

Abstract
We tested the hypothesis that biotinylation of K12 in histone H4 plays a role in the cellular response to double-strand breaks (DSB) of DNA in human cells. DSB were caused by treating choriocarcinoma JAr cells with etoposide. Biotinylation of K12 in histone H4 decreased by 50% as early as 10 – 20 min after initiation of treatment with etoposide. Biotinylation returned to initial levels 30 – 40 min after addition of etoposide to the medium. Temporal patterns of K12-biotinylation were similar for human lymphoma cells. Phosphorylation of S14 of histone H2B and poly(ADP-ribosylation) of glutamate residues on histone H2A are known markers of DSB in DNA; these modifications increased 10 – 40 min after alterations in K12-biotinylation occurred. Decreased biotinylation of K12 of histone H4 was specific for DSB but was not detectable in response to single-strand breaks or formation of thymine dimers. Biotin-deficient choriocarcinoma cells exhibited a 40% decrease in rates of survival in response to etoposide compared with biotin-sufficient controls. These studies suggest that the lack of biotinylation of K12 in histone H4 is an early signaling event in response to DSB.
Keywords: Biotin, DNA damage, double strand breaks, histone H4
In eukaryotic cell nuclei, DNA associates with histones H2A, H2B, H3, and H4 to form nucleosomal core particles. Histones may undergo various post-translational modifications in their N-termini. These modifications include acetylation (1–3), methylation (4), phosphorylation (4), ubiquitination (4), and poly(ADP-ribosylation) (5–7) of ɛ-amino groups, guanidino groups, carboxyl groups, and hydroxyl groups. Distinct patterns of modifications of individual histones, nucleosomal core particles, and chromosomal domains play unique roles in chromatin structure, cell signaling, and genomic stability (8, 9). Histone modifications may affect each other in synergistic or antagonistic ways. For example, acetylation of K23 mediates methylation of R17 in histone H3 (10).
There is increasing evidence that histones are also covalently modified by the vitamin biotin (11–13), mediated by holocarboxylase synthetase (14) and biotinidase (11). The following biotinylation sites have been identified in human histones: K9 and K13 in histone H2A (15); K4, K9, and K18 in histone H3 (16); and K8 and K12 in histone H4 (13). Biotinylation of histones plays a role in processes such as cell proliferation (12, 14), gene silencing (17), and the cellular response to UV – induced DNA damage (17).
Various cellular events and environmental insults may cause DNA damage. For example UV light may trigger formation of thymine dimers, whereas the antineoplastic agent etoposide inhibits topoisomerase II, mediating double strand breaks (DSB) of DNA. Damaged DNA causes chromosomal abnormalities, potentially leading to malignant transformations. The risk for propagation of pre-malignant cells is reduced by activation of the following pathways in response to DNA damage: (i) activation of DNA repair mechanisms, which mediate removal of DNA lesions (18); and (ii) activation of apoptotic cascades, which mediate elimination of pre-malignant cells if DNA damage is beyond repair (19). Histone modifications play a crucial role in these processes. For example, DNA damage causes poly(ADP-ribosylation) of histones, recruiting DNA repair proteins to sites of DNA damage (20). These proteins participate in base excision repair (21) and nucleotide repair pathways (22).
Here, we tested the hypothesis that biotinylation of histone H4 plays a role in the cellular response to DSB of DNA. Specifically we determined (i) whether etoposide-induced DSB are associated with alterations of K12 biotinylation in histone H4 in cells originally derived from various human tissues; (ii) whether putative changes of K12 biotinylation in histone H4 are specific for DSB or whether similar changes are observed in response to formation of thymine dimers and single strand breaks; and (iii) whether biotin concentrations in culture media affect cell survival in response to DSB in DNA.
MATERIALS AND METHODS
Cell culture
Human choriocarcinoma (JAr) cells and lymphoid (Jurkat) cells (ATCC, Manassas, VA) were cultured in biotin-supplemented medium (2 μmol/L) as described (23, 24). JAr cells were harvested at 60–80% confluence for subsequent analyses. Culture media were supplemented with sodium butyrate (5 mmol/L) for 24 h to inhibit histone deacetylases if samples were analyzed for acetylated histones. For survival studies, biotin-deficient JAr cells were obtained by culturing cells in biotin-deficient medium (0.025 nmol/L) for 3 weeks as described (23); cells cultured in medium containing a physiological concentration of biotin (0.25 nmol/L) were used as control.
DNA damage
Cells were treated with 60 nmol/L etoposide for up to 2 h in order to cause DSB in DNA (25). Specificity controls were treated as follows: 20 nmol/L doxorubicin for up to 2 h in order to cause DSB in DNA (26); 0.15 mmol/L zeocin in order to cause single-stranded DNA breaks (27); and UV light (500 mJ/m2) for 30 sec (28) in order to cause formation of thymine dimers. At timed intervals cells were collected for analyses as described below.
Etoposide is an inhibitor of topoisomerase II, causing DSB at the replication fork and G2 phase arrest (25). Theoretically, etoposide will not cause DSB until the replication fork is formed in the S phase of the cell cycle. Hence, etoposide-resistant controls were generated by treating cells with hydroxyurea (2 mmol/L) for 30 h to cause G1 arrest (29) prior to treatment with 60 nmol/L etoposide. G1 arrest was confirmed by staining with propidium iodide and flow cytometry (Cell Analysis Facility, University of Nebraska Medical Center) as described (30).
In some experiments, we determined whether biotin deficiency decreases the survival of etoposide-treated JAr cells. In these experiments, biotin-deficient cells and biotin-sufficient controls were treated with 6 nmol/L etoposide, and cell viability was quantified at timed intervals using trypan blue exclusion (31).
Histone analysis
Nuclear histones were extracted from JAr and Jurkat cells by using 1 mol/L HCl (12). For analysis of phosphorylated histones, 1 mmol/L sodium vanadate was added to the HCl used for extraction. Equal amounts of histones (as judged by gel densitometry after staining with Coomassie blue) were resolved by using 18% Tris glycine gels (Invitrogen, Carlsbad, CA). Proteins were electroblotted onto polyvinylidene fluoride membranes; histone modifications were probed using the following primary antibodies: rabbit anti-human biotinyl-K12 histone H4 (13), rabbit anti-human acetyl-K12 histone H4, rabbit anti-human dimethyl-K4 histone H3, rabbit anti-human phospho-S14 histone H2B, and mouse anti-human ubiquitinyl-histone H2A (Upstate, Waltham, MA). Antibody binding was visualized using appropriate horseradish peroxidase-labeled secondary antibodies and chemiluminescence; gel densitometry was used for quantification (32). If bands were too faint for detection by gel densitometry, we used half the detection limit for data analysis.
Poly(ADP-ribosylation) of histones is mediated by poly(ADP-ribose) polymerase using NAD as a substrate (20). For analysis of poly(ADP-ribosylation) of histones, JAr cell nuclei were isolated as described (33). Nuclei were were incubated with [32P]NAD for 5 min, followed by incubation with 60 nmol/L etoposide for up to 2 h (33) The reaction was quenched using 10 mmol/L nicotinamide (33). Histones were isolated using acid extraction (12), and poly(ADP)ribosylated histones were quantified by autoradiography and gel densitometry.
Analysis of DNA breaks
Cells were grown on microscope cover slips for treatment with etoposide as described above. Positive controls were treated with 1 μmol/L DNase for 10 min at room temperature. DNA breaks were visualized by TDT-mediated dUTP nick end labeling (TUNEL) using a commercial kit (ApoAlert – DNA Fragmentation Assay; BD Biosciences, Palo Alto, CA). DNA strand breaks appear in green color due to the incorporation of Fluorescein-labeled nucleotides at nicked ends. Images were obtained using an Olympus FV500 confocal microscope (Microscopy Core Facility, University of Nebraska – Lincoln).
Immunocytochemistry
Intracellular histone H4 was visualized by using standard procedures of immunocytochemistry (34). Anti-human biotinyl-K12 histone H4 antibody (13) and Cy™2-conjugated donkey anti-rabbit IgG (Jackson ImmunoResearch, West Grove, PA) were used for staining. The cytoplasmic compartment was stained using rhodamine phalloidin (Molecular Probes, Eugene, OR), and nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). S14-phosphorylated histone H2B is a known marker for DNA damage (34) and was visualized using rabbit anti-human phospho-S14 H2B and Cy™2-conjugated donkey anti-rabbit IgG (positive control). Images were obtained by using an Olympus FV500 confocal microscope (Microscopy Core Facility, University of Nebraska – Lincoln).
Biotin-dependent carboxylase
Biotin-dependent carboxylases are good markers for cellular biotin (35). Biotinylated carboxylases in cell extracts were resolved by polyacrylamide gel electrophoresis and probed using streptavidin peroxidase (24)
Statistics
Homogeneity of variances among groups was tested using Bartlett’s test (36). If variances were heterogeneous data were log-transformed before further statistical analysis. Significance of differences among groups was tested by one-way ANOVA. Fisher’s Protected Least Significant Difference procedure was used for posthoc testing (36). Effects of treatment over time (survival experiment) were analyzed by repeated measures ANOVA, followed of pairwise comparison of time points (e.g., biotin-deficient versus biotin-sufficient cells at time 6 h) by paired t-test (36). StatView 5.0.1 (SAS Institute; Cary, NC) was used to perform all calculations. Differences were considered significant if P < 0.05.
RESULTS
Biotinylation of K12 in histone H4 decreased rapidly but transiently in response to treatment with etoposide in JAr cells. Specifically, biotinylation of K12 in histone H4 decreased by about 50% as early as 10 – 20 min after initiation of treatment with etoposide (Figure 1 and online Supplemental Table 1). Biotinylation of K12 returned to pretreatment levels within 30 – 40 min after addition of etoposide to culture media.
Fig. 1.
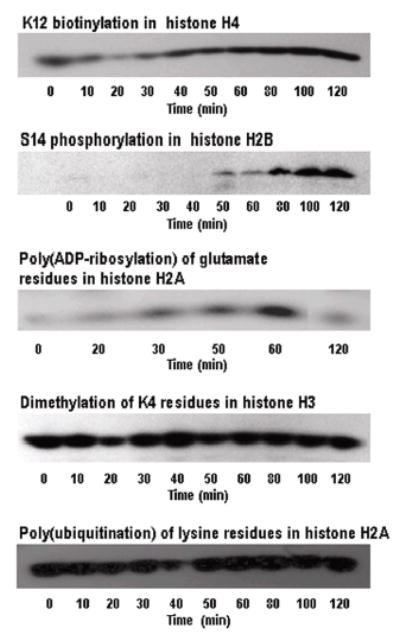
Biotinylation of K12 in histone H4 decreases rapidly and transiently in response to treatment with etoposide in JAr cells. Biotinylation of K12 in histone H4, phosphorylation of S14 in histone H2B, dimethylation of K4 in histone H3, and poly-ubiquitination of histone H2A were visualized by Western blot analysis at timed intervals after addition of etoposide to culture medium. Poly(ADP-ribosylation) of histone H2A was visualized using autoradiography after etoposide addition. Representative gels from at least three repeats are depicted.
ONLINE SUPPLEMENTAL TABLE 1.
The abundance of modified histone in response to treatment with etoposide in JAr cells. Histones were quantified by Western blot analysis and gel densitometry at timed intervals after addition of etoposide to culture medium.1
| Time after etoposide (min)
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Histone2 | 0 | 10 | 20 | 30 | 40 | 50 | 60 | 80 | 100 | 120 |
| % of zero-time control | ||||||||||
| K12bio H4 | 100 | 45 ± 8.6* | 53 ± 10* | 91 ± 22 | 96 ± 20 | 120 ± 20 | 99 ± 18 | 139 ± 21 | 100 ± 8.3 | 100 ± 17 |
| S14p H2B | 100 | 112 ± 46 | 73 ± 13 | 126 ± 59 | 321 ± 154 | 511 ± 299 | 580 ± 389 | 880 ± 335* | 1250 ±122* | 1474 ± 314* |
| K4Me2 H3 | 100 | 101 ± 7.4 | 98 ± 15 | 98 ± 18 | 106 ± 25 | 103 ± 14 | 104 ± 15 | 103 ± 15 | 87 ± 3.2 | 97 ± 11 |
| Ub H2A | 100 | 101 ± 7.5 | 98 ± 15 | 100 ± 11 | 93 ± 13 | 96 ± 10 | 98 ± 9.3 | 88 ± 5.9 | 100 ± 2.5 | 114 ± 14 |
Values are means ± SEM; n = 3 – 5.
Different from zero-time control, P < 0.05.
Abbreviations used: K12bio H4, K12-biotinylated histone H4; S14p H2B, S14-phosphorylated histone H2B; K4Me2 H3, K4-dimethylated histone H3; Ub H2A, poly(ubiquitinated) histone H2A.
Biotinylation of K12 in histone H4 appears to be an early signaling event in response to DNA damage. This notion is based on a comparison of the time course of K12-biotinylation with the time courses of other known markers of DNA damage (7, 34). First, phosphorylation of S14 in histone H2B increased from barely detectable levels at zero time to detectable levels about 50 min after addition of etoposide to culture media (Figure 1 and online Supplemental Table 1). Second, poly(ADP-ribosylation) of histone H2A increased from barely detectable levels at zero time to maximal levels 60 min after addition of etoposide to culture media. Hence, these two markers of DNA damage were detectable 10 – 40 min after alterations in K12 biotinylation occurred.
DSB of DNA caused specific time-dependent modifications of histones as opposed to global changes in histone modifications. For example, treatment of JAr cells with etoposide did not affect the abundance of K4-dimethylation of histone H3 and poly(ubiquitination) of histone H2A (Figure 1 and online Supplemental Table 1), strongly suggesting that changes in biotinylation of K12 on histone H4 was an early indicator of DSB in DNA in response to etoposide treatment. The occurrence of DSB of DNA was confirmed by TUNEL assay. DNA breaks were detectable about 60 min after addition of etoposide to culture media, but some damage probably occurred earlier (Figure 2).
Fig. 2.
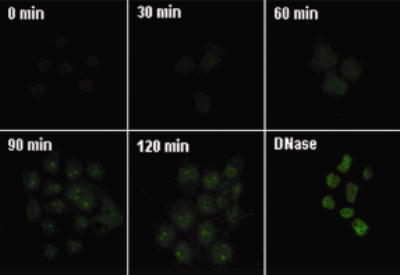
Treatment with etoposide causes DNA breaks in JAr cells. Cells were treated with etoposide and DNA breaks were visualized at timed intervals using TUNEL assay. Positive controls were treated with DNase I for 10 min.
Biotinylation of K12 in histone H4 did not change in response to etoposide treatment in G1 phase-arrested JAr cells (Figure 3). Here, we compared temporal patterns of K12-biotinylation in G1 phase-arrested JAr cells and a mixed population of asynchronous JAr cells. G1 arrest by hydroxyurea treatment was confirmed using flow cytometry (Fig. 3, top row). Note that etoposide causes DSB only during S phase of the cell cycle (25). Consistent with this notion, treatment with etoposide did not affect biotinylation of K12 in histone H4 in G1-arrested cells, but caused a substantial decrease in K12-biotinylated histone H4 in asynchronous cells (Fig. 3, bottom row).
Fig. 3.
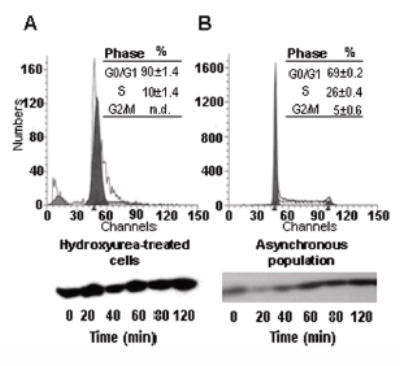
Etoposide does not affect biotinylation of K12 in histone H4 from G1 phase-arrested JAr cells. Cells were arrested in G1 phase using hydroxyurea before treatment with etoposide. Panel A: G1-arrested cells; panel B: asynchronous cells (control). Cell cycle arrest was confirmed using flow cytometry (top row), and biotinylation of K12 in histone H4 was visualized by Western analysis at timed intervals after etoposide treatment (bottom row); n.d. = not detectable.
Immunocytochemistry experiments were consistent with the hypothesis that decreased biotinylation of K12 in histone H4 is an early signaling event in response to DSB. First, we confirmed the localization of K12-biotinylated histone H4 in untreated JAr cells (Figure 4A). Pre-immune serum did not produce a detectable signal for K12-biotinylated histone H4 (data not shown). Second, we monitored the temporal patterns of K12-biotinylated histone H4 and S14-phosphorylated histone H2B (control) in response to etoposide. Decreased biotinylation of K12 was detectable as early as 20 min after addition of etoposide to culture media (Figure 4B). The abundance of S14-phosphorylated histone H2B increased about 60 min after addition of etoposide to culture media.
Fig. 4.
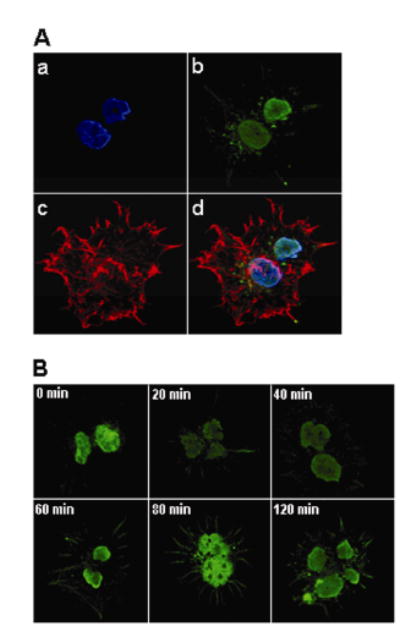
Immunocytochemistry experiments are consistent with a rapid and transient decrease in K12-biotinylated histone H4 in JAr cell nuclei in response to treatment with etoposide. Panel A: Staining of biotinylation of K12 in histone H4 and cellular compartments in etoposide-free JAr cells: a = staining of nuclei using DAPI; b = staining of K12-biotinylation of histone H4, using a site-specific antibody; c = staining of cytoplasmic compartment using rhodamine phalloidin; d = merged image. Panel B: Time course of K12-biotinylation in histone H4 after addition of etoposide to culture media.
Decreased K12-biotinylation of histone H4 in response to DSB is a ubiquitous phenomenon as opposed to being a tissue-specific event. For example, if lymphoid (Jurkat) cells were treated with etoposide, we observed a decrease of K12-biotinylation similar to the decrease observed in JAr cells. K12-biotinylation of histone H4 in Jurkat cells reached a low about 40 min after addition of etoposide to the culture medium (Figure 5A).
Fig. 5.
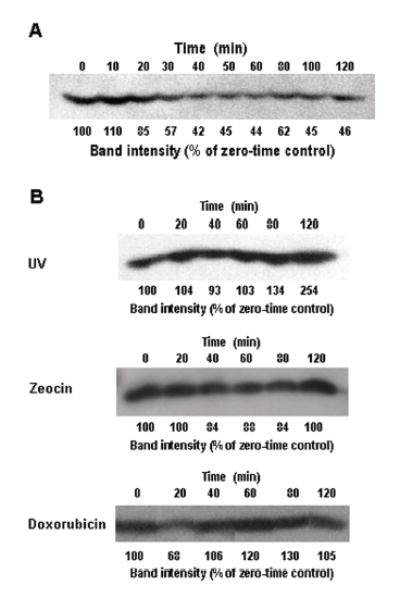
Alterations of K12-biotinylation in histone H4 are specific for DSB, but are not specific for JAr cells. (A) Jurkat cells were treated with etoposide, and K12-biotinylation in histone H4 was monitored at timed intervals using Western blot analysis; band intensities were quantified by gel densitometry. (B) JAr cells were treated with UV, zeocin and doxorubicin to cause formation of thymine dimers, single-strand breaks, and DSB, respectively. Biotinylation of K12 in histone H4 was monitored at timed intervals by using Western blot analysis; band intensities were quantified by gel densitometry.
Decreased biotinylation of K12 in histone H4 is specific for DSB as opposed to being a global marker for various kinds of DNA damage. First, when formation of thymine dimers in DNA was caused by UV treatment of JAr cells, biotinylation of K12 in histone H4 increased for at least up to 120 min (Figure 5B). Second, if single strand breaks of DNA were caused by treatment of JAr cells with zeocin, K12-biotinylation in histone H4 did not change substantially (Figure 5B). In contrast, if DSB of DNA were caused by treatment of JAr cells with doxorubicin, the temporal pattern of K12-biotinylation in histone H4 was similar to the pattern seen after treatment with etoposide (Figure 5B). These observations are consistent with the notion that alterations of K12-biotinylation are specific for the kind of DNA damage, but not for the agent used to cause the damage.
Biotin deficiency was associated with decreased cell survival in response to treatment with etoposide. Here, cells were cultured in biotin-sufficient or biotin-deficient media for 21 days. First, we confirmed that biotin concentrations in culture media affected intracellular biotin concentrations. The abundance of biotinylated pyruvate carboxylase, propionyl-CoA carboxylase (α-chain), and 3-methylcrotonyl-CoA carboxylase (α-chain) was substantially greater in cells from biotin-sufficient medium compared to cells from biotin-deficient medium (Figure 6A). Note that propionyl-CoA carboxylase and 3-methylcrotonyl-CoA carboxylase were not resolved by the electrophoresis procedure used and migrated as a single band. Acetyl-CoA carboxylase was not detectable in JAr cells, consistent with previous studies in this cell line (23). Next, biotin-defined cells were exposed to 6 nmol/L etoposide, and cell survival was monitored at timed intervals. Cells cultured in biotin-sufficient medium had greater survival rates than cells cultured in biotin-deficient medium (Figure 6B). For example, 6 h after addition of etoposide to culture media, 64 ± 14% of biotin-sufficient cells were still alive whereas only 38 ± 11% of biotin-deficient cells were still alive. These effects cannot be explained by effects of biotin deficiency on proliferation rates, which are known to be moderate (23, 24, 37).
Fig. 6.
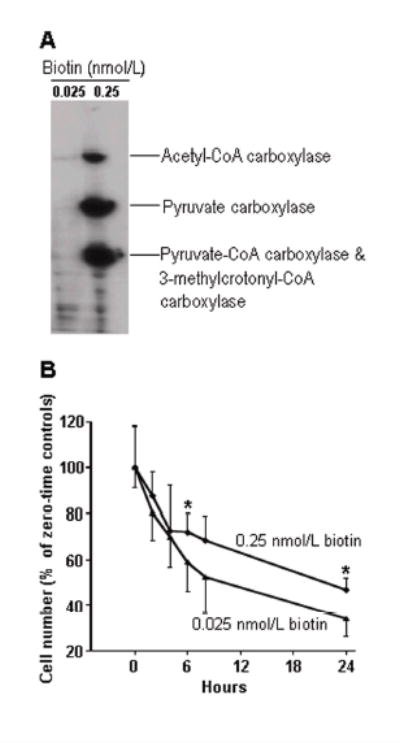
Biotin deficiency decreases survival of JAr cells in response to treatment with etoposide. (A) Extracts from etoposide-free cells grown in media containing 0.025 nmol/L and 0.25 nmol/L biotin were resolved by gel electrophoresis; transblots were probed with streptavidin to visualize biotin-dependent carboxylases. (B) JAr cell were treated with etoposide and cell survival was monitored at timed intervals using trypan blue exclusion. Values are means ± SD (* P<0.05 versus cells cultured in medium containing 0.025 nmol/L biotin; n = 10)
DISCUSSION
Cells are constantly exposed to environmental insults that may cause DSB of DNA. Cells respond to DSB by activating repair pathways that depend on alterations of chromatin structure (38). Histone modifications play a key role in mediating these chromatin remodeling events. Previous studies have identified some of the histone modifications that occur in response to DSB in mammalian cells. For e.g., phosphorylation of S14 in histone H2B, phosphorylation of S139 in histone H2AX, and poly(ADP-ribosylation) of glutamate residues in histone H2A (20, 34, 39). The present study expands the list of damage-dependent histone modifications by unveiling a temporal pattern of K12-biotinylation in histone H4 in response to DSB in human cells.
This is the first time that a correlation has been shown between the biotinylation of a distinct lysine residue in a human histone and a physiological event that requires chromatin remodeling. In previous studies, streptavidin peroxidase has been used as a probe for biotin to provide circumstantial evidence for biological functions of histone biotinylation in cell proliferation and UV-induced DNA damage (12, 17). Note that binds to biotin in general and does not permit pinpointing distinct biotinylation sites in histones, e.g., K8 and K12 in histone H4 (13). In contrast, the site-specific antibody to K12-biotinylated histone H4 as a probe used in this study indicates the physiological relevance of biotinylation of K12 in histone H4 in DNA repair signaling.
Specifically, this study provides evidence (i) that a transient decrease of K12-biotinylation in histone H4 is an early signaling event in response to DSB; (ii) that signaling by biotinylation of K12 is a universal rather than a tissue-specific mechanism in humans; (iii) that biotinylation of K12 in histone H4 decreases specifically in response to DSB but does not decrease in response to single-strand breaks or formation of thymine dimers; and (iv) that biotin deficiency is associated with decreased cell survival in response to DSB.
The decreased biotinylation of K12 in histone H4 in response to DSB occurs before phosphorylation of S14 in histone H2B and potentially ahead of or in concert with poly(ADP-ribosylation) of glutamate residues in histone H2A. Currently it is unknown whether phosphorylation of S14 and poly(ADP-ribosylation) of glutamate residues directly depend on biotinylation of K12 in histone H4. This uncertainty is being addressed in ongoing investigations in our laboratory. Moreover, it is uncertain as to whether biotinylation of K12 itself depends on other damage-induced modifications of histones such as phosphorylation of H2AX (40). Note that histone H2AX also is a potential target for biotinylation (15). Theoretically, biotinylation of K12 in histone H4 might mediate either DNA repair or apoptosis in response to DSB. We favor the former explanation, given that biotin deficiency was associated with decreased survival of etoposide-treated cells compared with biotin-sufficient controls in the present study. In this context the following question needs to be addressed. Why would biotin deficiency decrease cell survival in response to DSB, although the damage-related signaling event actually causes a decrease in K12-biotinylation? We speculate that a sufficient supply of biotin is essential to allow for an efficient re-biotinylation of K12 following its rapid transient debiotinylation. We further speculate that biotinylation of K12 plays a role in the reassembly of nucleosomes following DNA repair (41).
Current models of chromatin remodeling in response to DSB are consistent with the findings reported here. Specifically, it has been proposed that DSB are associated with a rapid, yet transient, relaxation of chromatin structures mediated by histone modifications (42). Relaxation of chromatin gives DNA repair factors access to sites of DNA breaks. Subsequently, chromatin re-condensation prevents dissociation of the two broken ends of DNA before repair is mediated by ligases. Note that K12-biotinylated histone H4 is known to be associated with chromatin condensation such as in pericentromeric heterochromatin (A.M. Oommen and J. Zempleni, submitted for publication). Hence, a rapid decrease in K12-biotinylation in response to DSB is likely associated with an opening of chromatin structures, giving repair proteins access to sites of damage. Subsequent re-biotinylation of histone H4 is likely associated with re-condensation of chromatin. Note, however, that this is an untested hypothesis.
Is the response to DSB breaks comparable in transformed human cell lines and in primary human cells? Previous studies suggested that the frequency of DNA breaks in response to X-rays is similar in transformed and primary cells (43). Also, the kinetics of joining DNA ends is similar in transformed and primary cells. These observations are consistent with the notion that transformed human cells maintain a normal response to DSB breaks. Nevertheless, it remains to be formally demonstrated that pathways of DSB repair are identical in the choriocarcinoma cells used in this study and in primary placental cells.
Taken together, the findings presented here are important for health professionals, based on the following lines of observation. Dietary biotin deficiency is relatively rare in humans, but certain subgroups in the general population are at increased risk for developing biotin deficiency. First, catabolism of biotin is increased during pregnancy (44); consistent with this observation is the fairly high prevalence of marginal biotin deficiency in pregnant women (45, 46). Second, some drugs are known to interfere with biotin metabolism. For example, certain anti-convulsants and lipoic acid impair biotin transport and distribution, potentially causing biotin deficiency (35). Third, biotin transporter deficiency may cause intracellular biotin depletion (47). Fourth, mutations of the two enzymes biotinidase and holocarboxylase synthetase that mediate biotinylation of histones are relatively prevalent in humans. For example, the prevalence of biotinidase deficiency is 1 in 60,000 live births in humans (48). Currently it is unknown whether dietary biotin deficiency or drug-induced biotin deficiency, and mutations of genes coding for biotin transporters, biotinidase, and holocarboxylase synthetase are associated with an impaired ability to repair damaged DNA in humans. Likewise, it is unknown whether supplementation with pharmacological dose of biotin enhances an individual’s DNA repair capabilities.
Footnotes
This work was supported by NIH grants DK 60447, DK 063945, and 1 P20 RR16469, and by NSF EPSCoR grant EPS-0346476. This paper is a contribution of University of Nebraska-Agricultural Research Division, Lincoln, NE 68583 (Journal Series No. 14530).
Supplemental Table 1 is available as Online Supporting Material with the online posting of this paper at www.nutrition.org.
This is an un-copyedited author manuscript that has been accepted for publication in the Journal of Nutrition, © American Society for Nutrition. This may not be duplicated or reproduced, other than for personal use or within the rule of “Fair Use of Copyrighted Materials” (section 107, Title 17, U.S. Code) without permission of the copyright owner. The final copy of the edited article, which is the version of record, can be found at http://www.nutrition.org. The American Society for Nutrition disclaims any responsibility or liability for errors of omissions in this version of the manuscript or in any version derived from it by the National Institutes of Health or other parties.
Abbreviations: DAPI, 4′,6-diamidino-2-phenylindole; DSB, double-strand breaks; TUNEL, TDT-mediated dUTP nick end labeling
References
- 1.Ausio J, van Holde KE. Histone hyperacetylation: its effect on nucleosome conformation and stability. Biochemistry. 1986;25:1421–8. doi: 10.1021/bi00354a035. [DOI] [PubMed] [Google Scholar]
- 2.Hebbes TR, Thorne AW, Crane-Robinson C. A direct link between core histone acetylation and transcriptionally active chromatin. EMBO J. 1988;7:1395–402. doi: 10.1002/j.1460-2075.1988.tb02956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee DY, Hayes JJ, Pruss D, Wolffe AP. A positive role for histone acetylation in transcription factor access to nucleosomal DNA. Cell. 1993;72:73–84. doi: 10.1016/0092-8674(93)90051-q. [DOI] [PubMed] [Google Scholar]
- 4.Wolffe A. Chromatin. 3th ed. San Diego, CA: Academic Press; 1998.
- 5.Chambon P, Weill JD, Doly J, Strosser MT, Mandel P. On the formation of a novel adenylic compound by enzymatic extracts of liver nuclei. Biochem Biophys Res Commun. 1966;25:638–43. [Google Scholar]
- 6.Boulikas T. At least 60 ADP-ribosylated variant histones are present in nuclei from dimethylsulfate-treated and untreated cells. EMBO J. 1988;7:57–67. doi: 10.1002/j.1460-2075.1988.tb02783.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boulikas T. DNA strand breaks alter histone ADP-ribosylation. Proc Natl Acad Sci USA. 1989;86:3499–503. doi: 10.1073/pnas.86.10.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–5. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 9.El-Osta A. Coordination of epigenetic events. Cell Mol Life Sci. 2004;61(17):2135–6. doi: 10.1007/s00018-004-4173-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Daujat S, Bauer UM, Shah V, Turner B, Berger S, Kouzarides T. Crosstalk between CARM1 methylation and CBP acetylation on histone H3. Curr Biol. 2002;12(24):2090–7. doi: 10.1016/s0960-9822(02)01387-8. [DOI] [PubMed] [Google Scholar]
- 11.Hymes J, Fleischhauer K, Wolf B. Biotinylation of histones by human serum biotinidase: assessment of biotinyl-transferase activity in sera from normal individuals and children with biotinidase deficiency. Biochem Mol Med. 1995;56:76–83. doi: 10.1006/bmme.1995.1059. [DOI] [PubMed] [Google Scholar]
- 12.Stanley JS, Griffin JB, Zempleni J. Biotinylation of histones in human cells: effects of cell proliferation. Eur J Biochem. 2001;268:5424–9. doi: 10.1046/j.0014-2956.2001.02481.x. [DOI] [PubMed] [Google Scholar]
- 13.Camporeale G, Shubert EE, Sarath G, Cerny R, Zempleni J. K8 and K12 are biotinylated in human histone H4. Eur J Biochem. 2004;271:2257–63. doi: 10.1111/j.1432-1033.2004.04167.x. [DOI] [PubMed] [Google Scholar]
- 14.Narang MA, Dumas R, Ayer LM, Gravel RA. Reduced histone biotinylation in multiple carboxylase deficiency patients: a nuclear role for holocarboxylase synthetase. Hum Mol Genet. 2004;13:15–23. doi: 10.1093/hmg/ddh006. [DOI] [PubMed] [Google Scholar]
- 15.Chew YC, Camporeale G, Kothapalli N, Sarath G, Zempleni J. Lysine residues in N-and C-terminal regions of human histone H2A are targets for biotinylation by biotinidase. J Nutr Biochem. 2005 (in press). [DOI] [PMC free article] [PubMed]
- 16.Kobza K, Camporeale G, Rueckert B, Kueh A, Griffin JB, Sarath G, Zempleni J. K4, K9, and K18 in human histone H3 are targets for biotinylation by biotinidase. FEBS J (in press). [DOI] [PMC free article] [PubMed]
- 17.Peters DM, Griffin JB, Stanley JS, Beck MM, Zempleni J. Exposure to UV light causes increased biotinylation of histones in Jurkat cells. Am J Physiol Cell Physiol. 2002;283:C878–C84. doi: 10.1152/ajpcell.00107.2002. [DOI] [PubMed] [Google Scholar]
- 18.Rouse J, Jackson SP. Interfaces between the detection, signaling, and repair of DNA damage. Science. 2002;297(5581):547–51. doi: 10.1126/science.1074740. [DOI] [PubMed] [Google Scholar]
- 19.Rich T, Allen RL, Wyllie AH. Defying death after DNA damage. Nature. 2000;407:777–83. doi: 10.1038/35037717. [DOI] [PubMed] [Google Scholar]
- 20.D’Amours D, Desnoyers S, D’Silva I, Poirier GG. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J. 1999 Sep 1;342(Pt 2):249–68. [PMC free article] [PubMed] [Google Scholar]
- 21.Dantzer F, Nasheuer HP, Vonesch JL, de Murcia G, Menissier-de Murcia J. Functional association of poly(ADP-ribose) polymerase with DNA polymerase alpha-primase complex: a link between DNA strand break detection and DNA replication. Nucl Acids Res. 1998;26(8):1891–8. doi: 10.1093/nar/26.8.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flohr C, Burkle A, Radicella JP, Epe B. Poly(ADP-ribosyl)ation accelerates DNA repair in a pathway dependent on Cockayne syndrome B protein. Nucl Acids Res. 2003;31(18):5332–7. doi: 10.1093/nar/gkg715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crisp SERH, Camporeale G, White BR, Toombs CF, Griffin JB, Said HM, Zempleni J. Biotin supply affects rates of cell proliferation, biotinylation of carboxylases and histones, and expression of the gene encoding the sodium-dependent multivitamin transporter in JAr choriocarcinoma cells. Eur J Nutr. 2004;43:23–31. doi: 10.1007/s00394-004-0435-9. [DOI] [PubMed] [Google Scholar]
- 24.Manthey KC, Griffin JB, Zempleni J. Biotin supply affects expression of biotin transporters, biotinylation of carboxylases, and metabolism of interleukin-2 in Jurkat cells. J Nutr. 2002;132:887–92. doi: 10.1093/jn/132.5.887. [DOI] [PubMed] [Google Scholar]
- 25.Bozko P, Larsen AK, Raymond E, Skladanowski A. Influence of G2 arrest on the cytotoxicity of DNA topoisomerase inhibitors toward human carcinoma cells with different p53 status. Acta Biochim Pol. 2002;49(1):109–19. [PubMed] [Google Scholar]
- 26.Sargent J, Williamson C, Yardley C, Taylor C, Hellmann K. Dexrazoxane significantly impairs the induction of doxorubicin resistance in the human leukaemia line, K562. Br J Cancer. 2001;84:959–64. doi: 10.1054/bjoc.2001.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Makiniemi M, Hillukkala T, Tuusa J, Reini K, Vaara M, Huang D, Pospiech H, Majuri I, Westerling T, et al. BRCT domain-containing protein TopBP1 functions in DNA replication and damage response. J Biol Chem. 2001;276:30399–406. doi: 10.1074/jbc.M102245200. [DOI] [PubMed] [Google Scholar]
- 28.Jacobson EL, Antol KM, Juarez-Salinas H, Jacobson MK. Poly(ADP-ribose) metabolism in ultraviolet irradiated human fibroblasts. J Biol Chem. 1983 Jan 10;258(1):103–7. [PubMed] [Google Scholar]
- 29.Borel F, Lacroix FB, Margolis RL. Prolonged arrest of mammalian cells at the G1/S boundary results in permanent S phase stasis. J Cell Sci. 2002;115:2829–38. doi: 10.1242/jcs.115.14.2829. [DOI] [PubMed] [Google Scholar]
- 30.Vindelov LL. Flow microfluorometric analysis of DNA in cells from solid tumors and cell suspensions: A new method for rapid isolation and staining of nuclei. Virchows Arch B Cell Path. 1977;24:227–42. [PubMed] [Google Scholar]
- 31.Zempleni J, Mock DM. Uptake and metabolism of biotin by human peripheral blood mononuclear cells. Am J Physiol Cell Physiol. 1998;275:C382–C8. doi: 10.1152/ajpcell.1998.275.2.C382. [DOI] [PubMed] [Google Scholar]
- 32.Wiedmann S, Eudy JD, Zempleni J. Biotin supplementation causes increased expression of genes encoding interferon-γ, interleukin-1β, and 3-methylcrotonyl-CoA carboxylase, and causes decreased expression of the gene encoding interleukin-4 in human peripheral blood mononuclear cells. J Nutr. 2003;133:716–9. doi: 10.1093/jn/133.3.716. [DOI] [PubMed] [Google Scholar]
- 33.Boulikas T. Poly(ADP-ribosylated) histones in chromatin replication. J Biol Chem. 1990;265(24):14638–47. [PubMed] [Google Scholar]
- 34.Cheung WL, Ajiro K, Samejima K, Kloc M, Cheung P, Mizzen CA, Beeser A, Etkin LD, Chernoff J, et al. Apoptotic phosphorylation of histone H2B is mediated by mammalian sterile twenty kinase. Cell. 2003;113:507–17. doi: 10.1016/s0092-8674(03)00355-6. [DOI] [PubMed] [Google Scholar]
- 35.Zempleni J. Biotin. In: Bowman BA, Russell RM, editors. Present Knowledge in Nutrition. 8th ed. Washington, D.C.: ILSI Press; 2001. p. 241–52.
- 36.SAS Institute. StatView Reference. 3th ed. Cary, NC: SAS Publishing; 1999.
- 37.Scheerger SB, Zempleni J. Expression of oncogenes depends on biotin in human small cell lung cancer cells NCI-H69. Int J Vitam Nutr Res. 2003;73:461–7. doi: 10.1024/0300-9831.73.6.461. [DOI] [PubMed] [Google Scholar]
- 38.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003 Jan 30;421(6922):499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 39.Olive PL. Detection of DNA damage in individual cells by analysis of histone H2AX phosphorylation. Methods Cell Biol. 2004;75:355–73. doi: 10.1016/s0091-679x(04)75014-1. [DOI] [PubMed] [Google Scholar]
- 40.Bradshaw PS, Stavropoulos DJ, Meyn MS. Human telomeric protein TRF2 associates with genomic double-strand breaks as an early response to DNA damage. Nat Genet. 2005;37:193–7. doi: 10.1038/ng1506. [DOI] [PubMed] [Google Scholar]
- 41.Kosmoski JV, Ackerman EJ, Smerdon MJ. DNA repair of a single UV photoproduct in a designed nucleosome. Proc Natl Acad Sci USA. 2001;98:10113–8. doi: 10.1073/pnas.181073398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fernandez-Capetillo O, Allis CD, Nussenzweig A. Phosphorylation of histone H2B at DNA double-strand breaks. J Exp Med. 2004;199:1671–7. doi: 10.1084/jem.20032247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rothkamm K, Lobrich M. Misrejoining of DNA double-strand breaks in primary and transformed human and rodent cells: a comparison between the HPRT region and other genomic locations. Mutat Res. 1999;433:193–205. doi: 10.1016/s0921-8777(99)00008-7. [DOI] [PubMed] [Google Scholar]
- 44.Wang K-S, Mock NI, Mock DM. Biotin biotransformation to bisnorbiotin is accelerated by several peroxisome proliferators and steroid hormones in rats. J Nutr. 1997;127:2212–6. doi: 10.1093/jn/127.11.2212. [DOI] [PubMed] [Google Scholar]
- 45.Mock DM, Stadler D, Stratton S, Mock NI. Biotin status assessed longitudinally in pregnant women. J Nutr. 1997;127:710–6. doi: 10.1093/jn/127.5.710. [DOI] [PubMed] [Google Scholar]
- 46.Zempleni J, Mock DM. Marginal biotin deficiency is teratogenic. Proc Soc Exp Biol Med. 2000;223:14–21. doi: 10.1046/j.1525-1373.2000.22303.x. [DOI] [PubMed] [Google Scholar]
- 47.Mardach R, Zempleni J, Wolf B, Cannon MJ, Jennings ML, Cress S, Boylan J, Roth S, Cederbaum S, Mock DM. Biotin dependency due to a defect in biotin transport. J Clin Invest. 2002;109:1617–23. doi: 10.1172/JCI13138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wolf B. Worldwide survey of neonatal screening for biotinidase deficiency. J Inher Metab Dis. 1991;14:923–7. doi: 10.1007/BF01800475. [DOI] [PubMed] [Google Scholar]