

Abstract
Each cell division in Caulobacter crescentus is asymmetric, yielding a swarmer cell with several polar pili and a non-piliated stalked cell. To identify factors contributing to the asymmetric biogenesis of polar pili, cytological studies of pilus assembly components were performed. We show here that the CpaC protein, which is thought to form the outer membrane pilus secretion channel, and its assembly factor, CpaE, are localized to the cell pole prior to the polymerization of the pilus filament. We demonstrate that the PleC histidine kinase, a two-component signal transduction protein shown previously to localize to the piliated cell pole before and during pilus assembly, controls the accumulation of the pilin subunit, PilA. Using an inactive form of PleC (PleCH610A) that lacks the catalytic histidine residue, we provide evidence that PleC activity is responsible for the asymmetric distribution of CpaE and itself to only one of the two cell poles. Thus, a polar signal transduction protein controls its own asymmetric location as well as that of a factor assembling a polar organelle.
Keywords: Caulobacter/cell cycle/histidine kinase/pilus assembly/secretin
Introduction
Asymmetric cell division is a fundamental biological mechanism by which both eukaryotes and prokaryotes give rise to genetically identical daughter cells that perform different functions and often have distinct morphologies (Jacobs and Shapiro, 1998; Shapiro and Losick, 2000; Knoblich, 2001). The elucidation of this process is facilitated by the study of proteins assembling into daughter cell-specific morphological structures and their regulators.
The dimorphic bacterium Caulobacter crescentus undergoes an asymmetric cell division, yielding a motile swarmer cell and a sessile stalked cell. These two cell types differ by the presence of distinct polar organelles and by their ability to initiate DNA replication (Jensen et al., 2002). The larger stalked cell is replication proficient and therefore functionally analogous to eukaryotic S-phase cells (S). It bears the stalk, a cylindrical extension of the cell envelope, at one of its poles. The smaller swarmer cell is characterized by the presence of a single polar flagellum and several polar pili at the same pole and its inability to initiate DNA replication. By this criterion swarmer cells correspond to G1-phase cells. The DNA replication block is relieved only once the obligate differentiation step into a stalked cell (reflecting the G1–S transition) has occurred, a step that is accompanied by the release of the flagellum and the loss of polar pili.
Caulobacter crescentus pili are extracellular surface appendages that are polymerized from a processed pilin subunit (PilA) into a filament 1–4 µm in length and ∼4 nm in diameter (Skerker and Shapiro, 2000). It is believed that the pilus filament is anchored in the inner membrane, spans the periplasm and exits through an outer membrane channel (Russel, 1998; Soto and Hultgren, 1999). Caulobacter crescentus pili serve as receptor sites for the DNA bacteriophage φCbK. This property has been used as a screen for the isolation of a cluster of genes, all of which are required for Caulobacter pilus assembly (cpaA–F, Figure 1A) (Skerker and Shapiro, 2000). Several Cpa proteins are homologs of those required for the formation of polar pili in other bacteria, such as Pseudomonas aeruginosa and Myxococcus xanthus (Lory, 1998; Wall and Kaiser, 1999; Skerker and Shapiro, 2000; Sandkvist, 2001). These include CpaA, a homologue of pre-pilin peptidases, which process immature pilin into its mature form; CpaF, a putative ATPase that could provide the energy for pilus assembly; and CpaC, a homologue of secretins, which assemble into a multimeric secretion channel in the outer membrane (Bitter et al., 1998; Russel, 1998; Marciano et al., 1999, 2001; Nouwen et al., 1999). Others, like the pilus assembly factor CpaE, that are not represented in the pilus biogenesis machinery of P.aeruginosa and M.xanthus, are of unknown biochemical function. CpaE lacks a signal sequence and predicted membrane-spanning regions, suggesting that it may associate peripherally with the cytoplasmic side of the inner membrane (Skerker and Shapiro, 2000).

Fig. 1. Cell cycle-dependent accumulation of pilus assembly proteins. (A) Genetic organization of the pilA–cpaF locus. Filled colored boxes indicate genes relevant for this study (pilA, black; cpaC, magenta and cpaE, blue). The perpendicular arrow in front of pilA indicates the location of the pilA transcriptional start site (Skerker and Shapiro, 2000). Horizontal arrows denote the orientation of the genes. The three small black boxes represent the regions upstream of pilA that have been footprinted by CtrA∼P (dashed arrows) (Skerker and Shapiro, 2000). The perpendicular arrows in front of cpaA and cpaB indicate other putative promoters in the pilA–cpaF cluster (Laub et al., 2000). (B) Steady-state level of pilus assembly proteins during the cell cycle. Samples of equal volume were collected from a synchronized culture every 20 min, subjected to SDS–PAGE and immunoanalysis. The top panel shows a schematic drawing of the cell cycle stage at which the samples were collected. The middle panel shows the quantified graphs of PilA (black), CpaE (blue) and the PleC (gold) steady-state levels during the cell cycle. Immunoblots are shown in the lower panel. The CtrA immunoblots serve as a control for the cell cycle (Domian et al., 1997).
Differential biogenesis of polar pili during the C.crescentus cell cycle facilitates the study of both the spatial determinants directing pilus assembly at the cell pole and temporal control mechanisms that restrict pili to the swarmer cell (Sommer and Newton, 1988; Skerker and Shapiro, 2000; Jensen et al., 2002). Genome-wide analysis of gene expression performed during the Caulobacter cell cycle using DNA microarrays, revealed a strict temporal order of transcription of pilA–cpaF (Laub et al., 2000). Transcription of cpaB–F is induced first in the late predivisional cell, followed by cpaA transcription and then pilA, in dividing cells and progeny swarmer cells. The timing of pilus assembly can be shifted from the swarmer cell stage to the predivisional cell stage by transcribing pilA from a constitutive promoter, suggesting that temporal regulation of pilA transcription prevents premature assembly of pili in predivisional cells (Skerker and Shapiro, 2000).
A gene identified as a temperature-sensitive mutant conferring resistance to φCbK was shown to encode an essential two-component response regulator, CtrA, that coordinates cell cycle progression with polar morphogenesis (Quon et al., 1996). Both microarray analysis and direct footprinting with CtrA demonstrated that pilA transcription is dependent on CtrA (Laub et al., 2000, 2002; Skerker and Shapiro, 2000).
An additional gene identified by mutations conferring resistance to bacteriophage φCbK is pleC, encoding a sensor histidine kinase (Sommer and Newton, 1989; Wang et al., 1993). The molecular basis for the pili-less phenoype of pleC mutants (Sommer and Newton, 1989) is unknown. An additional phenotype of pleC mutant strains is the apparent loss of asymmetry. Cell division in pleC mutants yields two daughter cells that are similar in size and both bear a paralyzed flagellum, but lack pili at their poles (Ohta et al., 2000). PleC belongs to a subgroup of histidine kinases in Caulobacter, coordinating cell cycle progression and polar morphogenesis, whose polar locali zation varies as a function of the cell cycle (Jacobs et al., 1999; Wheeler and Shapiro, 1999). PleC localizes to the flagellated pole in predivisional and progeny swarmer cells, maintaining this location until the swarmer-to-stalked cell transition, when it is released from the pole (Wheeler and Shapiro, 1999).
We show here that the CpaE protein, which is required for pilus assembly, has the same localization pattern as PleC, and that CpaE is required for the localization of the secretin CpaC at the pole where it forms a pilus secretion channel. Our results using a mutant lacking the site of autophosphorylation in PleC support the interpretation that the ability of PleC to autophosphorylate is critical for asymmetric pilus assembly. Not only is active PleC required for PilA accumulation, but in a strain in which PleC is present but inactive, CpaE and PleC fail to be released from the flagellated swarmer cell pole during the transition to a stalked cell. As a consequence, predivisional cells are formed in which CpaE and inactive PleC are symmetrically located at both cell poles. Thus, a polarly localized signal transduction protein controls the accumulation of the subunit of the pilus filament and coordinates its own release and that of a pilus assembly factor from the piliated pole at the G1–S transition.
Results
Expression of pili proteins as a function of the cell cycle
To study the temporal and spatial determinants of pilus assembly at the flagellated pole of C.crescentus swarmer cells, antibodies were raised against the structural subunit of the pilus filament, PilA, and the CpaC and CpaE components of the pilus assembly machinery (Figure 1A and B). Steady-state levels of these proteins were assessed in synchronized populations by isolating swarmer cells (G1) and allowing them to proceed through the cell cycle. Immunoblots of cell samples collected at regular intervals are shown in Figure 1B. PilA steady-state levels are high in swarmer cells and decrease after the swarmer-to-stalked cell (G1–S) transition (60 min). PilA accumulates, again coincident with cell division, reflecting the time of induction of pilA transcription (Laub et al., 2000; Skerker and Shapiro, 2000) and the appearance of pili at the progeny swarmer cell (Sommer and Newton, 1988; Skerker and Shapiro, 2000). Previous experiments showed that constitutive transcription of pilA is not sufficient to trigger pilus assembly in stalked cells (Skerker and Shapiro, 2000). These experiments did not reveal whether the PilA protein accumulated as a consequence of constitutive transcription of pilA. The immunoblots in Figure 1B show that PilA is present in wild-type stalked cells, even without artificially transcribing pilA. Since stalked cells are non-piliated, these results indicate that conditions other than PilA abundance restrict pili assembly to swarmer cells.
Both cpaC and cpaE are part of a locus required for polar pilus biogenesis in Caulobacter (Figure 1A) (Skerker and Shapiro, 2000). CpaC is a member of a large superfamily of proteins, known as the secretins (Russel, 1998), that form outer membrane channels for the export of substrates such as virulence factors, filamentous phage or assembled pilin subunits (Bitter et al., 1998; Marciano et al., 1999, 2001; Nouwen et al., 1999).
Immunoblots using an antibody raised against the CpaC secretin detected two bands in wild-type cell extracts (Figure 1B), both of which were absent in those from the ΔcpaC mutant (Figure 3C). The faster-migrating protein had an apparent molecular mass of ∼50 kDa, the predicted molecular mass of the CpaC monomer. The other band migrated with an apparent molecular mass of ∼110 kDa and is likely to represent a modified form of CpaC (referred to as CpaC*). Secretins can form stable multimers that are not dissociated by boiling and SDS–PAGE (Nouwen et al., 2000); however, due to their large size, these aggregates fail to enter the separating gel during SDS–PAGE and remain in the stacking gel (Hardie et al., 1996). The ability of CpaC* to enter the separating gels and its smaller apparent molecular mass are inconsistent with it representing a secretin-like multimeric aggregate of the CpaC monomer. Moreover, prior to dissociation the secretin aggregate is far more abundant than the monomer (Hardie et al., 1996; Schmidt et al., 2001). Our immunoblots show that the CpaC monomer is more abundant than CpaC* (Figures 1B and 3C). At present the nature of CpaC* is mysterious and speculative at best, and will be the subject of future investigations. The steady-state levels of the two CpaC forms were found to have different profiles during the cell cycle (Figure 1B). Unlike the CpaC monomer, which gradually increases concomitant with the accumulation of biomass, CpaC* is absent in swarmer cells, begins to increase in abundance at the G1–S transition, and reaches high levels in predivisional cells, before decreasing in abundance as the predivisional cells undergo cell division. CpaE, on the other hand, is highly abundant in swarmer cells, but decreases in abundance at the G1–S transition coincident with the loss of pili (Figure 1B). Thus, the temporal correlation of CpaE abundance with the piliated swarmer cells and the predivisional cells that are thought to assemble the pilus secretion machinery, raises the possibility that CpaE may determine when and/or where pili are formed.
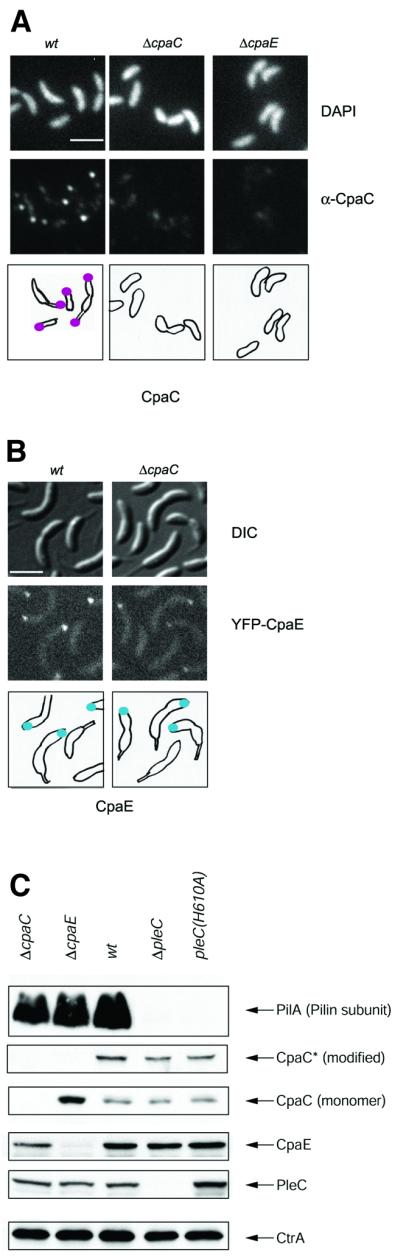
Fig. 3. CpaE is required for polar localization of CpaC and accumulation of CpaC*. (A) Subcellular localization of CpaC in wild-type, ΔcpaC and ΔcpaE mutant cells determined by indirect immunofluorescence microscopy using affinity-purified CpaC antibody as described in the legend for Figure 2A. Shown are images of DAPI-stained chromosomal DNA (upper row), immunofluorescence microscopy images of the cells showing the subcellular location of CpaC (middle row) and drawings showing the location of CpaC as magenta dots. The scale bar in (A) and (B) represents 2 µm. (B) Subcellular localization of CpaE in wild-type and ΔcpaC mutant cells expressing YFP–CpaE. Shown are DIC images (top panel), fluorescence images (middle panel) and schematics (lower panel) of the location of YFP–CpaE as blue dots. The scale bar represents 2 µm. (C) Steady-state levels of proteins required for pilus assembly (PilA, CpaC*, CpaC, CpaE, the histidine kinase PleC and CtrA) in wild-type, ΔcpaC, ΔcpaE, ΔpleC and pleC(H610A) mutant cells as determined by immunoblotting. Note that, unlike in Figure 1B where the CpaC and CpaC* lanes shown are from the same exposure, the CpaC and CpaC* lanes in this figure are taken from shorter exposure of the same blot, in order to demonstrate the difference in abundance of the CpaC monomer in the ΔcpaE strain compared with the wild-type strain, and a longer exposure to visualize CpaC* appropriately.
CpaC and CpaE localize to the site of pilus assembly in a cell cycle-dependent manner
Pili are assembled at the swarmer cell pole that also bears the single flagellum and the chemosensory apparatus (Sommer and Newton, 1988; Alley et al., 1992). To determine the locations of CpaC and CpaE, we employed indirect immunfluorescence microscopy of fixed samples from synchronized cell populations or direct visualization of YFP-tagged proteins in living cells.
Swarmer cells analyzed by immunofluorescence microscopy using affinity-purified CpaC antibodies yielded distinct unipolar foci (93/94 cells) (Figure 2A). By the time swarmer cells had differentiated into stalked cells (60 min), the CpaC focus was no longer associated with the cell periphery as determined by 4′,6-diamidino-2-phenylindole (DAPI) staining, but lay slightly offset from the pole. The observation that ΔcpaC mutant cells were devoid of foci (Figure 3A), indicates that these signals were CpaC derived. We considered the possibility that the foci beyond the cell periphery were located at the tip of the stalk, which is devoid of DNA and, therefore, not visible by DAPI staining. Since immunostaining involves permeabilizing the cells with lysozyme, resulting in some loss of integrity, similar experiments were performed with a modified procedure omitting the lysozyme treatment. Owing to its location in the outer membrane, CpaC should still be accessible to the antibody even without lysozyme treatment. These experiments revealed that in wild-type predivisional cells with visible stalks, CpaC foci were located at the tip of the stalk as well as the pole opposite the stalk (Figure 2B).

Fig. 2. Cell cycle-dependent polar localization of the CpaC outer membrane channel and the CpaE pilus assembly protein. (A) Subcellular location of CpaC in cells at different stages of the Caulobacter cell cycle was determined using indirect immunofluorescence microscopy. Swarmer cells were isolated and allowed to progress synchronously through the cell cycle. When the cells reached the indicated stages of the cell cycle (0, 30, 60, 90 or 120 min, as indicated above the images) the cells were fixed, treated with lysozyme and the subcellular location of the CpaC outer membrane channel was visualized by indirect immunofluorescence microscopy using affinity-purified CpaC antibodies. A low level of background intracellular signal was observed with a ΔcpaC strain, demonstrating that the polar signal is specific for CpaC (see Figure 3A). Shown are DAPI-stained images of the chromosomal DNA in the cells (upper row), immunofluorescence microscopy images of the cells showing the subcellular locations of CpaC (middle row), and drawings of the location of CpaC within the cells as magenta dots (bottom row). The scale bar in (A–C) represents 2 µm. Shown underneath is a schematic of the location of CpaC during the cell cycle. (B) Location of CpaC at the swarmer cell pole and the tip of the stalk in wild-type and ΔcpaC predivisional cells by immunofluorescence using affinity-purified CpaC antibody. Cells were processed as in (A), except that the lysozyme treatment and DAPI staining were omitted. Shown are images taken by Nomarski differential interference contrast (DIC) microscopy (upper panel) and by immunofluorescence microscopy (lower panel). The positions of CpaC in the cell on the left are indicated by arrowheads and in the cell on the right by arrows. The markings point to the location of CpaC at the swarmer cell pole (white) and at the tip of the stalk (yellow). The control experiments on the right show that the polar foci are absent in the ΔcpaC strain. (C) Subcellular location of CpaE in cells at different stages of the Caulobacter cell cycle as determined by direct fluorescence microscopy in a strain expressing YFP–CpaE. Swarmer cells were isolated and allowed to progress synchronously through the cell cycle. When the cells reached the indicated stages of the cell cycle (0, 30, 70, 100 or 130 min, as indicated above the images), samples of cells were withdrawn and placed on a thin layer of agarose, and images were acquired. Shown are DIC images (top panel), fluorescence images (middle panel) and schematics (lower panel) of the subcellular location of YFP–CpaE shown as blue dots. Shown underneath is a schematic of the location of CpaE during the cell cycle.
As shown in Figure 2A, by the time the cells reached the predivisional cell stage the distance of the ‘stalked’ CpaC focus had moved further from the cell body, presumably due to continued elongation of the stalk as the cell cycle progressed. Concomitantly, in some cells (15/96) a second CpaC focus appeared at the incipient swarmer pole opposite the stalked pole. By 120 min most of the late predivisional cells (53/75) had two CpaC foci, one at the tip of the stalk and one at the swarmer cell pole (Figure 2A). Thus, the appearance of the CpaC channel opposite the stalked pole in late predivisional cells and swarmer cells is consistent with the time of pilus biogenesis. The unexpected retention of CpaC at the tip of the stalk could reflect an additional role, perhaps in the secretion of a stalk-specific substrate. Alternatively, it could simply be a consequence of not disassembling the localized CpaC channels.
Cells in which chromosomal cpaE was replaced by a functional yfp–cpaE fusion (see Materials and methods) were examined by fluorescence microscopy at regular intervals during a synchronized cell cycle (Figure 2C). YFP–CpaE was detected as a unipolar focus in swarmer cells (60/63) that disappeared during the G1–S transition (59/60 cells). By the early predivisional cell stage (100 min) a fluorescent focus appeared at the incipient swarmer cell pole (60/66 cells), where it remained during and after cell division. Thus, as anticipated for components of the pilus secretion machinery, CpaE and CpaC localize to the site of pilus assembly in a temporally regulated manner, preceding the appearance of pili at the swarmer cell pole.
CpaC localization and modification is dependent on CpaE
To explore the possibility that CpaE is involved in controlling the assembly of the CpaC secretion channel at the swarmer cell pole, we examined the subcellular location of CpaC in ΔcpaE mutant cells by immunofluorescence microscopy using the affinity-purified CpaC antibody. As shown in Figure 3A, ΔcpaE mutant cells (50/50) are devoid of the polar CpaC foci, suggesting that CpaC localization requires CpaE. As a control, we show that ΔcpaC mutant cells (50/50) lack immunofluorescent foci in the presence of the CpaC antibody. Fluorescence microscopy of ΔcpaC mutant cells harboring a yfp–cpaE fusion in place of the wild-type version of cpaE revealed YFP–CpaE foci at the pole opposite the stalk (Figure 3B). Thus polar localization of CpaE is not dependent on CpaC.
Immunoblot analyses of wild-type cell extracts revealed the presence of both the CpaC monomer and CpaC*. However, only the monomeric form of CpaC was detected in ΔcpaE extracts (Figure 3C), suggesting that CpaE is required for the accumulation of CpaC*. The fact that the CpaC monomers were much more abundant in the ΔcpaE strain as opposed to the wild-type strain suggests that the accumulation of monomers is due to the failure to modify CpaC monomers into CpaC*.
Together, these results are consistent with the notion that CpaE is required for the modification and localization of the CpaC secretion channel at the swarmer cell pole.
The activity of the PleC histidine kinase is required for PilA accumulation
Strains with loss-of-function mutations in pleC are pili-less, stalkless and have a non-motile flagellum (Sommer and Newton, 1989; Wang et al., 1993). To determine the role of the PleC histidine kinase in the biogenesis of pili at the swarmer cell pole, we probed immunoblots of cell extracts from the ΔpleC mutant with antibodies against PilA, CpaC and CpaE. Steady-state levels of CpaE, monomeric CpaC and CpaC* in the ΔpleC mutant were comparable to those detected in the wild type (Figure 3C). However, PilA was absent in this mutant (Figure 3C).
To investigate whether this function of PleC requires its kinase activity, a mutant pleC allele [pleC(H610A)] was constructed in which the putative site of autophosphorylation (H610) was mutated to an alanine residue. Histidine kinases such as PleC use ATP as a phosphodonor for autophosphorylation at a conserved histidine residue, forming a transient phospho-histidine residue. The phosphoryl group is then transferred from the phospho-histidine residue to the aspartate residue in the response regulator (Parkinson, 1993). Lack of the site of autophos phorylation is thought to render the kinase inactive. Consistent with this notion, strains bearing the pleC(H610A) allele in place of wild-type pleC displayed a phenotype (non-motile, stalkless and φCbK resistant) indistinguishable from ΔpleC strains (Figure 4A and C; data not shown). Immunoblots of cell extracts from the pleC(H610A) mutant revealed that PilA accumulation depends on the ability of PleC to carry out autophosphorylation (Figure 3C).

Fig. 4. PleC kinase activity controls its own release and that of CpaE at the swarmer-to-stalked cell transition. (A) Time-lapse fluorescence microscopy analysis showing the retention of PleCH610A–GFP at the pole and its accumulation at the opposite pole later in the cell cycle in cells lacking PleC kinase acitivity. PleCH610A–GFP-expressing cells were placed on a thin layer of M2G agarose and images of the same cells were acquired every 30 min. Small cells with a single polar PleC–GFP focus were selected and their progression through the cell cycle was followed. DIC images (top panel) and fluorescence images (lower panel) are shown. Arrowheads indicate the position of PleCH610A–GFP. The black scale bar in (A) and (C) represents 2 µm. (B) Schematics showing the location of PleC (gold dots), PleCH610A (gold circles) and CpaE (blue dots) during the cell cycle in wild-type and various pleC mutant strains. (C) Time-lapse fluorescence microscopy analysis showing the retention of YFP–CpaE at the pole and its accumulation at the opposite pole later in the cell cycle in cells lacking PleC kinase activity. YFP–CpaE-expressing cells were placed on a thin layer of agarose containing nutrients and images of the same cells were acquired every 45 min. Small cells with a single polar YFP–CpaE focus were selected and their progression through the cell cycle was followed. DIC images (top panel) and fluorescence images (lower panel) are shown. Arrowheads indicate the position of YFP–CpaE.
The activity of the PleC histidine kinase is required for the loss of itself and CpaE from the flagellated pole at the swarmer-to-stalked cell transition
Strains with loss of function mutations in pleC form predivisional cells that are morphologically similar at the two poles (Ohta et al., 2000). Several two-component histidine kinases controlling cell cycle progression and/or polar morphogenesis in Caulobacter, including PleC, localize to either one or both cell poles in a cell cycle-dependent manner (Jacobs et al., 1999; Wheeler and Shapiro, 1999). Consistent with its role in pilus biogenesis and motility, PleC was shown previously to localize to the flagellated pole in swarmer cells and predivisional cells (Wheeler and Shapiro, 1999), and to be released from its polar location at the G1–S transition (Figure 4B). This pattern of polar localization is remarkably similar to that observed for the CpaE pilus assembly protein. Moreover, immunoblots using an antibody raised against PleC showed that the reduction in steady-state levels were similar to those of CpaE (Figure 1B). The increase in PleC steady-state levels preceded that of CpaE, presumably reflecting the difference in the timing of transcription during the cell cycle (Laub et al., 2000).
To explore the relationship between PleC and CpaE localization, fluorescence microscopy was performed on ΔpleC strains bearing a chromosomal yfp–cpaE fusion replacing wild-type cpaE. Because pleC mutant cells are stalkless and cannot be synchronized, we followed individual cells as they progressed through the cell cycle by time-lapse fluorescence microscopy (data not shown). Cells containing a single polar YFP–CpaE focus, retained the signal at its original location during the entire period. However, a second focus appeared at the opposite pole in predivisional cells, giving rise to two daughter cells, each with a unipolar YFP–CpaE focus. These experiments suggest that in the absence of PleC, CpaE is not released from the swarmer cell pole at the G1–S transition. Time-lapse fluorescence microscopy of a pleC(H610A) strain containing chromosomal yfp–cpaE in place of the wild-type cpaE gene showed that, as was the case in the absence of PleC, PleCH610A is unable to release YFP–CpaE from the flagellated pole (Figure 4C), yielding predivisional cells with CpaE foci located at both poles.
To determine whether catalytically inactive PleC retains the wild-type PleC localization pattern, we observed PleCH610A–GFP localization (Figure 4A). As is the case for YFP–CpaE, PleCH610A–GFP is not released from the swarmer cell pole and bipolar signals were observed in predivisional cells. The retention of both PleC and CpaE at the cell pole following the G1–S transition yields a symmetric predivisional cell, when new PleC and CpaE are deposited at the opposite incipient swarmer cell pole. By a process that is dependent on the ability of PleC to carry out autophosphorylation, PleC triggers the disappearance of two dissimilar proteins from the flagellated cell pole at the swarmer-to-stalked cell transition. Thus, PleC determines the subsequent asymmetric distribution of itself and CpaE in the predivisional cell.
Discussion
The studies reported here on the cascade of events leading to the asymmetric assembly of polar pili during a bacterial cell cycle, revealed that the CpaC pilus outer membrane channel and its assembly factor CpaE, are localized to the incipient swarmer pole in predivisional cells, prior to the synthesis and polymerization of the pilin subunit, PilA, into the pilus filament. Polar localization of CpaC and CpaE occurred at approximately the same time in the cell cycle. However, genetic and cytological experiments revealed a hitherto unknown CpaE-dependent pathway for the assembly of the polar CpaC pilus secretion channel in predivisional cells. In the absence of CpaE, CpaC is delocalized and pili are not formed, suggesting that localization of CpaC to the pole is a prerequisite for it to assemble into a channel competent for pilus assembly. Thus, CpaE is required for a critical step in channel assembly by triggering localization of CpaC at the pole.
When pili are lost or retracted at the swarmer-to-stalked cell transition, CpaE, but not CpaC, is lost from the cell pole. CpaC retains its original position and, once stalk growth initiates, is incorporated into the tip of the elongating stalk. CpaC may be required at this position for the secretion of a new substrate in stalked cells, after the pili are lost. The same machinery can be used for pilus formation and protein secretion in bacteria (Sauvonnet et al., 2000). Caulobacter crescentus cells bear an adhesive organelle at the tip of the stalk, the holdfast, raising the possibility that holdfast components may be secreted from the CpaC channel located at the tip of the stalk. However, functional and cytological assays failed to reveal defects in the holdfast (P.H.Viollier and L.Shapiro, unpublished data) in the ΔcpaC mutant, arguing against this possibility, but not excluding the possibility of secreting a substrate other than a holdfast component in stalked cells.
The PleC histidine kinase, which is required for pili biosynthesis, is also lost from the piliated pole at the swarmer-to-stalked cell transition (Wheeler and Shapiro, 1999). Using time-lapse fluorescence microscopy, we showed that swarmer cells with an inactive form of PleC, PleCH610A, fail to lose CpaE and PleCH610A from the cell pole, resulting in predivisional cells that are symmetric with respect to the polar location of CpaE and PleCH610A. Lack of PleC has been shown to result in cells that are apparently symmetric with respect to the location of the polar flagellum (Ohta et al., 2000). Moreover, PleC has been implicated in controlling the delocalization of the response regulator DivK from the incipient swarmer cell pole during cell division (Jacobs et al., 2001). Thus, PleC may serve as a regulator of polar asymmetry by triggering the pole-specific loss of polar proteins at specific times in the cell cycle by a signaling process that is dependent on its ability to autophosphorylate.
Lack of PleC activity also results in the failure to accumulate PilA, providing an explanation for the pili-less phenotype of pleC loss-of-function mutants (Sommer and Newton, 1989). Transcription of pilA is dependent on CtrA∼P (Skerker and Shapiro, 2000), raising the possibility that in vivo, PleC, like the CckA histidine kinase (Jacobs et al., 1999), controls the phosphorylation state of CtrA, perhaps via CckA or a distinct phosphorelay. However, since the phenotype of the pleC mutant only partially overlaps with that of ctrA and cckA mutants, PleC may only be one of several histidine kinases affecting CtrA∼P levels, each of which may act at a distinct phase in the cell cycle. For example, PleC may act late in the cell cycle to control CtrA∼P levels and therefore pilA transcription as cells undergo division. The single domain response regulator DivK lies in the pathway leading to CtrA phosphorylation (Wu et al., 1998) and the PleC-dependent release of polar DivK occurs at approximately the same time as the PleC-dependent accumulation of PilA. Moreover, PleC controls the DivK phosphorylation state in vivo and in vitro (Wu et al., 1998; Wheeler and Shapiro, 1999), raising the possibility that PleC regulates PilA accumulation transcriptionally through a phosphorelay in which the phosphate is passed indirectly from PleC to CtrA via DivK. However, we cannot exclude the alternative possibility that PleC controls PilA accumulation via a CtrA-independent pathway.
The regulation of pilin accumulation by a polarly localized two-component histidine kinase-response regulator pair is not unprecedented. Expression of the pilin (encoded by pilA) that is assembled into polar pili in P.aeruginosa is controlled by the polar histidine kinase PilS (Boyd, 2000). PilS phosphorylates the response regulator PilR, which transcriptionally activates pilA (Boyd et al., 1994). Even though the biological sig nificance of localizing the sensor kinase to the pole(s) remains unresolved, it appears to be a characteristic feature of histidine kinases required for the assembly of polar pili.
PleC and PilS both control pilin accumulation and localize to the pole (Wheeler and Shapiro, 1999; Boyd, 2000). We have discovered an activity of PleC that the two kinases probably do not share. The Pseudomonas predivisional cell is not asymmetric with respect to the polar localization of PilS. Instead, PilS is localized to both cell poles in the predivisional cell. In Caulobacter, PleC resides at the piliated pole in swarmer cells and, later, is released from the pole at the swarmer-to-stalked cell transition. As a consequence, once new PleC coalesces at the incipient swarmer cell pole in the predivisional cell, PleC is asymmetrically located at one cell pole. Under conditions where PleC is unable to autophosphorylate at H610, it is located at both cell poles, suggesting that PleC controls its own asymmetric location. This same activity of PleC is required for the release of the pilus assembly protein CpaE. While in wild-type predivisional cells CpaE is asymmetrically localized to the incipient swarmer cell pole, in those lacking PleC kinase activity, CpaE is bipolar due to its retention at the pole at the swarmer-to-stalked cell transition. Since our results suggest that both of these functions of PleC are dependent on its ability to autophosphorylate, we propose that PleC, in addition to the phosphorelay acting at cell division for the accumulation of PilA, also controls a distinct phosphorelay at the swarmer-to-stalked cell transition, altering the phosphorylation state of an unknown response regulator, which triggers the release of PleC and CpaE. A reason why PleC is equipped with this activity may lie in the asymmetric cell division inherent to C.crescentus. Unlike P.aeruginosa, only one of the two daughter cells in C.crescentus is piliated. PleC serves to maintain this asymmetry by releasing critical pilus assembly proteins, such as CpaE and itself, from the swarmer cell pole as the swarmer cell differentiates into a stalked cell, resulting in an asymmetric predivisional cell that divides into a piliated swarmer cell and a non-piliated stalked cell. While this hypothesis explains the asymmetric location of PleC in C.crescentus, the question of how and why histidine kinases controlling pilin accumulation are localized to the cell pole remains to be explored.
Materials and methods
Bacterial strains and growth conditions
Escherichia coli strains S17-1, BL21 and DH5α were grown in Luria–Bertani medium at 37 or 30°C supplemented with ampicillin (200 µg/ml), kanamycin (50 µg/ml), tetracycline (12.5 µg/ml), chloramphenicol (30 µg/ml) and a mixture of spectinomycin/streptomycin (100/50 µg/ml) when required. Wild-type C.crescentus CB15N, CB15 and their derivatives were grown at 28°C in complex (peptone yeast extract; PYE) or minimal (M2G) medium supplemented with kanamycin (5 µg/ml) or spectinomycin/streptomycin (30/5 µg/ml) when required. Synchronization was performed as described (Evinger and Agabian, 1977). Caulobacter strains were grown in M2G for immunoblotting and fluorescence microscopy experiments.
The ΔpilA, ΔcpaC and ΔcpaE strains have been described previously (Skerker and Shapiro, 2000). The strain with an in-frame deletion of pleC (ΔpleC) was a gift from Urs Jenal. The ΔcpaC deletion in CB15 was constructed as described previously (Skerker and Shapiro, 2000).
The pleC(H610A) strain was made by a two-step process. First, a ΔpleC::Ω strain was constructed by double crossover using a two-step sacB counter-selection procedure (Stephens et al., 1996) and pHPV142, selecting for spectinomycin/streptomycin resistance. Then, pHPV307 was used to exchange the ΔpleC::Ω allele for pleC(H610A) by double crossover using the sacB counter-selection procedure by negatively selecting for the loss of the spectinomycin/streptomycin resistance gene.
The pleC(H610A)–gfp and pleC–gfp strains were made by fusing gfp to the 3′ end of chromosmal pleC(H610A) and wild-type pleC, respectively. This was accomplished by integration of pHPV290, harboring the pleC 3′ end fused to gfp, into the chromosome of the pleC(H610A) or wild-type strain by a single crossover event. The fact that the pleC–gfp strain had a wild-type phenotype indicated that PleC–GFP was functional.
The YFP–CpaE-expressing strains were made by replacing the chromosomal version of cpaE in wild-type, ΔcpaC and the ΔpleC strains with yfp–cpaE by the two-step sacB counter-selection procedure using pHPV227. YFP–CpaE was functional, as indicated by the fact that the yfp–cpaE strain was sensitive to φCbK.
Plasmid transfer and cloning
Plasmids were mobilized from E.coli S17-1 to C.crescentus by bacterial conjugation. Recombinant DNA techniques were performed according to Sambrook et al. (1989). Minipreps and recovery of DNA fragments from agarose gels were done using the Qiaprep or Qiaquick kits (Qiagen). PCR amplifications were performed using Pfu Turbo DNA polymerase from Stratagene. The sequences of all oligonucleotides used are available upon request.
YFP and GFP fusion plasmids
The plasmid (pHPV227) used to replace the chromosomal version of cpaE with yfp–cpaE was constructed by fusing a version of yfp lacking the start and stop codon after the cpaE start codon. First a ∼520 bp upstream fragment, containing the 3′ end of cpaD and ending after the start codon of cpaE, was amplified by PCR and cloned as a HindIII–EcoRV fragment into pNPTS138 (M.R.K.Alley, unpublished data), yielding pHPV218. A downstream fragment, extending from the cpaE start codon ∼570 nt downstream into cpaE, was amplified by PCR and digested with EcoRI and BamHI. A central fragment containing the yfp open reading frame, in which the start and stop codon have been replaced by an in-frame BamHI and EcoRV site, respectively, was amplified by PCR from pEYFP (Clontech). After restriction enzyme digestion, the central fragment was triple ligated along with the downstream fragment via BamHI into EcoRI–EcoRV-restricted pHPV218, yielding pHPV227.
The plasmid for making chromosmal fusions of gfp to pleC, pHPV290, is based on pPleC–GFP (Wheeler and Shapiro, 1999). The 3′ end of pleC–gfp was released from pPleC–GFP by digestion with EcoRI and XbaI and cloned into the suicide vector pNPT228 (M.R.K.Alley, unpublished data).
pleC disruption and allele replacement plasmids
The pleC disruption plasmid (pHPV142) was constructed by cloning the pleC gene, isolated from pSCW408 (Wang et al., 1993) by digestion with XhoI and HindIII, into SalI–HindIII-restricted pNPTS138. The resulting plasmid (pHPV140) was cut with BamHI, which liberates a 1.6 kb internal fragment of pleC. The larger fragment containing the vector and the ends of pleC was gel purified and ligated to the Ω cassette, harboring the aadA gene, which confers resistance to spectinomycin/streptomycin. The resulting plasmid was designated pHPV142.
The pleC(H610A) allele was constructed by PCR using a mutagenic oligonucleotide (5′-GAATTCCTGGCCAACATGTCTGCCGAGCTG CGGACACCGCTGAA-3′), in which the histidine codon (CAC) coding for H610 of PleC is mutated into one coding for alanine (GCC, underlined), and another oligonucleotide overlapping the stop codon of pleC. The mutagenic oligonucleotide extends 21 nt upstream of the histidine codon, where it overlaps with a natural EcoRI site (bold) in pleC. The amplified fragment was digested with EcoRI and BstEII, whose recognition sequence is located 467 nt downstream of the EcoRI site, and the fragment containing the pleC(H610A) mutation was exchanged for the wild-type fragment in pHPV140 using EcoRI–BstEII. The resulting plasmid was named pHPV307.
Overexpression plasmids
The CpaE and CpaC overexpression plasmids were constructed by cloning PCR fragments coding for full-length CpaE and residues 164–560 of CpaC into pET28a (Novagen), yielding pHPV106 and pHPV35, respectively. The PleC overexpression construct was made by digesting pSCW408 with EcoRI and HindIII, and ligating the liberated ∼810 bp fragment coding for residues 603–842 of PleC in-frame into pET28a, yielding pHPV319.
Immunoblots and quantification
Crude extracts (∼10 µg) were separated on 10% SDS–polyacrylamide gels. Proteins were blotted onto polyvinylidene difluoride (PVDF) membrane (Millipore) in transfer buffer (15 mM Tris, 120 mM glycine, 10% methanol). The membranes were incubated in blocking buffer [TBS (20 mM Tris–HCl pH 7.6, 130 mM NaCl) containing 5% non-fat dried milk and 0.1% Tween-20] for 1 h at room temperature. Primary antibody was added at the appropriate dilutions (α-PilA 1/5000, affinity-purified α-CpaC 1/2000, α-PleC 1/2000, α-CtrA 1/10 000 and α-CpaE 1/20 000) to the blocking buffer and incubated with the membrane for 1 h. Unbound antibody was removed by four 5 min washes in TBS. The membrane was immersed into blocking buffer containing horseradish peroxidase-conjugated donkey anti-rabbit IgG (Jackson ImmmunoResearch) at a dilution of 1/10 000, incubated for 1 h and then washed four times (5 min) in TBS. Reactive bands were visualized using chemoluminescence (Lightning, Perkin Elmer). Immunoblots were performed in triplicate and quantified using the Imagequant Software (Molecular Dynamics).
Live cell microscopy
Live cell microscopy with cells expressing GFP or YFP fusion proteins was performed as described by Jensen et al. (2001).
Immunofluorescence microscopy
For immunofluorescence microscopy, synchronized cells were fixed using 3% formaldehyde at different stages of the cell cycle. The fixation solution was removed by filtration. Immunofluorescence microscopy was performed as described (Domian et al., 1997). Affinity-purified CpaC antibodies were used at a 1:200 dilution and FITC-conjugated goat anti-rabbit secondary antibodies (Jackson ImmunoResearch Laboratories, Inc.) were used at a 1:200 dilution. Images were processed using Metamorph (Universal Imaging Corporation) and Photoshop (Adobe).
Antibody production and purification
Escherichia coli BL21(DE3)/pLysS containing a plasmid overexpressing either CpaC (pHPV35), CpaE (pHPV106) or PleC (pHPV319) was grown in LB medium supplemented with kanamycin and chloramphenicol overnight at room temperature. This culture was diluted 50-fold into 1 l of the same medium and grown to an OD600 of 0.4 at room temperature. Synthesis of the recombinant proteins was induced by the addition of isopropyl-β-d-thiogalactopyranoside (IPTG) to a final concentration of 0.4 mM for 3 h. The cells were harvested by centrifugation, washed once in TBS and frozen at –70°C. Recombinant proteins were purified by metal-chelate affinity chromatography using Ni2+-NTA–agarose (Qiagen) under denaturing conditions using a standard protocol (Qiagen). A total of 2 mg of each purified protein was used to immunize two rabbits (Covance, Richmond, CA, USA) for the production of polyclonal antibodies. To raise antibodies against PilA, the rabbits were immunized with purified pilin filaments (Skerker and Shapiro, 2000).
CpaC antibodies were affinity purified by treating the crude serum with acetone powders prepared from the ΔcpaC mutant according to standard procedures (Sambrook et al., 1989). The acetone powder-treated serum was then affinity purified using protein A–agarose (Roche) according to the manufacturer’s instructions.
The CtrA antibodies have been described previously (Quon et al., 1996).
Acknowledgments
Acknowledgements
We are indebted to Jeff Skerker for the preparation of the PilA antibodies and Urs Jenal for providing the ΔpleC strain. We thank members of the Shapiro Laboratory and J.Jackobsen for critical reading of the manuscript and present and former members of the Shapiro Laboratory for valuable discussions. This work was supported by National Institutes of Health (NIH) Grant GM 32506/5120M2 and Defense Advanced Research Projects Agency (DARPA) Grant MDA972-00-0032. P.H.V. is funded in part by a grant from the Swiss National Science Foundation and the Roche Research Foundation.
References
- Alley M.R., Maddock,J.R. and Shapiro,L. (1992) Polar localization of a bacterial chemoreceptor. Genes Dev., 6, 825–836. [DOI] [PubMed] [Google Scholar]
- Bitter W., Koster,M., Latijnhouwers,M., de Cock,H. and Tommassen,J. (1998) Formation of oligomeric rings by XcpQ and PilQ, which are involved in protein transport across the outer membrane of Pseudomonas aeruginosa. Mol. Microbiol., 27, 209–219. [DOI] [PubMed] [Google Scholar]
- Boyd J.M. (2000) Localization of the histidine kinase PilS to the poles of Pseudomonas aeruginosa and identification of a localization domain. Mol. Microbiol., 36, 153–162. [DOI] [PubMed] [Google Scholar]
- Boyd J.M., Koga,T. and Lory,S. (1994) Identification and characterization of PilS, an essential regulator of pilin expression in Pseudomonas aeruginosa. Mol. Gen. Genet., 243, 565–574. [DOI] [PubMed] [Google Scholar]
- Domian I.J., Quon,K.C. and Shapiro,L. (1997) Cell type-specific phosphorylation and proteolysis of a transcriptional regulator controls the G1-to-S transition in a bacterial cell cycle. Cell, 90, 415–424. [DOI] [PubMed] [Google Scholar]
- Evinger M. and Agabian,N. (1977) Envelope-associated nucleoid from Caulobacter crescentus stalked and swarmer cells. J. Bacteriol., 132, 294–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie K.R., Lory,S. and Pugsley,A.P. (1996) Insertion of an outer membrane protein in Escherichia coli requires a chaperone-like protein. EMBO J., 15, 978–988. [PMC free article] [PubMed] [Google Scholar]
- Jacobs C. and Shapiro,L. (1998) Microbial asymmetric cell division: localization of cell fate determinants. Curr. Opin. Genet. Dev., 8, 386–391. [DOI] [PubMed] [Google Scholar]
- Jacobs C., Domian,I.J., Maddock,J.R. and Shapiro,L. (1999) Cell cycle-dependent polar localization of an essential bacterial histidine kinase that controls DNA replication and cell division. Cell, 97, 111–120. [DOI] [PubMed] [Google Scholar]
- Jacobs C., Hung,D. and Shapiro,L. (2001) Dynamic localization of a cytoplasmic signal transduction response regulator controls morphogenesis during the Caulobacter cell cycle. Proc. Natl Acad. Sci. USA, 98, 4095–4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen R.B., Wang,S.C. and Shapiro,L. (2001) A moving DNA replication factory in Caulobacter crescentus. EMBO J., 20, 4952–4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen R.B., Wang,S.C. and Shapiro,L. (2002) Dynamic localization of proteins and DNA during a bacterial cell cycle. Nature Rev. Mol. Cell Biol., 3, 167–176. [DOI] [PubMed] [Google Scholar]
- Knoblich J.A. (2001) Asymmetric cell division during animal development. Nature Rev. Mol. Cell Biol., 2, 11–20. [DOI] [PubMed] [Google Scholar]
- Laub M.T., McAdams,H.H., Feldblyum,T., Fraser,C.M. and Shapiro,L. (2000) Global analysis of the genetic network controlling a bacterial cell cycle. Science, 290, 2144–2148. [DOI] [PubMed] [Google Scholar]
- Laub M.T., Chen,S.L., Shapiro,L. and McAdams,H.H. (2002) Genes directly controlled by CtrA, a master regulator of the Caulobacter cell cycle. Proc. Natl Acad. Sci. USA, 99, 4632–4637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lory S. (1998) Secretion of proteins and assembly of bacterial surface organelles: shared pathways of extracellular protein targeting. Curr. Opin. Microbiol., 1, 27–35. [DOI] [PubMed] [Google Scholar]
- Marciano D.K., Russel,M. and Simon,S.M. (1999) An aqueous channel for filamentous phage export. Science, 284, 1516–1519. [DOI] [PubMed] [Google Scholar]
- Marciano D.K., Russel,M. and Simon,S.M. (2001) Assembling filamentous phage occlude pIV channels. Proc. Natl Acad. Sci. USA, 98, 9359–9364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nouwen N., Ranson,N., Saibil,H., Wolpensinger,B., Engel,A., Ghazi,A. and Pugsley,A.P. (1999) Secretin PulD: association with pilot PulS, structure and ion- conducting channel formation. Proc. Natl Acad. Sci. USA, 96, 8173–8177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nouwen N., Stahlberg,H., Pugsley,A.P. and Engel,A. (2000) Domain structure of secretin PulD revealed by limited proteolysis and electron microscopy. EMBO J., 19, 2229–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta N., Grebe,T.W. and Newton,A. (2000) Signal transduction and checkpoints in developmental regulation of Caulobacter. In Brun,Y.V. and Shimkets,L.J. (eds), Prokaryotic Development. American Society for Microbiology, Washington, DC, pp. 341–359.
- Parkinson J.S. (1993) Signal transduction schemes of bacteria. Cell, 73, 857–871. [DOI] [PubMed] [Google Scholar]
- Quon K.C., Marczynski,G.T. and Shapiro,L. (1996) Cell cycle control by an essential bacterial two-component signal transduction protein. Cell, 84, 83–93. [DOI] [PubMed] [Google Scholar]
- Russel M. (1998) Macromolecular assembly and secretion across the bacterial cell envelope: type II protein secretion systems. J. Mol. Biol., 279, 485–499. [DOI] [PubMed] [Google Scholar]
- Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual. 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Sandkvist M. (2001) Biology of type II secretion. Mol. Microbiol., 40, 271–283. [DOI] [PubMed] [Google Scholar]
- Sauvonnet N., Vignon,G., Pugsley,A.P. and Gounon,P. (2000) Pilus formation and protein secretion by the same machinery in Escherichia coli. EMBO J., 19, 2221–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt S.A., Bieber,D., Ramer,S.W., Hwang,J., Wu,C.Y. and Schoolnik,G. (2001) Structure–function analysis of BfpB, a secretin-like protein encoded by the bundle-forming pilus operon of enteropathogenic Escherichia coli. J. Bacteriol., 183, 4848–4859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro L. and Losick,R. (2000) Dynamic spatial regulation in the bacterial cell. Cell, 100, 89–98. [DOI] [PubMed] [Google Scholar]
- Skerker J.M. and Shapiro,L. (2000) Identification and cell cycle control of a novel pilus system in Caulobacter crescentus. EMBO J., 19, 3223–3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer J.M. and Newton,A. (1988) Sequential regulation of developmental events during polar morphogenesis in Caulobacter crescentus: assembly of pili on swarmer cells requires cell separation. J. Bacteriol., 170, 409–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer J.M. and Newton,A. (1989) Turning off flagellum rotation requires the pleiotropic gene pleD: pleA, pleC and pleD define two morphogenic pathways in Caulobacter crescentus. J. Bacteriol., 171, 392–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto G.E. and Hultgren,S.J. (1999) Bacterial adhesins: common themes and variations in architecture and assembly. J. Bacteriol., 181, 1059–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens C., Reisenauer,A., Wright,R. and Shapiro,L. (1996) A cell cycle-regulated bacterial DNA methyltransferase is essential for viability. Proc. Natl Acad. Sci. USA, 93, 1210–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall D. and Kaiser,D. (1999) Type IV pili and cell motility. Mol. Microbiol., 32, 1–10. [DOI] [PubMed] [Google Scholar]
- Wang S.P., Sharma,P.L., Schoenlein,P.V. and Ely,B. (1993) A histidine protein kinase is involved in polar organelle development in Caulobacter crescentus. Proc. Natl Acad. Sci. USA, 90, 630–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler R.T. and Shapiro,L. (1999) Differential localization of two histidine kinases controlling bacterial cell differentiation. Mol. Cell, 4, 683–694. [DOI] [PubMed] [Google Scholar]
- Wu J., Ohta,N. and Newton,A. (1998) An essential, multicomponent signal transduction pathway required for cell cycle regulation in Caulobacter. Proc. Natl Acad. Sci. USA, 95, 1443–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]