer. Cell debris, doublets and fixation artifacts were gated out, and G0–G1, S, G2–M and apoptotic populations recorded on a logarithmic scale. Apoptotic cells were identified as the lower fluorescence peak (‘sub-G1 peak’ on DNA frequency histograms) due to their reduced DNA content.
EMSA
ES cells (5 × 105 cells/ml) were treated with or without UV 5 h prior to harvest. Nuclear lysates were prepared and EMSA was performed (Thomas et al., 1995). Briefly, 6 µg of protein was combined with 1 ng of double-stranded [γ-32P]dATP-labeled p53 consensus oligonucleotides probe (5′-AGCTTAGACATGCCTAGACATGCCTA-3′) and/or an excess of unlabeled oligonucleotides. Specific antibodies including polyclonal anti-p53, -Ets1, -Ets2 or -CBP (Santa Cruz), anti-FLAG (Integrated Science) and monoclonal anti-Ets1 (E.Sanij and E.Wolvetang, manuscript in preparation) were used to supershift specific proteins. All complexes were resolved on a 5% polyacrylamide gel (0.25× TBE; 4°C). The gels were dried and exposed on Biomax film with intensifying screen overnight.
Western blot analysis
Cells were rinsed with phosphate-buffered saline (PBS) and directly lysed in SDS–PAGE sample buffer. Approximately 50 µg of protein was electrophoresed on 8–12% SDS–polyacrylamide gels. Transfers were performed using standard methods. The blots were then probed with antibodies against p53, mdm2, Bax, Bcl-2, Bcl-XL or CBP (Santa Cruz). A primary antibody to β-tubulin was used to assess equal protein loading.
Immunoprecipitation
Cells were lysed with triple detergent lysis buffer (50 mM Tris–HCl pH 8.0, 150 mM NaCl, 0.02% sodium azide, 0.1% SDS, 100 µg/ml PMSF, 1 µg/ml aprotinin, 1% NP-40, 0.5% sodium deoxycholate), and 1 mg of lysates were incubated with antibodies (anti-CBP, anti-p53 or anti-FLAG) as indicated. Antibody complexes were isolated using protein A/G–agarose beads (Pharmacia Biotech) and washed three times with PBS. The beads were boiled in SDS sample buffer and analyzed by SDS–PAGE and western blotting using anti-p53, anti-CBP (Santa Cruz) and anti-FLAG antibody (Integrated Science).
ChIP assays
ChIP assays were performed using a modification of a published protocol (Takahashi et al., 2000). After immunoprecipitation with anti-p53 (Santa Cruz), anti-acetyl histone H3 (Upstate) or an irrelevant control antibody, the recovered DNA was analysed by PCR with primers flanking the defined mdm2 promoter p53 site: 5′-GGTCCAGGAGGTGACAGGT-3′ and 5′-CAGGAGAAGCGCGGTGAT-3′. Oligos for the GAPDH promoter were used for controls. To ensure that PCR was in the linear range, serial dilutions of DNA were amplified for a maximum of 30 cycles. The products were analysed by gel electrophoresis and Southern blotting with a mdm2 probe.
Southern and northern blotting
Genomic DNA was isolated using standard ES cell isolation protocols. For northern blot analysis, poly(A)+ mRNA was isolated by a modification of Owczarek et al. (1997). Southern and northern blot analyses were carried out as described previously (Hwang et al., 1995).
Luciferase assays
Mdm2 and cyclin G promoters were cloned into the pGL3Basic Vector (Promega). Transcription factor binding site mutants were generated by overlap extension PCR using the following primers: wt-mdm2, 5′-GGTCAAGTTGGGACACGTCC-3′ and Δmdm2 (mut-p53I) primers 5′-GGTGAACTTGGGACACGTCC-3′; wt-mdm2, 5′-GATGCGGCTTCCGGGACGGG-3′ and Δmdm2 (mut-ETSA) 5′-GATGCGGCTTCTGGGACGGG-3′; wt cyclin G: 5′-ACGCAAGCCCGGGCT AGTCT-3′ and Δcyclin G (mut-p53II) 5′-ACGCAAGCCCGGGATCCCGT-3′). ES cells were plated at 1 × 105 cells per well of a 12–well plate and 12 h later transfected with 840 ng of the pmdm2-luc, Δmdm2-luc, cyclin G-luc or Δcyclin G-luc reporter and β-gal vector encoding Renilla luciferase using Fugene 6 (Roche). Luciferase assays were performed using the chemiluminescent assay (Roche Molecular Biochemicals) and data normalized to the Renilla luciferase levels.
Immunofluorescence
Cells were grown for at least 16 h on gelatinized 18 mm coverslips (Sigma), then treated with UV. Cells were fixed on coverslips with acetone:methanol and stained with monoclonal anti-SSEA1 and anti-mouse-FITC. Nuclei were counterstained with 13 µg/ml Hoechst.
Acknowledgments
Acknowledgements
We thank Moshe Oren for kind gifts of the mdm2-luc and cyclin G-luc constructs, Dr Ricky Johnston for anti-acetyl histone H3 antibody and pCEP4-CMVp53 plasmid, Dr Ernst J.Wolvetang and Elaine Sanij for anti-Ets1 and anti-Ets2 monoclonal antibody. We thank Drs Ricky Johnston, Ross S.Thomas, Ernst J.Wolvetang, Rocco Iannello, Melanie Pritchard and Josephine Tkalcevic for critical reading of the manuscript. Paul Hutchinson for help with FACS analysis and Carol Wang, Jodee Gould, Anna Michalska and Irene Tellis for assistance with cell culture and karyotyping.
References
- Aladjem M.I., Spike,B.T., Rodewald,L.W., Hope,T.J., Klemm,M., Jaenisch,R. and Wahl,G.M. (1998) ES cells do not activate p53-dependent stress responses and undergo p53-independent apoptosis in response to DNA damage. Curr. Biol., 8, 145–155. [DOI] [PubMed] [Google Scholar]
- Ariumi Y., Kaida,A., Lin,J.Y., Hirota,M., Masui,O., Yamaoka,S., Taya,Y. and Shimotohno,K. (2000) HTLV-1 tax oncoprotein represses the p53-mediated trans-activation function through coactivator CBP sequestration. Oncogene, 19, 1491–1499. [DOI] [PubMed] [Google Scholar]
- Attardi L.D., Reczek,E.E., Cosmas,C., Demicco,E.G., McCurrach,M.E., Lowe,S.W. and Jacks,T. (2000) PERP, an apoptosis-associated target of p53, is a novel member of the PMP-22/gas3 family. Genes Dev., 14, 704–718. [PMC free article] [PubMed] [Google Scholar]
- Avantaggiati M.L., Ogryzko,V., Gardner,K., Giordano,A., Levine,A.S. and Kelly,K. (1997) Recruitment of p300/CBP in p53-dependent signal pathways. Cell, 89, 1175–1184. [DOI] [PubMed] [Google Scholar]
- Bannister A.J. and Kouzarides,T. (1995) CBP-induced stimulation of c-Fos activity is abrogated by E1A. EMBO J., 14, 4758–4762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlev N.A., Liu,L., Chehab,N.H., Mansfield,K., Harris,K.G., Halazonetis,T.D. and Berger,S.L. (2001) Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol. Cell, 8, 1243–1254. [DOI] [PubMed] [Google Scholar]
- Bories J.C., Willerford,D.M., Grevin,D., Davidson,L., Camus,A., Martin,P., Stehelin,D. and Alt,F.W. (1995) Increased T-cell apoptosis and terminal B-cell differentiation induced by inactivation of the Ets-1 proto-oncogene. Nature, 377, 635–638. [DOI] [PubMed] [Google Scholar]
- Bouvard V., Zaitchouk,T., Vacher,M., Duthu,A., Canivet,M., Choisy-Rossi,C., Nieruchalski,M. and May,E. (2000) Tissue and cell-specific expression of the p53-target gene: bax, fas, mdm2 and waf1/p21, before and following ionising irradiation in mice. Oncogene, 19, 649–660. [DOI] [PubMed] [Google Scholar]
- Canman C.E., Gilmer,T.M., Coutts,S.B. and Kastan,M.B. (1995) Growth factor modulation of p53-mediated growth arrest versus apoptosis. Genes Dev., 9, 600–611. [DOI] [PubMed] [Google Scholar]
- Chao C., Saito,S., Kang,J., Anderson,C.W., Appella,E. and Xu,Y. (2000) p53 transcriptional activity is essential for p53-dependent apoptosis following DNA damage. EMBO J., 19, 4967–4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke A.R., Purdie,C.A., Harrison,D.J., Morris,R.G., Bird,C.C., Hooper,M.L. and Wyllie,A.H. (1993) Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature, 362, 849–852. [DOI] [PubMed] [Google Scholar]
- Corbet S.W., Clarke,A.R., Gledhill,S. and Wyllie,A.H. (1999) P53-dependent and -independent links between DNA-damage, apoptosis and mutation frequency in ES cells. Oncogene, 18, 1537–1544. [DOI] [PubMed] [Google Scholar]
- Dai P., Akimaru,H., Tanaka,Y., Hou,D.X., Yasukawa,T., Kanei-Ishii,C., Takahashi,T. and Ishii,S. (1996) CBP as a transcriptional coactivator of c-Myb. Genes Dev., 10, 528–540. [DOI] [PubMed] [Google Scholar]
- Dulic V., Kaufmann,W.K., Wilson,S.J., Tlsty,T.D., Lees,E., Harper,J.W., Elledge,S.J. and Reed,S.I. (1994) p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell, 76, 1013–1023. [DOI] [PubMed] [Google Scholar]
- El-Deiry W.S., Kern,S.E., Pietenpol,J.A., Kinzler,K.W. and Vogelstein,B. (1992) Definition of a consensus binding site for p53. Nat. Genet., 1, 45–49. [DOI] [PubMed] [Google Scholar]
- El-Deiry W.S. et al. (1993) WAF1, a potential mediator of p53 tumor suppression. Cell, 75, 817–825. [DOI] [PubMed] [Google Scholar]
- Espinosa J.M. and Emerson,B.M. (2001) Transcriptional regulation by p53 through intrinsic DNA/chromatin binding and site-directed cofactor recruitment. Mol. Cell, 8, 57–69. [DOI] [PubMed] [Google Scholar]
- Gong J., Traganos,F. and Darzynkiewicz,Z. (1994) A selective procedure for DNA extraction from apoptotic cells applicable for gel electrophoresis and flow cytometry. Anal. Biochem., 218, 314–319. [DOI] [PubMed] [Google Scholar]
- Graeber T.G., Peterson,J.F., Tsai,M., Monica,K., Fornace,A.J.,Jr and Giaccia,A.J. (1994) Hypoxia induces accumulation of p53 protein, but activation of a G1-phase checkpoint by low-oxygen conditions is independent of p53 status. Mol. Cell. Biol., 14, 6264–6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W. and Roeder,R.G. (1997) Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell, 90, 595–606. [DOI] [PubMed] [Google Scholar]
- Hardy K., Handyside,A.H. and Winston,R.M. (1989) The human blastocyst: cell number, death and allocation during late preimplantation development in vitro. Development, 107, 597–604. [DOI] [PubMed] [Google Scholar]
- Haupt Y., Maya,R., Kazaz,A. and Oren,M. (1997) Mdm2 promotes the rapid degradation of p53. Nature, 387, 296–299. [DOI] [PubMed] [Google Scholar]
- Heyer B.S., MacAuley,A., Behrendtsen,O. and Werb,Z. (2000) Hypersensitivity to DNA damage leads to increased apoptosis during early mouse development. Genes Dev., 14, 2072–2084. [PMC free article] [PubMed] [Google Scholar]
- Hwang S.Y., Hertzog,P.J., Holland,K.A., Sumarsono,S.H., Tymms,M.J., Hamilton,J.A., Whitty,G., Bertoncello,I. and Kola,I. (1995) A null mutation in the gene encoding a type I interferon receptor component eliminates antiproliferative and antiviral responses to interferons α and β and alters macrophage responses. Proc. Natl Acad. Sci. USA, 92, 11284–11288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito A., Lai,C.H., Zhao,X., Saito,S., Hamilton,M.H., Appella,E. and Yao,T.P. (2001) p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J., 20, 1331–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman G. et al. (1999) p300/cAMP-responsive element-binding protein interactions with ets-1 and ets-2 in the transcriptional activation of the human stromelysin promoter. J. Biol. Chem., 274, 17342–17352. [DOI] [PubMed] [Google Scholar]
- Kamei Y. et al. (1996) A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell, 85, 403–414. [DOI] [PubMed] [Google Scholar]
- Kapoor M. and Lozano,G. (1998) Functional activation of p53 via phosphorylation following DNA damage by UV but not γ radiation. Proc. Natl Acad. Sci. USA, 95, 2834–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko L.J. and Prives,C. (1996) p53: puzzle and paradigm. Genes Dev., 10, 1054–1072. [DOI] [PubMed] [Google Scholar]
- Kola I., Brookes,S., Green,A.R., Garber,R., Tymms,M., Papas,T.S. and Seth,A. (1993) The Ets1 transcription factor is widely expressed during murine embryo development and is associated with mesodermal cells involved in morphogenetic processes such as organ formation. Proc. Natl Acad. Sci. USA, 90, 7588–7592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubbutat M.H., Jones,S.N. and Vousden,K.H. (1997) Regulation of p53 stability by Mdm2. Nature, 387, 299–303. [DOI] [PubMed] [Google Scholar]
- Lambert P.F., Kashanchi,F., Radonovich,M.F., Shiekhattar,R. and Brady,J.N. (1998) Phosphorylation of p53 serine 15 increases interaction with CBP. J. Biol. Chem., 273, 33048–33053. [DOI] [PubMed] [Google Scholar]
- Lee J.S., See,R.H., Deng,T. and Shi,Y. (1996) Adenovirus E1A downregulates cJun- and JunB-mediated transcription by targeting their coactivator p300. Mol. Cell. Biol., 16, 4312–4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelievre E., Lionneton,F., Soncin,F. and Vandenbunder,B. (2001) The Ets family contains transcriptional activators and repressors involved in angiogenesis. Int. J. Biochem. Cell Biol., 33, 391–407. [DOI] [PubMed] [Google Scholar]
- Levine A.J. (1997) p53, the cellular gatekeeper for growth and division. Cell, 88, 323–331. [DOI] [PubMed] [Google Scholar]
- Li R., Pei,H. and Papas,T. (1999) The p42 variant of ETS1 protein rescues defective Fas-induced apoptosis in colon carcinoma cells. Proc. Natl Acad. Sci. USA, 96, 3876–3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R., Pei,H., Watson,D.K. and Papas,T.S. (2000) EAP1/Daxx interacts with ETS1 and represses transcriptional activation of ETS1 target genes. Oncogene, 19, 745–753. [DOI] [PubMed] [Google Scholar]
- Lill N.L., Grossman,S.R., Ginsberg,D., DeCaprio,J. and Livingston,D.M. (1997) Binding and modulation of p53 by p300/CBP coactivators. Nature, 387, 823–827. [DOI] [PubMed] [Google Scholar]
- Linke S.P., Clarkin,K.C., Di Leonardo,A., Tsou,A. and Wahl,G.M. (1996) Reversible, p53-dependent G0/G1 cell cycle arrest induced by ribonucleotide depletion in the absence of detectable DNA damage. Genes Dev., 10, 934–947. [DOI] [PubMed] [Google Scholar]
- Louis J.M., McFarland,V.W., May,P. and Mora,P.T. (1988) The phosphoprotein p53 is down-regulated post transcriptionally during embryogenesis in vertebrates. Biochim. Biophys. Acta, 950, 395–402. [DOI] [PubMed] [Google Scholar]
- Lowe S.W., Schmitt,E.M., Smith,S.W., Osborne,B.A. and Jacks,T. (1993) p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature, 362, 847–849. [DOI] [PubMed] [Google Scholar]
- Lu H., Taya,Y., Ikeda,M. and Levine,A.J. (1998) Ultraviolet radiation, but not γ radiation or etoposide-induced DNA damage, results in the phosphorylation of the murine p53 protein at serine-389. Proc. Natl Acad. Sci. USA, 95, 6399–6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundblad J.R., Kwok,R.P., Laurance,M.E., Harter,M.L. and Goodman,R.H. (1995) Adenoviral E1A-associated protein p300 as a functional homologue of the transcriptional co-activator CBP. Nature, 374, 85–88. [DOI] [PubMed] [Google Scholar]
- Maroulakou I.G., Papas,T.S. and Green,J.E. (1994) Differential expression of ets-1 and ets-2 proto-oncogenes during murine embryogenesis. Oncogene, 9, 1551–1565. [PubMed] [Google Scholar]
- Matsuyama S., Xu,Q., Velours,J. and Reed,J.C. (1998) The mitochondrial F0F1-ATPase proton pump is required for function of the proapoptotic protein Bax in yeast and mammalian cells. Mol. Cell, 1, 327–336. [DOI] [PubMed] [Google Scholar]
- Meek D.W. (1998) Multisite phosphorylation and the integration of stress signals at p53. Cell Signal., 10, 159–166. [DOI] [PubMed] [Google Scholar]
- Muthusamy N., Barton,K. and Leiden,J.M. (1995) Defective activation and survival of T cells lacking the Ets-1 transcription factor. Nature, 377, 639–642. [DOI] [PubMed] [Google Scholar]
- Nakano T., Abe,M., Tanaka,K., Shineha,R., Satomi,S. and Sato,Y. (2000) Angiogenesis inhibition by transdominant mutant ets1. J. Cell Physiol., 184, 255–262. [DOI] [PubMed] [Google Scholar]
- Owczarek C.M., Hwang,S.Y., Holland,K.A., Gulluyan,L.M., Tavaria,M., Weaver,B., Reich,N.C., Kola,I. and Hertzog,P.J. (1997) Cloning and characterization of soluble and transmembrane isoforms of a novel component of the murine type I interferon receptor, IFNAR 2. J. Biol. Chem., 272, 23865–23870. [DOI] [PubMed] [Google Scholar]
- Perry M.E., Piette,J., Zawadzki,J.A., Harvey,D. and Levine,A.J. (1993) The mdm-2 gene is induced in response to UV light in a p53-dependent manner. Proc. Natl Acad. Sci. USA, 90, 11623–11627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabapathy K., Klemm,M., Jaenisch,R. and Wagner,E.F. (1997) Regulation of ES cell differentiation by functional and conformational modulation of p53. EMBO J., 16, 6217–6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi K., Herrera,J.E., Saito,S., Miki,T., Bustin,M., Vassilev,A., Anderson,C.W. and Appella,E. (1998) DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev., 12, 2831–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savatier P., Huang,S., Szekely,L., Wiman,K.G. and Samarut,J. (1994) Contrasting patterns of retinoblastoma protein expression in mouse embryonic stem cells and embryonic fibroblasts. Oncogene, 9, 809–818. [PubMed] [Google Scholar]
- Schmid P., Lorenz,A., Hameister,H. and Montenarh,M. (1991) Expression of p53 during mouse embryogenesis. Development, 113, 857–865. [DOI] [PubMed] [Google Scholar]
- Scholer H.R., Hatzopoulos,A.K., Balling,R., Suzuki,N. and Gruss,P. (1989) A family of octamer-specific proteins present during mouse embryogenesis: evidence for germline-specific expression of an Oct factor. EMBO J., 8, 2543–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth A., Giunta,S., Franceschil,C., Kola,I. and Venanzoni,M.C. (1999) Regulation of the human stress response gene GADD153 expression: role of ETS1 and FLI-1 gene products. Cell Death Differ., 6, 902–907. [DOI] [PubMed] [Google Scholar]
- Takahashi Y., Rayman,J.B. and Dynlacht,B.D. (2000) Analysis of promoter binding by the E2F and pRB families in vivo: distinct E2F proteins mediate activation and repression. Genes Dev., 14, 804–816. [PMC free article] [PubMed] [Google Scholar]
- Teruyama K., Abe,M., Nakano,T., Iwasaka-Yagi,C., Takahashi,S., Yamada,S. and Sato,Y. (2001) Role of transcription factor Ets1 in the apoptosis of human vascular endothelial cells. J. Cell Physiol., 188, 243–252. [DOI] [PubMed] [Google Scholar]
- Thomas R.S., Tymms,M.J., Seth,A., Shannon,M.F. and Kola,I. (1995) ETS1 transactivates the human GM-CSF promoter in Jurkat T cells stimulated with PMA and ionomycin. Oncogene, 11, 2135–2143. [PubMed] [Google Scholar]
- Thornborrow E.C. and Manfredi,J.J. (2001) The tumor suppressor protein p53 requires a cofactor to transcriptionally activate the human bax promoter. J. Biol. Chem., 276, 15598–15608. [DOI] [PubMed] [Google Scholar]
- Torii S., Egan,D.A., Evans,R.A. and Reed,J.C. (1999) Human Daxx regulates Fas-induced apoptosis from nuclear PML oncogenic domains (PODs). EMBO J., 18, 6037–6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger T., Juven-Gershon,T., Moallem,E., Berger,M., Vogt Sionov,R., Lozano,G., Oren,M. and Haupt,Y. (1999) Critical role for Ser20 of human p53 in the negative regulation of p53 by Mdm2. EMBO J., 18, 1805–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venanzoni MC., Robinson,L.R., Hodge,D.R., Kola,I. and Seth,A. (1996) ETS1 and ETS2 in p53 regulation: spatial separation of ETS binding sites (EBS) modulate protein: DNA interaction. Oncogene, 12, 1199–1204. [PubMed] [Google Scholar]
- Wasylyk C., Bradford,A.P., Gutierrez-Hartmann,A. and Wasylyk,B. (1997) Conserved mechanisms of Ras regulation of evolutionary related transcription factors, Ets1 and Pointed P2. Oncogene, 14, 899–913. [DOI] [PubMed] [Google Scholar]
- Watson D.K, Li,R., Sementchenko,V.I., Mavrothalassitis,G. and Seth,A. (2002) ETS family of transcription factors. In Bertino,J.R. (ed.), Encyclopedia of Cancer. Academic Press, San Diego, CA, pp. 189–196.
- Wills K.N. et al. (1994) Development and characterization of recombinant adenoviruses encoding human p53 for gene therapy of cancer. Hum. Gene Ther., 5, 1079–1088. [DOI] [PubMed] [Google Scholar]
- Wobus A.M., Holzhausen,H., Jakel,P. and Schoneich,J. (1984) Characterization of a pluripotent stem cell line derived from a mouse embryo. Exp. Cell Res., 152, 212–219. [DOI] [PubMed] [Google Scholar]
- Yang C., Shapiro,L.H., Rivera,M., Kumar,A. and Brindle,P.K., (1998) A role for CREB binding protein and p300 transcriptional coactivators in Ets-1 transactivation functions. Mol. Cell. Biol., 18, 2218–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yardley G., Zauberman,A., Oren,M. and Jackson,P. (1998) Individual promoter and intron p53-binding motifs from the rat Cyclin G1 promoter region support transcriptional activation by p53 but do not show co-operative activation. FEBS Lett., 430, 171–175. [DOI] [PubMed] [Google Scholar]
- Zauberman A., Lupo,A. and Oren,M. (1995a) Identification of p53 target genes through immune selection of genomic DNA: the cyclin G gene contains two distinct p53 binding sites. Oncogene, 10, 2361–2366. [PubMed] [Google Scholar]
- Zauberman A., Flusberg,D., Haupt,Y., Barak,Y. and Oren,M. (1995b) A functional p53-responsive intronic promoter is contained within the human mdm2 gene. Nucleic Acids Res., 23, 2584–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]