

Abstract
We previously showed that activation of the human endothelin A receptor (HETAR) by endothelin-1 (Et-1) selectively inhibits the response to mu opioid receptor (MOR) activation of the G-protein-gated inwardly rectifying potassium channel (Kir3). The Et-1 effect resulted from PLA2 production of an eicosanoid that inhibited Kir3. In this study, we show that Kir3 inhibition by eicosanoids is channel subunit-specific, and we identify the site within the channel required for arachidonic acid sensitivity. Activation of the G-protein-coupled MOR by the selective opioid agonist D-Ala2Glyol, enkephalin, released Gβγ that activated Kir3. The response to MOR activation was significantly inhibited by Et-1 activation of HETAR in homomeric channels composed of either Kir3.2 or Kir3.4. In contrast, homomeric channels of Kir3.1 were substantially less sensitive. Domain deletion and channel chimera studies suggested that the sites within the channel required for Et-1-induced inhibition were within the region responsible for channel gating. Mutation of a single amino acid in the homomeric Kir3.1 to produce Kir3.1(F137S)(N217D) dramatically increased the channel sensitivity to arachidonic acid and Et-1 treatment. Complementary mutation of the equivalent amino acid in Kir3.4 to produce Kir3.4(S143T)(D223N) significantly reduced the sensitivity of the channel to arachidonic acid- and Et-1-induced inhibition. The critical aspartate residue required for eicosanoid sensitivity is the same residue required for Na+ regulation of PIP2 gating. The results suggest a model of Kir3 gating that incorporates a series of regulatory steps, including Gβγ, PIP2, Na+, and arachidonic acid binding to the channel gating domain.
The G-protein-gated inwardly rectifying potassium channels (Kir3)1 provide essential regulation of neuronal and cardiac excitability (1). By mediating the effects of acetylcholine, monoamine, and peptide receptor activation, Kir3 channels respond to a wide range of transmitters. The Kir3 family of G-protein-gated inwardly rectifying potassium channels consists of subunits (Kir3.1–Kir3.5) that can assemble to form functional heteromultimers. Kir3 channels share a common design characterized by cytoplasmic N and C termini and two transmembrane domains M1 and M2 that surround a potassium selective pore region (P or H5) (2) (see Fig. 1 below). The activation of Kir3 is complex and not completely understood. Kir3 channels require the interaction of multiple components to produce channel activation, including Gβγ, Na+ the phospholipid PIP2, and ATP (3–7). Huang and colleagues proposed that Gβγ activates Kir3 by stabilizing interactions between PIP2 and the potassium channel. They showed that PIP2 depletion blocks activation of Kir3 by both Gβγ and Na+ (8). Both the stimulation and inhibition of Kir3 mediated by Gβγ were recently shown to depend on the type of β subunit in the dimer (9). ATP is another regulatory element in Kir3 gating that presumably acts indirectly by maintaining phosphorylation of PIP2. The response to activation of Kir3.1 and Kir3.4 heteromultimers is rapidly inactivated (run down) when internal ATP is depleted. Sui et al. (10) show that Kir3 activity is ATP-dependent and is mediated by PIP2; moreover, ATP hydrolysis enables both Na+ and Gβγ activation (10). These studies support the hypothesis that Kir3 gating components responsible for channel activation are interdependent.
Fig. 1. Diagram of Kir3.
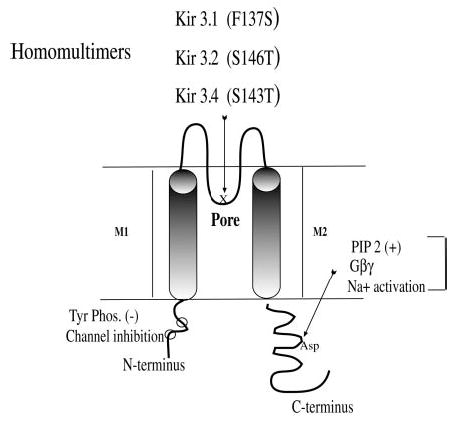
Kir3 channels share a common design characterized by a cytoplasmic N terminus (~90 amino acids) and C terminus with two transmembrane domains M1 and M2, which surround the ion-selective pore region (P or H5). Kir3 channels function as active heteromultimers (e.g. Kir3.1 pairs with other subtypes). Mutations in the P region enhance the activity of homomers. This diagram shows the P region with the site of specific point mutations that produce functional homomeric channels: Kir3.1(F137S), Kir3.2(S146T), and Kir3.4(S143T). The presumed PIP2-Na+ 1-Gβγ interaction site in the C terminus is indicated. Kir3 is also inhibited by tyrosine phosphorylation (11) with phosphorylation sites in the N terminus noted (○). Other sites of Gβγ interaction have been identified, but are not illustrated.
Kir3 normally exists as a heterotetramer, but the gating properties of individual subunits may be studied by using channel mutants able to form functional homomeric channels: Kir3.1(F137S) (11), Kir3.2(S146T) (12), and Kir3.4(S143T) (13). Although members of Kir3 are similar in structure, one primary difference in the subunits is the presence of a Na+ activation site found in Kir3.2 and Kir3.4, which is not found in Kir3.1. Ho and Murrell-Lagnado (14, 15) used chimeras of the Na+-insensitive Kir3.1 and the Na+-sensitive Kir3.2 to define the site on the channel that was sensitive to Na+ activation. Substitution of asparagine for aspartate 226 in Kir3.2(D226N) abolished Na+-dependent activation for both the Kir3.2 homomer and Kir3.1/Kir3.2 heteromultimers without altering the amplitude of receptor activation (14, 15). Ho and Murrell-Lagnado proposed that Na+ binding to an aspartic acid residue in Kir3.2 masks a nearby charged amino acid and permits PIP2 binding.
In contrast to the process of channel activation by PIP2, Na+, and Gβγ, the mechanism of arachidonic acid inhibition of Kir3 is less well defined. Arachidonic acid and its metabolites modulate several ion channels, including Kir3.1 and Kir3.4 heteromultimers in cardiac myocytes (16). Unsaturated free fatty acids such as oleic, linoleic, and arachidonic acids inhibit Kir3.1 and Kir3.4 heteromultimers by blocking ATP-dependent gating in atrial cells (7). We previously showed that arachidonic acid inhibits the potassium channel response to the mu opioid agonist DAMGO in Xenopus oocytes expressing MOR and Kir3 heteromultimers (17). Channel sensitivity to arachidonic acid depended on the channel subtype: heteromultimers consisting of Kir3.1 and Kir3.2 or Kir3.1 and Kir3.4 were more sensitive to arachidonic acid than heteromultimers consisting of Kir3.1 and Kir3.5. These results suggest that eicosanoids have direct effects on G-protein-gated inwardly rectifying potassium channels by modification of the channel conformation, but the molecular basis for eicosanoid inhibition of Kir3 was not defined. In the present study, we explored the hypothesis that eicosanoids generated by HETA activation may directly modulate Kir3 gating. Using site-directed mutagenesis to identify the eicosanoid-sensitive regulatory site on the Kir3 channel, we provide evidence that eicosanoid-induced inhibition of Kir3 requires the Na+-dependent gating site.
EXPERIMENTAL PROCEDURES
Complementary DNA Clones and cRNA Synthesis
The rat mu opioid receptor clone was obtained from Dr. Lei Yu (GenBank™ accession number L13069). cDNA for the human endothelin A (HETA) receptor (GenBank™ accession number S67127) was obtained from Dr. Richard Kris. cDNAs for the Kir 3.1 (GIRK1) (GenBank™ accession number U01071) and Kir 3.2 (GIRK2) (GenBank™ accession number U11859) were obtained from Drs. Cesar Lebarca and Henry Lester. Dr. John Adelman provided the Kir 3.4 (GIRK4) clone (GenBank™ accession number X83584). Kir3 chimeras were the kind gift of Dr. Diomedes Logothetis. Point mutations to produce functional homomeric channels; Kir3.1(F137S) (11), Kir3.2(S146T) (12), and Kir3.4(S143T) (13) were constructed. Mutations were introduced by polymerase chain reaction amplification using Pfu turbo DNA polymerase with complementary oligonucleotide primers incorporating the desired mutation. Positive clones were confirmed by automated sequencing. Plasmid templates for constructs were linearized prior to in vitro cRNA synthesis using mMESSAGE mMACHINE (Ambion Inc., TX).
Oocyte Maintenance and Injection
Healthy stage V and VI oocytes were harvested from mature anesthetized Xenopus laevis (Nasco, Ft. Atkinson, WI) and defolliculated enzymatically as described previously (18). The oocytes were maintained at 18 °C in standard oocyte buffer, ND96 (96 mm NaCl, 2 mm KCl, 1 mm CaCl2, 1 mm MgCl2, 5 mm HEPES, pH 7.5) supplemented with 2.5 mm sodium pyruvate and 50 μg/ml gentamicin (Sigma Chemical Co.). One day after harvest, each oocyte was injected with 50 nl of cRNA for the mu opioid receptor (MOR), human endothelin A receptor (HETA), and G-protein inwardly rectifying potassium channels. Recordings were made at least 48 h after cRNA injection.
Electrophysiological Studies
A Geneclamp 500 amplifier was used for standard two-electrode voltage-clamp experiments. The pCLAMP program (Axon Instruments) was used for data acquisition and analysis. Oocytes were removed from incubation medium, placed in the recording chamber containing ND96 medium, and clamped at −80 mV. Recordings were made in hK buffer (2 mm NaCl, 96 mmKCl, 1 mm CaCl2, 1 mm MgCl2, 5 mm HEPES, pH 7.5). Microelectrodes were filled with 3 M KCl and had resistances of 0.5–1.0 MΩ. Currents were measured without leak subtraction. Individual comparisons of drug effects on Kir3 were conducted using oocytes from the same harvest and injection batch. Pharmacologic agents were perfused or placed directly into the bath from freshly made stock solutions.
Materials
Stock solutions of arachidonic acid were dissolved in dimethyl sulfoxide (Me2SO); the final concentration of Me2SO applied to the oocytes was <0.02%. Arachidonic acid was stored at −70 °C until use. Nitrogen was bubbled through water prior to dissolving endothelin. Endothelin-1 and DAMGO were obtained from Phoenix Pharmaceuticals, Belmont, CA and were stored at −20 °C until use. Arachidonic acid and U73122 were from Calbiochem, La Jolla, CA. Me2SO was from Sigma Chemical Co., St. Louis, MO.
Statistical Analysis
Data are presented as means ± S.E. The statistical significance of differences between results was calculated using ANOVA followed by a student’s unpaired two-tailed t test. A probability of p < 0.05 was considered statistically significant.
RESULTS
Effects of Endothelin-1 on Kir3 Activation by MOR
Heterologous expression of MOR and Kir3 in Xenopus oocytes generated a malleable system for the study of channel activation (Fig. 2). Activation of the Gi/Go-coupled opioid receptors by the agonist DAMGO (1 μm) released Gβγ that activated Kir3 and produced a robust inward current (Fig. 2A). Activation of the heterologously expressed Gq-coupled endothelin receptor produced a cascade of signals that resulted in the reduction of the DAMGO-evoked response.
Fig. 2. Effects of endothelin-1 on the DAMGO elicited mu opioid response mediated by functional heteromultimers and homomers of Kir3 expressed in Xenopus oocytes.
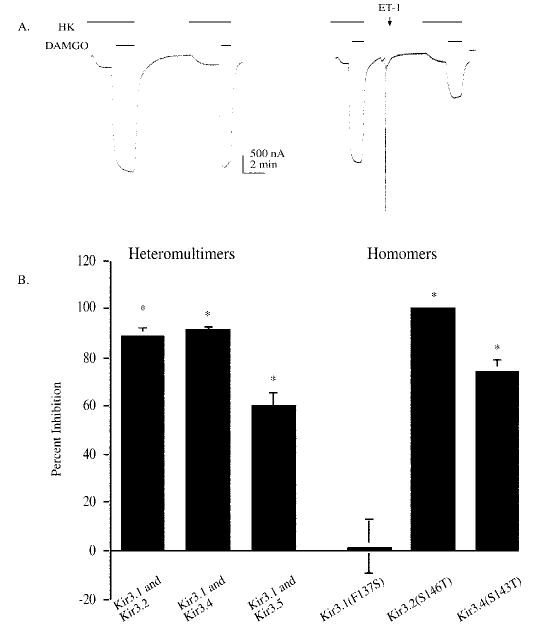
Oocytes were injected with a mixture of the following cRNAs: MOR (1 ng), HETAR (1 ng), and 0.05–1 ng of the Kir subunits listed below the bar. A, representative current traces from oocytes stimulated by perfusion with the mu opioid agonist (1 μm) DAMGO in oocyte buffer containing 96 mm KCl in two applications. Oocytes receiving a second application of 1 μm DAMGO showed a similar response to DAMGO; the amplitude of the second opioid response was 99 ± 2.9% (n = 34) of the first response. The left panel shows the response without Et-1. The right panel includes a pulse of Et-1 (50 nm) in ND96 applied directly to the oocyte in the recording chamber. When endothelin was applied after the first DAMGO application, there was a robust inhibition of the second DAMGO-evoked response (right trace). Application of vehicle had no effect, and the responses to endothelin were absent in oocytes not expressing the receptor. For these traces, oocytes were clamped at −80 mV in normal saline buffer. Horizontal bars indicate the duration of drug perfusion. The opioid response was only observed in oocytes injected with both receptor and channel cRNA. The endothelin-induced activation of the endogenous chloride current (24, 33) was only observed in oocytes injected with the endothelin A receptor cRNA. Horizontal bars indicate the duration of drug perfusion. The opioid response was only observed in oocytes injected with both receptor and channel cRNA. The endothelin-induced activation of the endogenous chloride current (24, 33) was only observed in oocytes injected with the endothelin A receptor cRNA. B, the percent inhibition of the second opioid receptor response is compared with the first response. Bars represent mean ± S.E. of 18–20 oocytes from three batches. Recordings were performed 2–3 days post-injection. Oocytes used were injected with MOR, HETAR, and the Kir3 cRNA listed on the x axis.
To identify the Kir3 subunit conferring sensitivity to endothelin, we used the strategy developed by Logothetis and colleagues to generate functional homomeric Kir3 channels. Mutation in the pore of these channels, Kir3.1(F137S) and Kir3.4(S143T) greatly increased expression and the current evoked by the channel homomers. In agreement with Vivaudou and colleagues (13), we noted that the wild type channel Kir3.4 does not produce functional currents when expressed alone in Xenopus oocytes. In contrast, the homomer Kir3.4(S143T) (1 ng of cRNA/oocyte) produced a robust current with an average receptor-activated current of 1–2 μA in response to agonist. Although the wild type channel Kir3.1 (1 ng) forms functional channels with the intrinsic Xenopus subunit Kir3.5, the current response produced was small (50–100 nA) compared with the large currents (1–2 μA) produced by the functional homomer Kir3.1(F137S) (1 ng). These results are consistent with the conclusions of Vivaudou et al. (13) that the Kir3.1 and Kir3.4 currents are produced by the functional homomeric channel without combining with endogenous Kir3.5. Similarly, Kir3.2(S146T) (1 ng) produced larger currents (500 nA to 1 μA) compared with wild type Kir3.2 (100 nA) (1 ng), which normally forms a homomeric channel. Moreover, the point mutation of Kir3.2(S146T) served as a control as this mutation in the pore was similar to Kir3.1(F137S) and Kir3.4(S143T). Thus, oocytes expressing presumptive homomeric forms of Kir3.1, 3.2, and 3.4 were functional and were robustly activated by Gβγ released by mu opioid receptor activation.
Oocytes expressing either Kir3.2(S146T) or Kir3.4(S143T) treated with Et-1 prior to the second DAMGO challenge showed a marked inhibition of the second opioid response (Fig. 2B). The amplitude of the second opioid response in Kir3.2(S146T) homomers after Et-1 treatment was inhibited by 100 ± 0% (n = 9). The amplitude of the second opioid response in Kir3.4(S143T) homomers after Et-1 treatment was inhibited by 74 ± 5% (n = 18) (p < 0.01) (Fig. 2B). In contrast, the second opioid response in oocytes injected with Kir3.1(F137S) and treated with Et-1 prior to the second DAMGO challenge was not significantly inhibited. The amplitude of the second opioid response in Kir3.1(F137S) homomers after Et-1 treatment was 1 ± 11% (n = 16). The difference in endothelin sensitivity of the Kir3.1–3.5 heteromeric channel and the Kir3.1(F137S) channel supports the conclusion that the latter formed a functional homomer under these expression conditions. In addition, the channel type selectivity evident from this experiment further supports the conclusion that the endothelin-induced suppression of the DAMGO-activated response was caused by a direct modification of the channel.
We previously showed that the PLA2 inhibitor AACOCF3 selectively blocked the endothelin receptor-mediated effect (17). These results suggested that an eicosanoid such as arachidonic acid produced by PLA2 activation following HETA receptor activation was responsible for channel inhibition. Another interpretation of our data was that the endothelin effect was caused by PLC-mediated PIP2 depletion. To test the latter hypothesis, oocytes were pretreated with the PLC inhibitor U73122 (5 μm) (19) for 10 min and tested for endothelin sensitivity under conditions previously shown to inhibit PLC activity in oocytes (20). The amplitude of the second opioid response in Kir3.4(S143T) homomers after Et-1 treatment was inhibited by 61 ± 5% (n = 8). Pretreatment with the PLC inhibitor U73122 (5 μm) did not block the inhibition of the second opioid response to Et-1. The amplitude of the second opioid response of Kir3.4(S143T) homomers after Et-1 treatment was inhibited by 66 ± 5% (n = 8) (not significantly different than in the absence of U73122). Thus, PLA2 activation and subsequent eicosanoid production likely caused the endothelin-induced inhibition. Furthermore, the results suggest that Kir3.2 and Kir3.4 were sensitive to the eicosanoid generated by PLA2 activation whereas Kir3.1 was not.
Endothelin Effects on Kir3 Channel Truncations and Chimeras
To determine the basis for the insensitivity of Kir3.1(F137S) to endothelin receptor activation, the Et-1-insensitive Kir3.1 and Et-1-sensitive Kir3.4 sequences were aligned and compared. These Kir3 subunits share the most homology in the pore region and the greatest heterogeneity in the tail regions. Macica and colleagues (21, 22) identified a serine residue in the distal N terminus in the inward rectifier ROMK1 that confers sensitivity to arachidonic acid. We wanted to test the hypothesis that an N-terminal residue was responsible for eicosanoid sensitivity of Kir3. Sequential truncations of the N terminus were made, and the effect of Et-1 on Kir3 activation was tested. Oocytes expressing a truncated Kir3.4(S143T) lacking amino acids 1–57 produced strong potassium currents and a robust response following DAMGO activation of MOR. The amplitude of the second opioid response in the channel with the N-terminal truncation Kir3.4(S143T)(Δ 1–57) after Et-1 treatment was inhibited, a result not significantly different than the parent (p >0.05) (Fig. 3B). Truncation of the first 23 amino acids in Kir3.2 also did not block either the DAMGO activation or endothelin sensitivity (data not shown).
Fig. 3. Endothelin effects on channel mutants.
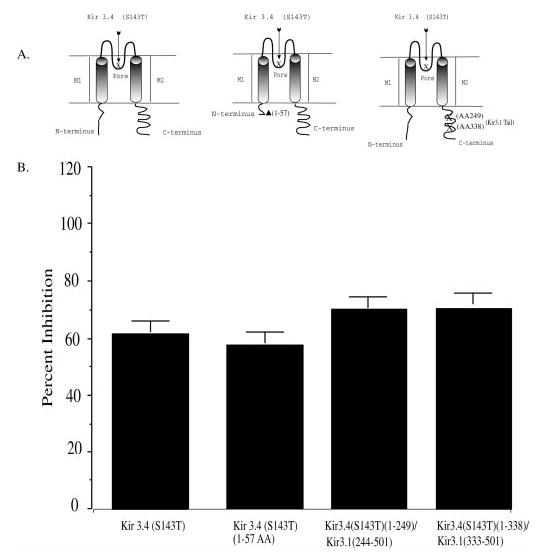
A, diagrams depict channel mutants: Kir3.4 (S143T) parent, N-terminal truncation, and chimeras. We produced the Kir3.4(S143T) using a cDNA template for cRNA coding a Kir3.4(S143T) having a truncated (1–57) N terminus. We used Kir3 chimeras: Kir3.4(S143T)-(1–338)/Kir3.1-(333–501) and Kir3.4(S143T)-(1–249)/Kir3.1-(244–501). B, oocytes were injected with 1 ng of MOR and 1 ng of HETAR, and either 1 ng of Kir3.4(S143T), 1 ng of Kir3.4(S143T)-(Δ1–57), 1 ng of Kir3.4(S143T)-(1–338)/Kir3.1-(333–501), or 1 ng of Kir3.4(S143T)-(1–249)/Kir3.1-(244–501). The percent inhibition of the second mu opioid response after Et-1 pretreatment is shown. The bar graph summarizes the effects of Et-1 on the DAMGO activation of MOR compared with untreated controls from the same batch. Data are means ± S.E. from four to seven oocytes and two to three independent experiments.
In contrast, the C terminus of Kir3.1 and Kir3.4 differed significantly in both amino acid length and amino acid sequence. Chimeras composed of the Et-1-sensitive Kir3.4(S143T) N terminus and pore region with the Et-1-insensitive Kir3.1 C-terminal tail, Kir3.4(S143T)-(1–338)/Kir3.1-(333–501) and Kir3.4(S143T)-(1–249)/Kir3.1-(244–501), were used to define the region of endothelin sensitivity. The response to DAMGO of oocytes expressing the channel chimera Kir3.4(S143T)-(1–338)/Kir3.1-(333–501) was inhibited following Et-1 treatment. The sensitivity of the chimera was not significantly different from the parent channel Kir3.4(S143T) (p >0.05) (Fig. 3B). Similarly, the DAMGO response of oocytes expressing the chimera Kir3.4(S143T)-(1–249)/Kir3.1-(244–501) was inhibited following Et-1 treatment. The sensitivity of this chimera was also not significantly different from Kir3.4(S143T) (p >0.05) (Fig. 3B). Thus, neither the N terminus nor the distal C terminus of Kir3 contained elements required for the Et-1-induced inhibition. The chimera and truncation data suggest that the channel domain responsible for endothelin sensitivity was within the proximal C-terminal region near the transmembrane domain M2.
The proximal C-terminal domain of the channel contains sites responsible for Gβγ, PIP2, and Na+ gating (5, 9, 14, 15). After comparing the different Kir3 channels, we noted a correlation between endothelin sensitivity and Na+ gating sensitivity. Kir3.1 was substantially less sensitive to endothelin, whereas Kir3.2 and Kir3.4 were very sensitive (Fig. 2B). Moreover, Kir3.1 is not gated by Na+, whereas Kir3.2 and Kir3.4 are gated by Na+ (5, 14, 15). Based on this correlation, we explored the hypothesis that the Kir3 gating domain was important in the observed eicosanoid inhibition of Kir3. Na+ sensitivity was reported to depend on the presence of a critical aspartate residue in the gating domain (5, 14, 15). Kir3.1(F137S) and Kir3.4(S143T)(D223N) lack the aspartate thought to be responsible for Na+ gating, and the corresponding channels Kir3.4(S143T) and Kir3.1(F137S)(N217D) contain this residue. These channel variants were produced and expressed in oocytes (Fig. 4).
Fig. 4. The effect of the sodium binding site on the endothelin-1 inhibition of Kir3 homomers.
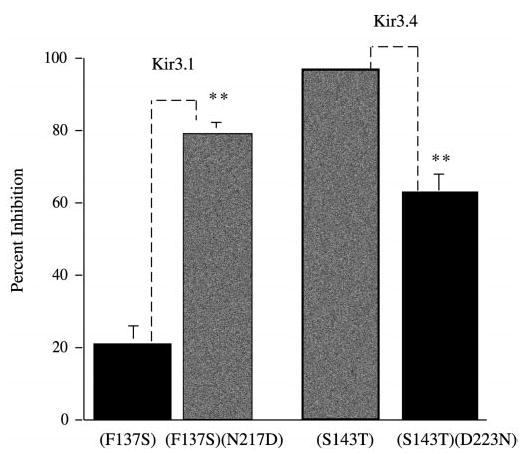
Oocytes were injected with a mixture of the following cRNAs: MOR (1 ng), HETAR (1 ng), and the Kir form listed below the bar (1 ng each). Recordings were performed 2–3 days post-injection. The experiment was performed as in Fig. 2. The bar graph compares Kir3.1(F137S) versus Kir3.1(F137S)(N217D) with addition of a site for Na+-dependent gating. The second set of bars are Kir3.4(S143T) compared with Kir3.4(143T)(D223N); a mutant with a deletion of the Na+ activation site. Data are means ± S.E. from four to seven oocytes and two to three independent experiments (**, p < 0.01).
The Effect of the Sodium Binding Site on the Endothelin-1 Inhibition of Kir3
We coexpressed either Na+-sensitive or Na+-insensitive channel mutants of Kir3.1 or Kir3.4 with cRNA for the MOR and cRNA for the human endothelin A receptor. The amplitude of the second opioid response in Kir3.1(F137S) (Na+-insensitive) homomers after Et-1 treatment was inhibited by 22 ± 5% (n = 18) (Fig. 4). The slightly greater sensitivity to Et-1 shown by Kir3.1(F137S) in this set of recordings compared with the data presented in Fig. 2 was not statistically different from the data shown in Fig. 1. The slight difference is attributed to normal seasonal variation in oocytes, and the increased sensitivity to Et-1 was also shown by Kir3.4(S143T). Introduction of the aspartate residue that confers Na+ sensitivity also dramatically increased endothelin sensitivity. Oocytes injected with Kir3.1(F137S)(N217D) (Na+-sensitive) and then treated with Et-1 prior to the second DAMGO challenge showed a marked inhibition of the second opioid response. The second opioid response after Et-1 treatment was inhibited by 82 ± 5% (n = 16) (p < 0.01).
For Kir3.4, removal of the critical aspartate significantly reduced endothelin sensitivity (Fig. 4). The second mu opioid response after Et-1 activation in oocytes expressing the HETA receptor, the MOR and Kir3.4(S143T) (Na+-sensitive) was inhibited by 97 ± 1% (n = 7). Oocytes expressing Kir3.4(S143T)(D223N) (Na+-insensitive) treated with Et-1 prior to the second DAMGO challenge showed significantly less inhibition of the second opioid response; endothelin reduced the response to DAMGO by 63 ± 6% (n = 12). Kir3.4(S143T)(D223N) remains partially sensitive to endothelin treatment, but the sensitivity was significantly reduced compared with Kir3.4(S143T). These data indicate that a significant component of the endothelin sensitivity of Kir3 depended on the presence of the aspartate residue also responsible for Na+ gating. The residual sensitivity suggests that endothelin receptor activation may also inhibit Kir3 at other sites.
Arachidonic Acid Effects on Channel Homomers
The results suggested that endothelin activation of PLA2 produced an eicosanoid that inhibited Kir3 channels by interacting at the Na+ gating domain. Prior work showed that arachidonic acid was the most potent of the eicosanoids at inhibiting Kir3 in cardiac myocytes (7). Based on this finding, we tested the effectiveness of arachidonic acid application on the Na+-sensitive and -insensitive Kir3 variants. In oocytes expressing the channel homomer Kir3.1(F137S), arachidonic acid (20 μm) did not inhibit the second DAMGO-elicited response of MOR (0 ± 3%). In oocytes expressing the channel with a site for Na+ activation, Kir3.1(F137S)(N217D), arachidonic acid produced an inhibition of the second opioid response; the second DAMGO response was inhibited by (26 ± 1%) (p < 0.05) (Fig. 5). Moreover, arachidonic acid inhibited the Na+-sensitive Kir3.4(S143T) (18 ± 2%). compared with oocytes with the analogous Na+-site removed Kir3.4(143T)(D223N) (−2 ± 60%). These data support the hypothesis that eicosanoid inhibition of Kir3 was dependent on the presence of the Na+ gating site.
Fig. 5. The effects of arachidonic acid Na+ activation site mutants in homomers Kir 3.1(F137S) and Kir3.4(S143T).
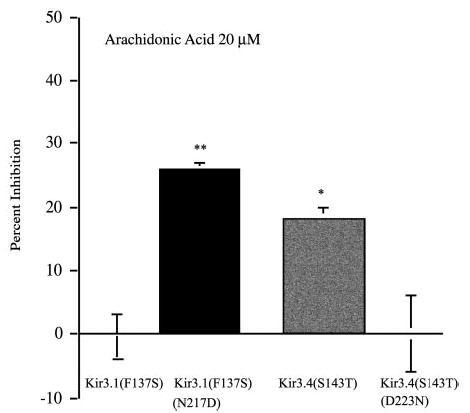
Oocytes were injected with a mixture of the following cRNAs: 1 ng of MOR and either 1 ng of Kir 3.1(F137S), 1 ng of Kir 3.1(F137S)(N217D), 1 ng of Kir3.4(S143T), or 1 ng of Kir3.4(S143T)(D223N). Recordings were performed 2–3 days post-injection. Oocytes expressing MOR and the channel variants were challenged with DAMGO then treated with 20 μm arachidonic acid placed directly into the bath for 6 min prior to activation of the second opioid response. The bar graph compares arachidonic acid effects on Kir3.1(F137S) versus Kir3.1(F137S)(N217D) with addition of a site for Na+-dependent gating. The second set of bars are Kir3.4(S143T) compared with Kir3.4(143T)(D223N); a mutant with a deletion of the Na+ activation site (**, p < 0.01; *, p < 0.05).
DISCUSSION
The principal finding of this study is that arachidonic acid inhibited G-protein-gated potassium channels at the Na+/PIP2 gating domain. The identification of this mechanism helps clarify the effects of eicosanoids generated by phospholipase A2 activation, and the results provide new insights into the gating process controlling Kir3 function. Previous studies established that the elevation of Gβγ concentration following Gi/o-coupled receptor activation increases Kir3 conductance (23). Gβγ was shown to work in concert with PIP2 to induce a conformational change in the channel and open the K+-selective pore. In addition to Gβγ, Kir3.2, and Kir3.4 channel subunits are activated by Na+ (14, 15). The results provide support for the hypothesis that arachidonic acid is an additional component, directly regulating Kir3 gating.
The results obtained provide evidence that the production of an eicosanoid by endothelin receptor activation was responsible for the observed inhibition of Kir3 in this expression system. The HETAR is a Gq-coupled receptor that activates both PLC and PLA2 (24). As PLA2 metabolizes PIP2 to arachidonic acid (Fig. 6), depletion of PIP2 could potentially result from endothelin receptor activation. Either mechanism could result in Kir3 channel inhibition, because the production of an eicosanoid (7) or the depletion of PIP2 (20) could reduce channel conductance. Meyer et al. (25) recently showed that endothelin receptor activation inhibits IK(ACh) in atrial myocytes, and this effect could be blocked by increasing the intracellular concentration of PIP2. They interpret their data to suggest that endothelin receptor activation of PLC results in PIP2 depletion and IK(ACh) inhibition; however the role of an eicosanoid in the endothelin effect was not considered and competition between activation by PIP2 and inhibition by arachidonic acid could explain their result (Fig. 6).
Fig. 6. Proposed mechanism of lipid modulation of Kir3.
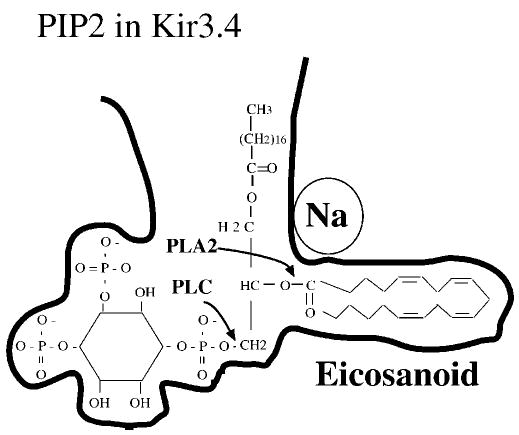
The figure shows a proposed binding site for PIP2 in the proximal domain of the C terminus of Kir3.4. The site for Na+ activation may shield adjacent charged residues, which is permissive for the PIP2/arachidonic acid complex to bind the channel. The diagram also shows the sites of PLA2 and PLC cleavage of PIP2.
Several lines of evidence exclude the explanation that depletion of PIP2 was responsible for the endothelin effect in oocytes. We found that PLC inhibition by U73122 did not block endothelin action. Our prior study showed that the PLA2 inhibitor AACOCF3 blocked endothelin effects (17). The third observation is that the Kir3 subunit sensitivity to arachidonic acid matches endothelin sensitivity. Thus, our results support the conclusion that endothelin activation of PLA2 produces an eicosanoid that inhibits Kir3 rather than depletion of PIP2 required for channel activation. The specific eicosanoid generated by endothelin receptor activation in Xenopus oocytes was not identified; however, arachidonic acid is a reasonable candidate based on its high potency shown by Kim and Pleumsamran (7). In addition, arachidonic acid exhibited an inhibition profile that was similar to that endothelin receptor activation (Fig. 5).
We note that Kir3.4(S143T)(D223N) was not significantly affected by arachidonic acid application whereas Kir3.4(S143T)(D223N) had residual sensitivity to endothelin inhibition. The difference could either result from the possibility that endothelin acted by additional mechanisms or that the activation of the HETAR was a more efficient means to deliver the active eicosanoid than extracellular application of arachidonic acid. Consistent with the latter interpretation, the data showed that the inhibition produced by endothelin was more robust than that produced by arachidonic acid. These results suggest that the critical aspartate residue was not the sole determinant of endothelin effect. Additional site-directed mutagenesis would be required to define other residues in the binding pocket required for eicosanoid sensitivity.
The basis for the inhibitory effects of eicosanoids on Kir3 conductance was not defined, but the study provides clues. The correlation between Na+ sensitivity and arachidonic acid sensitivity suggests that the binding site of eicosanoids is part of the domain controlled by the Na+ binding site. Moreover, the Na+ binding site also regulates PIP2 binding. Because arachidonic acid shares structural features with PIP2, the PIP2 binding region of Kir3 may also bind arachidonic acid. The inhibition of gating caused by arachidonic acid may result from a competition with PIP2 for binding at this site. In the absence of structural data, we suggest the physical basis for the interaction between these gating components and the ion channel in a diagram (Fig. 6). Lei et al. (9) used mammalian cells transfected with Kir3 heterotetramers to show that Gβ5-containing dimers could inhibit Kir3. They suggested that Gβ5-containing dimers could competitively displace Gβγ binding in the proximal C terminus, a region near the putative eicosanoid binding site suggested in by the present study. Additional work will be required to further define the interaction between eicosanoids, Gβγ, and PIP2 at Kir3; however, our data show that the mechanism of Kir3 sensitivity to arachidonic acid differs from that reported for ROMK channels by Macica et al. (21, 22).
The large Kir3.1(F137S) currents clearly showed that the channel was formed as a homomer consistent with prior reports (11, 13). The DAMGO-evoked currents produced in oocytes expressing the Kir3.1(F137S)(N217D) mutant were smaller (387 ± 142 nA) than those produced by Kir3.1(F137S) (1170 ± 98 nA). We interpret the increased sensitivity of Kir3.1(F137S)(N217D) to endothelin and arachidonic acid as resulting from the introduction of the critical aspartate residue required for eicosanoid binding.
Although Kir 3.1 may form channels with endogenous Kir3.5, we did not exclude the less likely interpretation that the Kir3.1(F137S)(N217D) channel formed endothelin-sensitive heteromers with Kir3.5. We used a sequence alignment program to compare the Na+-insensitive Kir3.1 with the Xenopus homologue, Kir3.5. When Kir3.5 is aligned with Kir3.1, we note that Kir3.5 contains the critical aspartate that is required for Na+ activation of the channel (BCM Search Launcher, Baylor College of Medicine). Thus, we would expect that any heteromultimers composed of Kir3.1(F137S)(N217D) and Kir3.5 would be arachidonic acid-sensitive. This explains the sensitivity of Kir3.1 and Kir3.5 heteromultimers (Fig. 2). Future single channel recordings would resolve this question.
We explored the mechanism of endothelin receptor-induced inhibition of mu opioid receptor activation of Kir3 in vitro. Although Xenopus oocytes are a complex expression system with interacting signaling molecules, insights to Kir3 gating mechanisms are evident from these results. The endothelin receptor is one member of the Gq-coupled seven-transmembrane superfamily of receptors (24). Other Gq receptors that regulate Kir3 by eicosanoid production may also require specific gating components to exert effects on the channel. For example, activation of the Gq-coupled substance P receptors expressed in locus coeruleus neurons inhibits Kir3 currents (26, 27). Although the mechanism of inhibition was not established in that study, the results shown here may be relevant. Eicosanoid regulation of Kir3 as described in this study may be relevant to the pathological situations that occur during inflammation. Because Kir3 has in important role in cardiac excitability, cardiovascular disease may elicit an inflammatory response to produce eicosanoids that inhibit Kir3 (28–32). Nevertheless, the physiological significance of these findings needs to be directly established.
Acknowledgments
We thank Tracy Sherertz for assistance and Drs. Todd Scheuer and Tooraj Mirshahi for helpful discussion. We thank Dr. Lei Yu for the rat mu opioid receptor clone, Dr. Richard Kris for the human endothelin A receptor, Drs. Cesar Lebarca and Henry Lester for the Kir 3.1 and Kir 3.2 clones, Dr. John Adelman for the Kir 3.4 clone, and Dr. Diomedes Logothetis for the two Kir3 chimeras: Kir3.4(S143T) (1–338)/Kir3.1-(333–501) and Kir3.4(S143T)-(1–249)/Kir3.1-(244–501).
Footnotes
This study was supported by United States Public Health Services Grant DA 04123 from the National Institute on Drug Abuse.
The abbreviations used are: Kir3, G-protein-gated inwardly rectifying potassium channel; PIP2, phosphatidylinositol 4,5-bisphosphate; HETAR, human endothelin A receptor; MOR, mu opioid receptor; DAMGO, D-Ala2Glyol, enkephalin; Et-1, endothelin-1; Gβ2, β- 2 subunit of the G-protein; PLC, phospholipase C.
References
- 1.Jan LY, Jan YN. Curr Opin Cell Biol. 1997;9:155–160. doi: 10.1016/s0955-0674(97)80057-9. [DOI] [PubMed] [Google Scholar]
- 2.Dascal N. Cell Signal. 1997;9:551–573. doi: 10.1016/s0898-6568(97)00095-8. [DOI] [PubMed] [Google Scholar]
- 3.Krapivinsky G, Kennedy ME, Nemec J, Medina I, Krapivinsky L, Clapham DE. J Biol Chem. 1998;273:16946–16952. doi: 10.1074/jbc.273.27.16946. [DOI] [PubMed] [Google Scholar]
- 4.Sui JL, Chan KW, Logothetis DE. J Gen Physiol. 1996;108:381–391. doi: 10.1085/jgp.108.5.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang H, He C, Yan X, Mirshahi T, Logothetis DE. Nat Cell Biol. 1999;1:183–188. doi: 10.1038/11103. [DOI] [PubMed] [Google Scholar]
- 6.Logothetis DE, Zhang H. J Physiol (Lond) 1999;520:630. doi: 10.1111/j.1469-7793.1999.00630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim D, Pleumsamran A. J Gen Physiol. 2000;115:287–304. doi: 10.1085/jgp.115.3.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang CL, Feng SY, Hilgemann DW. Nature. 1998;391:803–806. doi: 10.1038/35882. [DOI] [PubMed] [Google Scholar]
- 9.Lei Q, Jones MB, Talley EM, Schrier AD, McIntire WE, Garrison JC, Bayliss DA. Proc Natl Acad Sci U S A. 2000;97:9771–9776. doi: 10.1073/pnas.97.17.9771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sui JL, Petit-Jacques J, Logothetis DE. Proc Natl Acad Sci U S A. 1998;95:1307–1312. doi: 10.1073/pnas.95.3.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chan KW, Sui JL, Vivaudou M, Logothetis DE. Proc Natl Acad Sci U S A. 1996;93:14193–14198. doi: 10.1073/pnas.93.24.14193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rogalski SL, Appleyard SM, Pattillo A, Terman GW, Chavkin C. J Biol Chem. 2000;275:25082–25088. doi: 10.1074/jbc.M000183200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vivaudou M, Chan KW, Sui JL, Jan LY, Reuveny E, Logothetis DE. J Biol Chem. 1997;272:31553–31560. doi: 10.1074/jbc.272.50.31553. [DOI] [PubMed] [Google Scholar]
- 14.Ho IH, Murrell-Lagnado RD. J Physiol (Lond) 1999;520:645–651. doi: 10.1111/j.1469-7793.1999.00645.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ho IH, Murrell-Lagnado RD. J Biol Chem. 1999;274:8639–8648. doi: 10.1074/jbc.274.13.8639. [DOI] [PubMed] [Google Scholar]
- 16.Kurachi Y, Ito H, Sugimoto T, Shimizu T, Miki I, Ui M. Nature. 1989;337:555–557. doi: 10.1038/337555a0. [DOI] [PubMed] [Google Scholar]
- 17.Rogalski SL, Cyr C, Chavkin C. J Neurochem. 1999;72:1409–1416. doi: 10.1046/j.1471-4159.1999.721409.x. [DOI] [PubMed] [Google Scholar]
- 18.Leonard, J. P., and Snutch, T. P. (1991) Molecular Neurobiology: A Practical Approach (Chad, J., and Wheal, H., eds) pp. 161–182, Oxford University Press, New York
- 19.Vickers JD. J Pharmacol Exp Ther. 1993;266:1156–1163. [PubMed] [Google Scholar]
- 20.Kobrinsky E, Mirshahi T, Zhang H, Jin T, Logothetis DE. Nat Cell Biol. 2000;2:507–514. doi: 10.1038/35019544. [DOI] [PubMed] [Google Scholar]
- 21.Macica CM, Yang Y, Hebert SC, Wang WH. Am J Physiol. 1996;271:F588–594. doi: 10.1152/ajprenal.1996.271.3.F588. [DOI] [PubMed] [Google Scholar]
- 22.Macica CM, Yang Y, Lerea K, Hebert SC, Wang W. Am J Physiol. 1998;274:F175–F181. doi: 10.1152/ajprenal.1998.274.1.F175. [DOI] [PubMed] [Google Scholar]
- 23.Henry DJ, Grandy DK, Lester HA, Davidson N, Chavkin C. Mol Pharmacol. 1995;47:551–557. [PubMed] [Google Scholar]
- 24.Rubanyi GM, Polokoff MA. Pharmacol Rev. 1994;46:325–415. [PubMed] [Google Scholar]
- 25.Meyer T, Wellner-Kienitz M, Biewald A, Bender K, Eickel A, Pott L. J Biol Chem. 2001;276:5650–5658. doi: 10.1074/jbc.M009179200. [DOI] [PubMed] [Google Scholar]
- 26.Koyano K, Grigg JJ, Velimirovic BM, Nakajima S, Nakajima Y. Neurosci Res. 1994;20:345–354. doi: 10.1016/0168-0102(94)90057-4. [DOI] [PubMed] [Google Scholar]
- 27.Velimirovic BM, Koyano K, Nakajima S, Nakajima Y. Proc Natl Acad Sci U S A. 1995;92:1590–1594. doi: 10.1073/pnas.92.5.1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Henrich CJ, Simpson PC. J Mol Cell Cardiol. 1988;20:1081–1085. doi: 10.1016/0022-2828(88)90588-3. [DOI] [PubMed] [Google Scholar]
- 29.Sugden PH, Clerk A. Adv Enzyme Regul. 1998;38:87–98. doi: 10.1016/s0065-2571(97)00010-1. [DOI] [PubMed] [Google Scholar]
- 30.Sugden PH, Clerk A. Cell Signal. 1997;9:337–351. doi: 10.1016/s0898-6568(96)00191-x. [DOI] [PubMed] [Google Scholar]
- 31.Yamazaki T, Komuro I, Shiojima I, Yazaki Y. Ann N Y Acad Sci. 1999;874:38–48. doi: 10.1111/j.1749-6632.1999.tb09223.x. [DOI] [PubMed] [Google Scholar]
- 32.Van der Vusse GJ, Reneman RS, van Bilsen M. Prostaglandins Leukot Essent Fatty Acids. 1997;57:85–93. doi: 10.1016/s0952-3278(97)90497-x. [DOI] [PubMed] [Google Scholar]
- 33.Barish ME. J Physiol (Lond) 1983;342:309–325. doi: 10.1113/jphysiol.1983.sp014852. [DOI] [PMC free article] [PubMed] [Google Scholar]