

Abstract
MicroRNAs (miRNAs) constitute a novel, phylogenetically extensive family of small RNAs (∼22 nucleotides) with potential roles in gene regulation. Apart from the finding that miRNAs are produced by Dicer from the precursors of ∼70 nucleotides (pre-miRNAs), little is known about miRNA biogenesis. Some miRNA genes have been found in close conjunction, suggesting that they are expressed as single transcriptional units. Here, we present in vivo and in vitro evidence that these clustered miRNAs are expressed polycistronically and are processed through at least two sequential steps: (i) generation of the ∼70 nucleotide pre-miRNAs from the longer transcripts (termed pri-miRNAs); and (ii) processing of pre-miRNAs into mature miRNAs. Subcellular localization studies showed that the first and second steps are compartmentalized into the nucleus and cytoplasm, respectively, and that the pre-miRNA serves as the substrate for nuclear export. Our study suggests that the regulation of miRNA expression may occur at multiple levels, including the two processing steps and the nuclear export step. These data will provide a framework for further studies on miRNA biogenesis.
Keywords: microRNA/nuclear export/processing/subcellular localization/transcription
Introduction
Recent discoveries of an abundant class of ∼22 nucleotide (nt) RNAs, microRNAs (miRNAs), have widened the scope of eukaryotic gene regulation (Lagos-Quintana et al., 2001; Lau et al., 2001; Lee and Ambros, 2001; Mourelatos et al., 2002). Over a hundred miRNA genes have been discovered so far from biochemical and bioinformatic studies of Caenorhabditis elegans, Drosophila melanogaster and Homo sapiens. The number is expected to increase sharply as the survey for miRNAs continues. Only nine out of 40 miRNAs identified by Dreyfuss et al. (Mourelatos et al., 2002) overlapped with those charted by Tuschl et al. (Lagos-Quintana et al., 2001), although both groups used the same cell line (HeLa cells) for the cloning. Moreover, many miRNAs appeared in the cloning only once, implying that there remain rare unidentified miRNAs.
The paradigm for the function and biogenesis of miRNAs has been provided by lin-4 and let-7, which were originally identified by genetic analysis of C.elegans developmental timing (Lee et al., 1993; Reinhart et al., 2000). lin-4 and let-7 act as post-transcriptional repressors of their target genes when bound to their specific sites in the 3′ untranslated region of the target mRNA (Lee et al., 1993; Wightman et al., 1993; Moss et al., 1997; Olsen and Ambros, 1999; Slack et al., 2000). The functions of other miRNAs are currently unknown. However, given the diversity in sequence and expression pattern, miRNAs are expected to play various roles in a wide range of regulatory pathways (Lai, 2002).
Little is known about the biogenesis of miRNAs. RNAs of ∼70 nt as well as ∼22 nt have been detected from northern blot analyses for most miRNAs, suggesting that these ∼70 nt RNAs are the precursors of ∼22 nt miRNAs (Lee et al., 1993; Pasquinelli et al., 2000; Reinhart et al., 2000; Grishok et al., 2001). An interesting, common feature of miRNAs is that in silico secondary structure prediction indicates that the ∼70 nt putative precursor forms a stem–loop structure with some bulges (Lagos-Quintana et al., 2001; Lau et al., 2001; Lee and Ambros, 2001; Mourelatos et al., 2002). Because Dicer, the multi-domain RNase III-type protein, was found to function in generating small interfering RNAs (siRNAs) that are similar in size (21–25 nt) (Hammond et al., 2000; Bernstein et al., 2001), a prediction was made that Dicer also functions in the processing of ∼70 nt RNAs into mature miRNAs. Indeed, it was later shown that immunoprecipitated Dicer generates ∼22 nt miRNAs from in vitro synthesized ∼70 nt RNAs (Ketting et al., 2001; Knight and Bass, 2001). Moreover, when the human (Helicase-MOI) and C.elegans (dcr-1) homologs of Dicer were knocked down or knocked out, the ∼70 nt RNAs accumulated while ∼22 nt mature miRNAs disappeared (Grishok et al., 2001; Hutvagner et al., 2001; Ketting et al., 2001; Knight and Bass, 2001). Therefore, the ∼70 nt RNAs are the precursors of mature miRNAs and are accordingly referred to as ‘pre-miRNAs’. Apart from Dicer, Alg-1 and Alg-2 in C.elegans appear to be involved in miRNA maturation, although their precise roles in processing are not clear (Grishok et al., 2001). More recently, a number of human miRNAs have been found in association with eIF2C2, Gemin3 and Gemin4 to form an ∼15S RNP complex (Mourelatos et al., 2002). The function of this complex and each component protein remains to be determined.
Several miRNA genes exist as clusters of 2–7 genes with intervals as short as a few nucleotides long (Lagos-Quintana et al., 2001; Lau et al., 2001; Mourelatos et al., 2002). The expression profiles of clustered genes are highly similar, raising the possibility that transcription of such miRNAs is controlled by common regulatory sequences. One possibility is that a single promoter drives transcription of the clustered miRNA genes. If so, the nascent transcripts must be polycistronic and therefore would require an additional unknown processing step to produce the ∼70 nt pre-miRNAs before Dicer can participate and generate mature miRNAs. In other words, maturation of miRNAs may occur through at least two steps: (i) processing of the nascent transcript to the ∼70 nt precursor; and (ii) processing of the ∼70 nt to the ∼22 nt mature miRNA. Alternatively, it is also plausible that the ∼70 nt precursor itself is the nascent transcript. In this case, each miRNA in the cluster is transcribed from its own promoter, but the same enhancer controls transcription for coordinated expression.
Another open question regarding miRNA biogenesis is about the subcellular localization and transport of miRNAs. Nucleocytoplasmic transport (especially export) is critical in the expression and functions of RNAs (Wolin and Matera, 1999; Warner, 2001; Will and Luhrmann, 2001; Dreyfuss et al., 2002; Reed and Hurt, 2002). For instance, pre-mRNAs are retained in the nucleus until splicing is successfully carried out, so that only correctly processed mRNAs can pass the ‘quality control’ and become available for cytoplasmic translation (Chang and Sharp, 1989; Legrain and Rosbash, 1989; Luo and Reed, 1999; Maquat and Carmichael, 2001). Biogenesis of the UsnRNAs is also closely linked to the nucleocytoplasmic transport. The UsnRNAs, with the exception of U6 and U6atac, must first be exported to the cytoplasm where snRNP assembly is initiated (Hamm and Mattaj, 1990; Izaurralde et al., 1995; Fornerod et al., 1997). Following modifications and Sm core assembly, the UsnRNAs are re-imported to the nucleus to complete snRNP assembly and to participate in pre-mRNA splicing (Palacios et al., 1997; Huber et al., 1998; Yu et al., 1998). Studies of the localization and transport of miRNAs are likely to reveal important aspects of miRNA expression and function.
In this study we explore the mode of miRNA expression. By employing various in vivo and in vitro analytical methods, we demonstrate the polycistronic nature of clustered miRNAs and their stepwise processing in different subcellular locations.
Results
Primary precursors for miRNAs are longer than the ∼70 nt pre-miRNAs
Our initial approach to address this issue was to search for transcripts that are longer than the proposed ∼70 nt precursors. These long transcripts are likely to be the nascent miRNA transcripts that serve as the primary precursors for pre-miRNAs. Total RNA from HeLa cells was subject to RT–PCR. The primers were chosen to bind outside the boundary of the predicted ∼70 nt stem–loop clusters so that we could detect transcripts covering the whole cluster. Two miRNA clusters were examined in this study: mir-23∼27∼24-2 containing three miRNA genes and mir-17∼18∼19a∼20∼19b-1 containing five (Lagos-Quintana et al., 2001). Both the clusters reside in intergenic sequences, making it unlikely that the transcripts are part of or read-through of other gene transcripts. PCR products of the expected size (403 and 791 bp) were detected (Figure 1A and B, respectively), suggesting that the nascent transcripts are polycistronic and that these clusters may be transcribed as single transcriptional units. To show that the products of the RT–PCR originate from RNA but not from contaminated genomic DNA, reverse transcriptase was omitted during the first-strand synthesis (lanes 2). As an additional control, total RNA was pre-treated with RNase A before first-strand synthesis (lanes 3). The same approach was applied to mir-30a, which is a single gene and not part of a cluster. A PCR product of the expected size (171 bp) was detected (Figure 1C, lane 1). Different sets of primers were also used for RT–PCR to cover a broader region around miR-30a, resulting in PCR products of 301 nt, indicating that the long transcript is over 301 nt (data not shown). In agreement with our observation, several expressed sequence tag (EST) clones containing some miRNAs, such as miR-23∼27∼24-2, were found in public databases, as noted previously by Tuschl et al. (Lagos-Quintana et al., 2001). Therefore, miRNAs in general may exist as transcripts that are longer than the ∼70 nt pre-miRNAs.

Fig. 1. RT–PCR reveals the long transcripts containing miRNAs. (A) HeLa total RNA was used for reverse transcription using a primer that binds downstream of mir-23∼27∼24-2. The product from the reverse transcription reaction served as the template for PCR to amplify the indicated regions containing the miRNA cluster. As a control, reverse transcriptase was omitted (lane 2). For RNase treatment, DNase-free RNase A (Ambion) was added to a final concentration of 50 µg/ml and incubated at 37°C for 2 min before reverse transcription. The illustration on the right side shows the template miRNA genes, the primers for PCR and the PCR product. Gray and black boxes indicate the regions corresponding to ∼70 nt precursors and mature miRNAs, respectively. (B) A similar experiment as in (A) was performed for mir-17∼18∼19a∼20∼19b-1. (C) A similar experiment as in (A) was performed for mir-30a.
We next performed northern blot analysis for miR-30a using denaturing agarose gels in an attempt to characterize the full-length transcript (Figure 2). A transcript of ∼600 nt was detected from HeLa cells when using the probe of 301 nt surrounding miR-30a. This transcript does not seem to be abundant, since northern blotting never gave a strong signal. A long transcript for miR-23∼27∼24-2 has not been detected under similar conditions. Of note, we also tried 5′- and 3′-RACE (rapid amplification of cDNA ends) for miR-30a and miR-23∼27∼24-2, which has not been successful so far, supporting the notion that these transcripts are rare at steady-state level, possibly due to rapid processing.
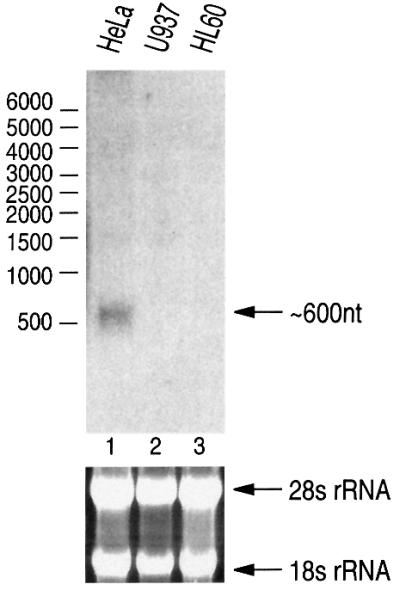
Fig. 2. Northern blot analysis of miR-30a. From HeLa total RNA, a band of ∼600 nt was detected when 20 µg of RNA were analyzed on 1% formaldehyde–agarose gel and the blot was probed with miR-30a-specific probe. The size was determined by using RNA Millennium size markers from Ambion. The lower panel shows ethidium bromide staining of the agarose gel.
Maturation of miRNAs occurs through at least two steps
In order to verify that these long transcripts are authentic precursors of mature miRNAs, we developed an in vitro processing system that is similar to the in vitro splicing system (Kim et al., 2001). Cell-free assay systems have proven invaluable for dissecting the molecular basis of physiological events, as elegantly exemplified by the in vitro splicing system originally reported by Krainer et al. (1984) and the cell-free system for RNA interference established by Tuschl et al. (1999).
In brief, the PCR products were inserted downstream of the T7 promoter and used for in vitro transcription. Uniformly radiolabeled RNA was then mixed with total cell extract prepared from HEK293T cells. Following incubation at 37°C for 90 min, the resulting RNA from the reaction mixture was extracted and analyzed on a denaturing polyacrylamide gel. The input RNA that contains miR-30a as well as its surrounding sequences resulted in four major fragments in the presence of cell extract (Figure 3A, lane 2). The sizes of two fragments were ∼65 and ∼23 nt, strikingly reminiscent of the pre-miRNAs and mature miRNAs, respectively, which have been observed previously by northern blot analyses of endogenous RNAs (Lagos-Quintana et al., 2001; Lau et al., 2001; Lee and Ambros, 2001). The antisense transcript did not produce any discrete fragments (Figure 3A, lane 5), indicating that the cleavage is not due to non-specific nuclease activity. To examine whether the ∼65 nt fragment is indeed the precursor of ∼23 nt fragment, as would be expected for miRNAs, the fragments from in vitro processing reaction were gel purified and used for an additional round of in vitro processing (Figure 3B). The ∼65 nt fragment, but not the others, was processed to generate the ∼23 nt fragments in the presence of cell extract (Figure 3B, lane 5), demonstrating that the ∼65 nt fragment is the precursor of the ∼23 nt fragments. Therefore, the input RNA was processed first into ∼65 nt and then into ∼23 nt fragments. The identities of the ∼195 and ∼110 nt fragments are currently under investigation. They may be the side products or the intermediates from the first-step processing.

Fig. 3. In vitro processing of miRNAs via two sequential steps. (A) Uniformly radiolabeled RNAs corresponding to 371 nt covering miR-30a were incubated either in the presence or absence of HEK293T cell extract. Following the indicated incubation time, RNA was extracted and analyzed on a 12.5% (v/v) denaturing polyacrylamide gel. The marker in the left lane is pBR322 digested with MspI and end-labeled with T4 polynucleotide kinase. (B) Each fragment from the in vitro processing reaction was gel purified and incubated with cell extract for 90 min. (C) Primer extension analysis. From both the HeLa total RNAs and the in vitro processing product, ∼23 nt bands were detected using the 5′-end-labeled antisense oligonucleotide to miR-30a-s. (D) Schematic representation of in vitro processing.
In order to confirm the identity of the ∼23 nt fragment, we carried out primer extention analysis (Figure 3C). The fragments of ∼23 nt were detected from the in vitro processed products (Figure 3C, lane 3) as well as from HeLa total RNA (Figure 3C, lane 1), verifying that the ∼23 nt fragment is the authentic mature miR-30a-s. When a similar experiment was carried out to detect miR-30a-as that is released from the opposite strand of pre-miR-30a, only a marginally detectable level of miR-30a-as was observed from both HeLa total RNA and in vitro processing reaction (data not shown). Two recent reports showed that miR-30a-as (originally named miR-30) is less abundant than miR-30a-s (previously termed miR-97) (Lagos-Quintana et al., 2002; Zeng et al., 2002). Thus, there seems to be a certain degree of asymmetry in processing of this miRNA.
Given that miRNA processing occurred correctly without the original 5′- and 3′-end sequences, it appears that the 5′- and 3′-end sequences are not required for processing. Furthermore, a shorter RNA substrate (171 nt) covering miR-30a was processed in vitro as efficiently as the 371-nt-long RNA substrate (data not shown), suggesting that the cis-acting element(s) for processing is located in close proximity to, and/or inside, pre-miRNA sequences.
To demonstrate that our observations are not restricted to a certain RNA substrate, we used the same assay conditions for miR-15∼16 and miR-23∼27∼24-2 (Figure 4A and B). The ∼70 nt fragments as well as the 21–23 nt fragments were observed from both the miRNA clusters. Thus, this cell-free system successfully recapitulates endogenous miRNA maturation and provides a general analysis tool for miRNA processing.

Fig. 4. The in vitro processing system is a general analysis tool for miRNA maturation. (A) In vitro processing of miR-15∼16. These clustered miRNAs can also be processed in vitro to result in ∼70 nt intermediates and 22–23 nt final products. (B) In vitro processing of miR-23∼27∼24-2.
Our data show that miRNA maturation consists of at least two sequential steps: (i) the long transcripts are first processed into the ∼70 nt intermediate (pre-miRNAs); and (ii) then into the final products of ∼22 nt (mature miRNAs). We suggest that the long transcript be designated ‘pri-miRNA’ (primary precursor for miRNA).
Three different forms of miRNAs
Pri-miRNAs could be detected by an additional method, RNase protection assay (RPA) (Figure 5). The probe was designed to cover part of the miRNA gene such that three different forms of miRNA (pri-, pre- and mature miRNAs) can be detected simultaneously as three fragments of different sizes. From HeLa total RNA, three specific bands were detected representing pri-miR-23, pre-miR-23 and mature miR-23 (Figure 5A, lanes 1 and 2). We cannot exclude the possibility that pri-miRNA might not be a single RNA species of the nascent transcript but might also represent some unidentified intermediates of the first-step processing. RT–PCR and RPA are extremely sensitive methods to detect the region to which the probes anneal. Therefore, if the nascent transcript is subject to rapid processing, the nascent transcript and the processing intermediates would appear as a discrete band in RT–PCR or RPA, whereas in northern blot analysis they would appear as one or more weak bands. We are currently investigating the mechanism and the factors of the first-step processing.

Fig. 5. Subcellular localization of the three different forms of miRNA. (A) RPA to detect miRNA. The indicated amounts of RNA from either HeLa or yeast cells were used for RPA. The asterisk indicates the self-protected probe. Schematically represented are the probes for the RPA and the protected fragments that are expected for each miRNA species. (B) Total, nuclear or cytoplasmic RNA from HeLa cells or yeast RNA was used for RPA.
Subcellular compartmentalization of miRNA processing
The RPA method enabled us to examine the subcellular localization of each miRNA form. From the fractionated nucleus and cytoplasm of HeLa cells, RNA was extracted and used for RPA (Figure 5B). Interestingly, pri- and pre-miR-23 are concentrated in the nucleus (Figure 5B, lane 2), while mature miR-23 is mainly in the cytoplasm (Figure 5B, lane 3). Thus, the first-step processing is likely to occur in the nucleus. The arena of the second-step processing is less clear. It may occur in the nucleus and then the resulting mature miRNAs may be rapidly exported to the cytoplasm. Alternatively, the pre-miRNAs are exported and subject to the second-step processing in the cytoplasm.
We tackled this issue by taking advantage of our in vitro processing system. Following subcellular fractionation, the nucleoplasmic and cytoplasmic fractions were used for in vitro processing, separately or in combination (Figure 6A). In the nucleoplasmic fraction, only the first-step reaction took place efficiently (Figure 6A, lanes 1–4), whereas in the cytoplasmic fraction neither of the reactions occurred effectively (Figure 6A, lane 8), showing that the processing machinery for the first step is restricted to the nuclear compartment. Only when the cytoplasmic fraction was added to the nucleoplasmic fraction was mature miRNA produced (Figure 6A, lane 7), which indicates that the second step is confined to the cytoplasm. With the cytoplasmic fraction only, the second step failed to occur because the cytoplasm lacks the enzymatic activity for the preceding reaction (Figure 6A, lane 8). Western blotting was performed to show the efficiency of the nucleocytoplasmic fractionation (Figure 6B). hnRNP C1/C2 and eIF4E are well known for their localization in the nucleus and the cytoplasm, respectively, at steady-state levels.
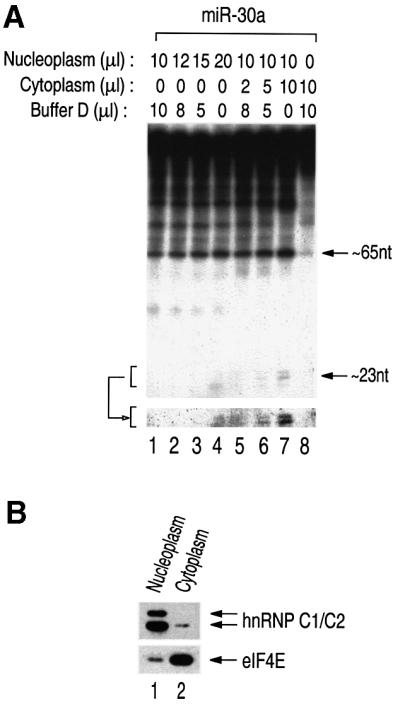
Fig. 6. Nucleocytoplasmic compartmentalization of miRNA processing. (A) In vitro processing of miRNA in fractionated HeLa cell extract. The indicated amounts of the nuclear or cytoplasmic fraction were used for in vitro processing. The lower panel shows the ∼22 nt fragments after longer exposure. (B) Western blot analysis. Nucleoplasmic and cytoplasmic fractions were probed with anti-hnRNP C1/C2 antibody and anti-eIF4E antibody.
Discussion
Our data, together with the recent identification of Dicer as an miRNA processing factor, suggest a model for how miRNA is transcribed, processed and exported (Figure 7). Clustered miRNA genes constitute single, and possibly autonomous, transcriptional units, although this may not be an absolute rule for all the clustered miRNA genes. The polycistronic nascent transcripts are processed into ∼70 nt pre-miRNAs while in the nucleus, by a yet-to-be-identified factor(s). Single miRNA genes that are not part of a cluster are also expressed as nascent transcripts that are longer than pre-miRNAs and need to be processed into pre-miRNA forms in the nucleus. It is the pre-miRNA form that is recognized and exported by an export factor. The second-step processing occurs in the cytoplasm where pre-miRNAs are met by Dicer, and possibly by other factors, and processed into the final products of ∼22 nt.

Fig. 7. A model for miRNA biogenesis. miRNA genes are transcribed by an unidentified polymerase to generate the primary transcripts, referred to as pri-miRNAs. Illustrated in the upper left is the clustered miRNA, such as miR-23∼27∼24-2, of which the pri-miRNA is polycistronic. Illustrated in the upper right is the miRNA, such as miR-30a, of which the pri-miRNA is monocistronic. The first-step processing (STEP 1) by an unknown factor(s) results in pre-miRNAs of ∼70 nt, which are recognized and exported by an unidentified export receptor. Upon export, Dicer, and possibly other factors, participates in the second-step processing (STEP 2) to produce mature miRNAs. The final product may function in a variety of regulatory pathways, such as translational control of certain mRNAs. The question marks indicate unidentified factors.
Grosshans and Slack (2002) have speculated that miRNA biogenesis may occur in a highly coordinated ‘channeling’ pathway, where miRNAs are handed over from one biogenesis factor to the next. Although it is clear from our data that the two main processing steps are separable (Figures 3B and 6A), we do not exclude the possibility that functional coupling of the steps along the miRNA biogenesis pathway is important in vivo, as shown previously in the case of mRNAs (Kim and Dreyfuss, 2001) and tRNAs (Wolin and Matera, 1999). Indeed, it may be noteworthy that the final product was produced more efficiently when the first- and the second-step reaction occurred simultaneously rather than when the second-step reaction occurred separately (compare Figure 3A, lane 2 with B, lane 5), implicating a functional coupling between the first and the second steps of miRNA processing. In light of this, it would be interesting to identify the processing factor for the first step, which is likely to be one of the key players in miRNA biogenesis.
It is intriguing that the two sequential steps of miRNA maturation are compartmentalized into the nucleus and the cytoplasm. This may leave additional room for fine regulation of miRNA expression. For instance, nuclear export of miRNA may be controlled in response to a certain signal. Based on our data, the ∼70 nt pre-miRNA is likely to be the substrate for the nuclear export factor. Export of small RNAs, such as tRNAs and UsnRNAs, is mediated by Exportin-t and Exportin-1/CRM1, respectively, members of the nuclear transport receptor (NTR) family (Nakielny and Dreyfuss, 1999; Conti and Izaurralde, 2001; Kuersten et al., 2001). It remains to be seen whether the unknown export factor for miRNAs also shares the common features of the NTR family, such as the requirements for the cofactor Ran.
Evidence suggests that miRNA expression may be regulated at the level of processing. Some miRNAs in D.melanogaster appear to be processed inefficiently at early stages of embryonic development (Hutvägner et al., 2001). In the sea urchin Strongylocentrotus purpuratus, let-7 transcripts of ∼100 nt are expressed throughout embryonic development, although mature let-7 appears only at a later stage (Pasquinelli et al., 2000). One interesting possibility is that the pri-miRNAs or pre-miRNAs remain in an inert state until a developmental signal triggers their processing. It is also tempting to speculate that some pre-miRNAs at a certain stage may be retained in the nucleus waiting for a signal for nuclear export.
Since little information is currently available on miRNA transcription and processing, our results lay a framework for further studies. Important questions remain as to what are the processing factors and mechanisms. It would also be of great interest to know whether processing is indeed regulated and, if so, what is the mechanism underlying the regulation. The human in vitro processing system developed in this study may provide a powerful tool for analysis of miRNA maturation. Understanding the mechanism of miRNA biogenesis may enable the development of new methods for functional genomics and gene therapy.
Materials and methods
RT–PCR
Total RNA was prepared from subconfluent HeLa cells with TRIzol reagent (Gibco-BRL). RNA (5 µg) was used for the first-strand cDNA synthesis with SUPERSCRIPT II (Gibco-BRL). As controls, RNA was pre-treated with 50 µg/ml RNase A at 37°C for 2 min before the first-strand synthesis, or the reverse transcriptase was omitted during the first-strand synthesis. Primers used for first-strand synthesis were as follows. For miR-30a, 5′-TTCAGCTTTGTAAAAATGTATCAAAGAGAT-3′ was used for first-strand synthesis, and 5′-ATTGCTGTTTGAATGAGGCTTCAGTACTTT-3′ (forward) and 5′-TTCAGCTTTGTAAAAATGTATCAAAGAGAT-3′ (reverse) were used for PCR amplification. For miR-23∼27∼24-2, 5′-GGCAGGGGCTGCAGGCTCCAAGGGGGCTTG-3′ was used for first-strand synthesis, and 5′-CGCCCGGTGCCCCCCTCACCCCTGTGCCAC-3′ (forward) and 5′-CCCTGTTCC TGCTGAACTGAGCCAGTGTAC-3′ (reverse) were used for PCR amplification. For miR-17∼18∼19a∼20∼19b-1, 5′-GGGGTTTGAGTTTCCCTTACTTTTCTACAG-3′ was used for first-strand synthesis, and 5′-TGCTGAATTTGTATGGTTTATAGTTGTTAG-3′ (forward) and 5′-CACTACCACAGTCAGTTTTGCATGGATTTG-3′ (reverse) were used for PCR amplification.
In vitro transcription
In vitro transcription was carried out as described previously (Pellizzoni et al., 1998), with the following modifications. To prepare templates for in vitro transcription, the PCR products from Figure 1 were subcloned into pGEM-T-easy (Promega) and clones with opposite directions were selected so that sense as well as antisense transcripts can be generated. The plasmids were designated as pGEM-T-30(+), pGEM-T-30(–), pGEM-T- 15-16(+), pGEM-T-15-16(–), pGEM-T-23(+) and pGEM-T-23(–). The plasmids were linearized with SpeI. In the transcription reaction, 1 mM GTP was added instead of 0.1 mM and the cap analog was omitted.
Northern blot analysis
Total RNA (20 µg) from HeLa, U937 and HL60 cells was loaded on a 1% formaldehyde–agarose gel. The resolved RNA was transferred to a Zeta-Probe GT blotting membrane (Bio-Rad) overnight. Probe was prepared by the random priming method using a fragment containing a 301 bp region of pri-miR-30a from pGEM-T-30(+) as a template. Pre-hybridization and hybridization were carried out using ExpressHyb Hybridization Solution (Clontech) following the manufacturer’s instruction. The most stringent wash was carried out in 0.1× SSC and 0.1% SDS at 50°C.
In vitro processing system
Preparation of cell extract and in vitro processing were performed with some modifications to our previously reported in vitro splicing method (Kim et al., 2001). Whole-cell extract was prepared from HEK293T cells in buffer D (20 mM HEPES–KOH pH 7.9, 100 mM KCl, 0.2 mM EDTA, 0.5 mM DTT, 0.2 mM PMSF, 5% glycerol) by sonication, followed by centrifugation. Ten microliters of processing reaction contained 5 µl of whole-cell extract, 1 µl of solution A (32 mM MgCl2, 5 mM ATP, 200 mM creatine phosphate), 1 U/µl RNase inhibitor (Takara) and the labeled transcripts of 1 × 104 – 1 × 105 c.p.m. The reaction mixture was incubated at 37°C for 90 min.
Primer extension analysis
The cold miR-30a precursor was synthesized in vitro in the presence of 1 mM each NTP and incubated with HEK293T cell extract for 90 min. Following phenol extraction and ethanol precipitation, the RNA was used for annealing to 5′-end-labeled primer at 37°C for 40 min and subsequently for extension using SUPERSCRIPT II (Gibco-BRL) at 50°C for 30 min. HeLa total RNA (10 µg) was used as a control. Primer sequences were 5′-GCTTCCAGTCGAGGATGT-3′.
Nucleocytoplasmic fractionation for RNA preparation
Subcellular fractionation of HeLa cells was carried out in RSB-100 buffer (10 mM Tris–HCl pH 7.5, 100 mM NaCl, 2.5 mM MgCl2, 35 µg/ml digitonin), as described previously (Siomi et al., 1997). RNA was extracted from each fraction using TRIzol.
Nucleocytoplasmic fractionation for in vitro processing
For in vitro processing with the fractionated extracts, the nuclear and the cytoplasmic fractions prepared in RSB-100 were dialyzed against buffer D before they were used for in vitro processing.
RPA
RNA (50–120 µg) was used for each assay using RNase T1 at a 1:50 dilution from the RPA III kit (Ambion). An equal amount of yeast RNA was used as a control. The probes were designed to hybridize to miRNA as well as the surrounding sequences so that the precursors will give rise to distinctly sized fragments. The probes were synthesized using T7 polymerase (Promega) directly from the PCR products that contain the T7 promoter. Primers for miR-23 were 5′-TAATACGACTCACTATA GGTAAGTTGGCAGCATCCTCGGTGGCAG-3′ (forward) and 5′-AAT TGTATACGTATTATGAAATCACATTGCCAGGGATTTC-3′ (reverse).
Western blot analysis
Anti-hnRNP C monoclonal antibody 4F4, a kind gift from Dr Gideon Dreyfuss, and anti-eIE4E (Sigma) were used for western blotting.
Acknowledgments
Acknowledgements
We are grateful to members of our laboratory and to Drs Kyriacos Mitrophanous, Jin Mo Park, Sara Nakielny and Zissimos Mourelatos for critical reading of this manuscript and helpful discussion, and to Hyunjung Yang for secretarial assistance. We also thank Dr Gideon Dreyfuss for kindly providing 4F4 antibody. This work was supported by the Ministry of Science and Technology (MI-9808-00-0040) and the BK21 Research Fellowship from the Ministry of Education and Human Resources Development of Korea.
References
- Bernstein E., Caudy,A.A., Hammond,S.M. and Hannon,G.J. (2001) Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature, 409, 363–366. [DOI] [PubMed] [Google Scholar]
- Chang D.D. and Sharp,P.A. (1989) Regulation by HIV Rev depends upon recognition of splice sites. Cell, 59, 789–795. [DOI] [PubMed] [Google Scholar]
- Conti E. and Izaurralde,E. (2001) Nucleocytoplasmic transport enters the atomic age. Curr. Opin. Cell Biol., 13, 310–319. [DOI] [PubMed] [Google Scholar]
- Dreyfuss G., Kim,V.N. and Kataoka,N. (2002) Messenger-RNA-binding proteins and the messages they carry. Nat. Rev. Mol. Cell Biol., 3, 195–205. [DOI] [PubMed] [Google Scholar]
- Fornerod M., Ohno,M., Yoshida,M. and Mattaj,I.W. (1997) CRM1 is an export receptor for leucine-rich nuclear export signals. Cell, 90, 1051–1060. [DOI] [PubMed] [Google Scholar]
- Grishok A. et al. (2001) Genes and mechanisms related to RNA interference regulate expression of the small temporal RNAs that control C.elegans developmental timing. Cell, 106, 23–34. [DOI] [PubMed] [Google Scholar]
- Grosshans H. and Slack,F.J. (2002) Micro-RNAs: small is plentiful. J. Cell Biol., 156, 17–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamm J. and Mattaj,I.W. (1990) Monomethylated cap structures facilitate RNA export from the nucleus. Cell, 63, 109–118. [DOI] [PubMed] [Google Scholar]
- Hammond S.M., Bernstein,E., Beach,D. and Hannon,G.J. (2000) An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature, 404, 293–296. [DOI] [PubMed] [Google Scholar]
- Huber J., Cronshagen,U., Kadokura,M., Marshallsay,C., Wada,T., Sekine,M. and Luhrmann,R. (1998) Snurportin1, an m3G-cap-specific nuclear import receptor with a novel domain structure. EMBO J., 17, 4114–4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutvägner G., McLachlan,J., Pasquinelli,A.E., Balint,E., Tuschl,T. and Zamore,P.D. (2001) A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science, 293, 834–838. [DOI] [PubMed] [Google Scholar]
- Izaurralde E., Lewis,J., Gamberi,C., Jarmolowski,A., McGuigan,C. and Mattaj,I.W. (1995) A cap-binding protein complex mediating U snRNA export. Nature, 376, 709–712. [DOI] [PubMed] [Google Scholar]
- Ketting R.F., Fischer,S.E., Bernstein,E., Sijen,T., Hannon,G.J. and Plasterk,R.H. (2001) Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C.elegans. Genes Dev., 15, 2654–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim V.N. and Dreyfuss,G. (2001) Nuclear mRNA binding proteins couple pre-mRNA splicing and post-splicing events. Mol. Cell, 12, 1–10. [PubMed] [Google Scholar]
- Kim V.N., Kataoka,N. and Dreyfuss,G. (2001) Role of the nonsense-mediated decay factor hUpf3 in the splicing-dependent exon–exon junction complex. Science, 293, 1832–1836. [DOI] [PubMed] [Google Scholar]
- Knight S.W. and Bass,B.L. (2001) A role for the RNase III enzyme DCR-1 in RNA interference and germ line development in Caenorhabditis elegans. Science, 293, 2269–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krainer A.R., Maniatis,T., Ruskin,B. and Green,M.R. (1984) Normal and mutant human β-globin pre-mRNAs are faithfully and efficiently spliced in vitro. Cell, 36, 993–1005. [DOI] [PubMed] [Google Scholar]
- Kuersten S., Ohno,M. and Mattaj,I.W. (2001) Nucleocytoplasmic transport: Ran, beta and beyond. Trends Cell Biol., 11, 497–503. [DOI] [PubMed] [Google Scholar]
- Lagos-Quintana M., Rauhut,R., Lendeckel,W. and Tuschl,T. (2001) Identification of novel genes coding for small expressed RNAs. Science, 294, 853–858. [DOI] [PubMed] [Google Scholar]
- Lagos-Quintana M., Rauhut,R., Yalcin,A., Meyer,J., Lendeckel,W. and Tuschl,T. (2002) Identification of tissue-specific microRNAs from mouse. Curr. Biol., 12, 735–739. [DOI] [PubMed] [Google Scholar]
- Lai E.C. (2002) Micro RNAs are complementary to 3′ UTR sequence motifs that mediate negative post-transcriptional regulation. Nat. Genet., 30, 363–364. [DOI] [PubMed] [Google Scholar]
- Lau N.C., Lim,L.P., Weinstein,E.G. and Bartel,D.P. (2001) An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science, 294, 858–862. [DOI] [PubMed] [Google Scholar]
- Lee R.C. and Ambros,V. (2001) An extensive class of small RNAs in Caenorhabditis elegans. Science, 294, 862–864. [DOI] [PubMed] [Google Scholar]
- Lee R.C., Feinbaum,R.L. and Ambros,V. (1993) The C.elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell, 75, 843–854. [DOI] [PubMed] [Google Scholar]
- Legrain P. and Rosbash,M. (1989) Some cis- and trans-acting mutants for splicing target pre-mRNA to the cytoplasm. Cell, 57, 573–583. [DOI] [PubMed] [Google Scholar]
- Luo M.J. and Reed,R. (1999) Splicing is required for rapid and efficient mRNA export in metazoans. Proc. Natl Acad. Sci. USA, 96, 14937–14942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maquat L.E. and Carmichael,G.G. (2001) Quality control of mRNA function. Cell, 104, 173–176. [DOI] [PubMed] [Google Scholar]
- Moss E.G., Lee,R.C. and Ambros,V. (1997) The cold shock domain protein LIN-28 controls developmental timing in C.elegans and is regulated by the lin-4 RNA. Cell, 88, 637–646. [DOI] [PubMed] [Google Scholar]
- Mourelatos Z., Dostie,J., Paushkin,S., Sharma,A., Charroux,B., Abel,L., Rappsilber,J., Mann,M. and Dreyfuss,G. (2002) miRNPs: a novel class of ribonucleoproteins containing numerous microRNAs. Genes Dev., 16, 720–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakielny S. and Dreyfuss,G. (1999) Transport of proteins and RNAs in and out of the nucleus. Cell, 99, 677–690. [DOI] [PubMed] [Google Scholar]
- Olsen P.H. and Ambros,V. (1999) The lin-4 regulatory RNA controls developmental timing in Caenorhabditis elegans by blocking LIN-14 protein synthesis after the initiation of translation. Dev. Biol., 216, 671–680. [DOI] [PubMed] [Google Scholar]
- Palacios I., Hetzer,M., Adam,S.A. and Mattaj,I.W. (1997) Nuclear import of U snRNPs requires importin β. EMBO J., 16, 6783–6792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquinelli A.E. et al. (2000) Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature, 408, 86–89. [DOI] [PubMed] [Google Scholar]
- Pellizzoni L., Kataoka,N., Charroux,B. and Dreyfuss,G. (1998) A novel function for SMN, the spinal muscular atrophy disease gene product, in pre-mRNA splicing. Cell, 95, 615–624. [DOI] [PubMed] [Google Scholar]
- Reed R. and Hurt,E. (2002) A conserved mRNA export machinery coupled to pre-mRNA splicing. Cell, 108, 523–531. [DOI] [PubMed] [Google Scholar]
- Reinhart B.J., Slack,F.J., Basson,M., Pasquinelli,A.E., Bettinger,J.C., Rougvie,A.E., Horvitz,H.R. and Ruvkun,G. (2000) The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature, 403, 901–906. [DOI] [PubMed] [Google Scholar]
- Siomi M.C., Eder,P.S., Kataoka,N., Wan,L., Liu,Q. and Dreyfuss,G. (1997) Transportin-mediated nuclear import of heterogeneous nuclear RNP proteins. J. Cell Biol., 138, 1181–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack F.J., Basson,M., Liu,Z., Ambros,V., Horvitz,H.R. and Ruvkun,G. (2000) The lin-41 RBCC gene acts in the C.elegans heterochronic pathway between the let-7 regulatory RNA and the LIN-29 transcription factor. Mol. Cell, 5, 659–669. [DOI] [PubMed] [Google Scholar]
- Tuschl T., Zamore,P.D., Lehmann,R., Bartel,D.P. and Sharp,P.A. (1999) Targeted mRNA degradation by double-stranded RNA in vitro. Genes Dev., 13, 3191–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner J.R. (2001) Nascent ribosomes. Cell, 107, 133–136. [DOI] [PubMed] [Google Scholar]
- Wightman B., Ha,I. and Ruvkun,G. (1993) Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C.elegans. Cell, 75, 855–862. [DOI] [PubMed] [Google Scholar]
- Will C.L. and Luhrmann,R. (2001) Spliceosomal UsnRNP biogenesis, structure and function. Curr. Opin. Cell Biol., 13, 290–301. [DOI] [PubMed] [Google Scholar]
- Wolin S.L. and Matera,A.G. (1999) The trials and travels of tRNA. Genes Dev., 13, 1–10. [DOI] [PubMed] [Google Scholar]
- Yu Y.T., Shu,M.D. and Steitz,J.A. (1998) Modifications of U2 snRNA are required for snRNP assembly and pre-mRNA splicing. EMBO J., 17, 5783–5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y., Wagner,E.J. and Cullen,B.R. (2002) Both natural and designed micro RNAs can inhibit the expression of cognate mRNAs when expressed in human cells. Mol. Cell, 9, 1327–1333. [DOI] [PubMed] [Google Scholar]