

Abstract
Eukaryotic DNA repair enzymes must interact with the architectural hierarchy of chromatin. The challenge of finding damaged DNA complexed with histone proteins in nucleosomes is complicated by the need to maintain local chromatin structures involved in regulating other DNA processing events. The heterogeneity of lesions induced by DNA-damaging agents has led us to design homogeneously damaged substrates to directly compare repair of naked DNA with that of nucleosomes. Here we report that nucleotide excision repair in Xenopus nuclear extracts can effectively repair a single UV radiation photoproduct located 5 bases from the dyad center of a positioned nucleosome, although the nucleosome is repaired at about half the rate at which the naked DNA fragment is. Extract repair within the nucleosome is >50-fold more rapid than either enzymatic photoreversal or endonuclease cleavage of the lesion in vitro. Furthermore, nucleosome formation occurs (after repair) only on damaged naked DNA (165-bp fragments) during a 1-h incubation in these extracts, even in the presence of a large excess of undamaged DNA. This is an example of selective nucleosome assembly by Xenopus nuclear extracts on a short linear DNA fragment containing a DNA lesion.
Eukaryotic DNA is complexed with histone proteins to form nucleosomes, the fundamental repeating unit of chromatin hierarchy (1, 2). Despite variation in DNA sequence, nucleosomes are quite similar in their folding of DNA around an octamer of core histones (3), although the structural hierarchy becomes more variable at higher levels of DNA packaging (2, 4). DNA processing events, such as transcription, replication, and repair, must take place in conjunction with various chromatin structures to gain access to DNA sequences (4). In the case of transcription, a small fraction of genomic DNA is maintained in a more open chromatin structure to facilitate gene expression (5). However, DNA repair enzymes must access the entire genome during DNA damage surveillance (6, 7) and, therefore, must interact with each level of the chromatin hierarchy.
When subjected to UV light, different photoproducts form at dipyrimidine sites in DNA with yields modulated by sequence and local structure (8–10). Formation of UV photoproducts may in turn modify local chromatin structures and subsequent processing (11). The major UV photoproduct, cis-syn cyclobutane pyrimidine dimer (CPD), forms with a striking 10.3-base (average) periodicity when mixed-sequence DNA is assembled into nucleosomes (12) and reflects the rotational setting of DNA on the histone octamer surface (13). However, CPDs appear to be removed at similar rates from all surfaces of the DNA helix within nucleosomes during the early rapid repair phase of nucleotide excision repair (NER) in human cells (reviewed in ref. 9). This uniform rate of removal indicates that an active process of nucleosome rearrangement may be present during NER and/or CPDs may trigger DNA helix rotation in a dynamic nucleosome (14) to accommodate the helix distortion such that CPDs face away from the histone surface.
To overcome limitations associated with heterogeneous samples obtained from UV-irradiated cells or isolated chromatin, we adopted a synthetic approach to design nucleosomes with a single UV photoproduct at only one site and one structural orientation (15). The most prevalent UV photoproduct, a cis-syn cyclobutane thymine dimer (CTD), was synthesized and incorporated near the center of a short (30 bp) DNA sequence flanked by nucleosome positioning elements (16). Both the damaged and undamaged 165-bp DNA molecules were assembled into nucleosomes by histone octamer exchange, yielding damaged and undamaged nucleosome substrates with rotationally positioned DNA for NER in cell extracts. In a previous report, we showed the CTD faces away from the histone surface in these constructs and is 5 bases from the dyad center of these designed nucleosomes (15).
Recently, a similar approach was used to study excision of the pyrimidine (6–4) pyrimidone photoproduct, located at the center of a 136-bp DNA fragment, in a reconstituted human excision nuclease system and in cell extracts (17). The authors observed that the (6–4) photoproduct is excised from nucleosome DNA at about 10% the rate of naked DNA in both systems and hypothesized that nucleosome packing of DNA is a primary determinant of slow repair in chromatin. Furthermore, because there was no marked difference between the apparent rates of incision with purified proteins or cell extracts, the authors suggested that “there are no nucleosome accessibility factors specific for nucleotide excision repair” (17). Interestingly, (6–4) photoproducts are removed 5- to 10-fold faster than CPDs from nucleosome core DNA in intact human cells (18). Therefore, the rapid removal of these photoproducts cannot be ascribed solely to their preferential location in nucleosome linker and nucleosome-free DNA (19). It is possible that the dramatic structural alterations at (6–4) photoproduct sites in DNA renders their rate of detection and/or removal exceptional to CPDs, even within nucleosomes (20, 21).
Although complex, NER mechanisms can be summarized by five basic steps: recognition of the DNA lesion; cleavage of the damaged strand 3′ and 5′ of the lesion; excision of the damaged strand, creating a gap; synthesis of new DNA to fill the gap; and ligation of the final nick (6, 22). Unlike human cell extracts, nuclear extracts from Xenopus oocytes have a robust NER activity in vitro (23, 24), making measurement of DNA damage removal and completion of NER possible. Indeed, we recently showed that the time course of CPD removal at 35 different sites in a 175-bp 5S rDNA fragment assembled into a nucleosome can be measured simultaneously after incubation with Xenopus oocyte nuclear extracts (25). These results demonstrated that nucleosome structure strongly inhibits NER at most CPD sites in the fragment, although nucleosome assembly had little effect on NER at a few CPD sites within the nucleosome. In addition, nucleosomes assembled with histones lacking their highly charged amino-terminal tails had little effect on this inhibition (25). Thus, the mechanism by which NER accesses damaged DNA within chromatin remains elusive.
In the present study, we have used a mononucleosome and naked DNA as templates for repair in Xenopus nuclear extracts. We find that these extracts can effectively repair a single UV photoproduct located 5 bases from the dyad center of the well-positioned nucleosome. Moreover, nucleosome formation occurs rapidly and specifically in the newly repaired naked DNA fragments, even in the presence of a huge excess of nonspecific DNA.
Materials and Methods
Synthetic DNA Substrates.
Our strategy for the synthesis and assembly of DNA substrates has been described (15). Briefly, the synthetic oligonucleotide sequence (5′-TCGGGTGTACAGGATGT^TCTAGCCTGTAAC-3′, where T^T denotes the CTD site) and its undamaged control were annealed to their complementary sequence to create a unidirectional 4-base 5′ overhang. This central insert was bracketed with oligonucleotides containing TG motifs (16) and ligated into 165 bp of dsDNA. Products were isolated by PAGE followed by elution of the desired band. The terminal 5′ was specifically end-labeled with [γ-32P]ATP on the same strand as the CTD by T4 kinase (GIBCO/BRL), with the use of the forward reaction. (Substrates that harbor CTDs at this site are denoted by a * in the text.) Typically one-half of the radiolabeled DNA samples were reconstituted into nucleosomes by salt gradient-mediated histone octamer exchange from chicken erythocyte core particles as described (11). The CTD photoproduct slowly reverts back to thymidine with increased storage time, and there is a small amount of 5′ end-labeling on the opposite strand in each experiment (see ref. 15). These features cause incomplete cutting by T4 endonuclease V (e.g., see Fig. 1), and, therefore, the uncut fraction is rigorously determined and corrected for in each reaction.
Figure 1.

Repair of CTDs in Xenopus oocyte nuclear extracts. (A) DNA at different times of repair incubation was isolated, cleaved at CTDs with T4 endonuclease V, and separated by denaturing gel electrophoresis. The gel picture is a PhosphorImage following repair for the times shown (in minutes) with DNA* (lanes 1–6) and NUC* (lanes 7–12). Damaged DNA (d) migrates faster, and repaired DNA (u) migrates as the full-length fragment (165 bp). (B) Average repair curves (mean ± 1 SD) for three different extract preparations and three separate trials with each extract. Images were analyzed with imagequant software, and the percentage of repair was determined for each experiment (see Materials and Methods).
Xenopus Extract and Repair.
Repair extracts were prepared from Xenopus oocyte nuclei as described by Ackerman et al. (26). The in vitro repair experiments were performed as described (24), with some modification (23). Briefly, in vitro repair reactions (30 μl final volume) included 0.1 pmol radiolabeled substrate; 0.7 mM dATP; 0.5 mM dTTP, dCTP, and dGTP; 1 μg double-digested (EcoRI, BamHI) calf thymus DNA averaging 2.5 kb; 0.1 nmol chicken erythrocyte core particles; fresh J buffer (8.5 mM MgCl2/7 mM Hepes, pH 7.4/70 mM KCl/0.1 mM EDTA/3 mM DTT/10% glycerol/1% pvp360); and 3 μl of the nuclear extract preparation adjusted to approximately one nucleus per microliter. Reactions were carried out at room temperature in the dark.
For kinetic experiments, 4.0 μl of the repair reaction was removed at the appropriate time, combined with 16 μl of the stop buffer (5 mM Tris, pH 7.6/10 mM EDTA/proteinase K, 40 μg/μl final concentration), and heated to 42°C for 30 min. Proteins were extracted with phenol/chloroform/isoamyl alcohol (50:48:2), and the DNA was precipitated and washed in 70% ethanol. Samples were assayed for CTDs by digestion with T4 endonuclease V followed by electrophoresis on 7 M urea/10% acrylamide denaturing gels (27). Gels were dried, exposed to PhosphorImager screens, and visualized on a Molecular Dynamics (model 455-P90) PhosphorImager. Images were analyzed with imagequant (Molecular Dynamics) software. Reactions were performed in triplicate with different extract preparations. The percentage of repair was determined for each experiment at the indicated times from the equation (F0 − Ft)/F0 × 100%, where F0 and Ft are the fractional intensities in the lower band (see Fig. 1A) at times zero and t, respectively.
Gel Analysis of NER Products.
After 1 h of repair, samples were directly loaded onto a 6% native acrylamide gel (0.25× TBE) (1× TBE = 90 mM Tris/90 mM borate/2.5 mM EDTA, pH 8.3) and run at 100 V for 1 h at room temperature. The glycerol in the reaction buffer eliminated the need to add loading buffer. Gels were dried and visualized on the PhosphorImager as above. Analysis of CTDs within the newly formed nucleosome complex was conducted by first separating the products of the extract repair reaction for DNA* after 1 h by native acrylamide gel electrophoresis as described. The wet gel was immediately exposed to the PhosphorImager screen and developed on the PhosphorImager, and the desired bands were excised, nebulized, and eluted from the gel. All proteins were removed with phenol/chloroform/isoamyl alcohol, and the DNA was precipitated with ethanol. The samples were subjected to T4 endonuclease V, as described above, for the analysis of the repair time course.
Nucleosome Characterization.
Newly assembled nucleosomes were subjected to DNase 1 footprinting (28) after the repair incubation by native gel isolation and denaturing gel footprinting. Samples were digested for 2 or 3 min (optimized for single hit kinetics) by addition of DNase 1 directly to the repair mix at the end of the 1-h repair reaction. Samples were immediately loaded onto native gels, and the newly formed nucleosomes (upper band), observed only with DNA*, were excised and eluted. These samples, along with appropriate controls, were analyzed on sequencing gels. G sequencing was conducted by Maxam–Gilbert chemical degradation.
Results
Repair of Damaged DNA.
Our previous work demonstrated that the noneukaryotic repair enzymes UV photolyase and T4 endonuclease V suffered a 100- to 1,000-fold loss of activity when DNA* (damaged DNA) was assembled into NUC* (nucleosome with damaged DNA) (15). Nuclear extracts from Xenopus oocytes, however, are capable of removing the centrally located CTD in the nucleosome at almost half the rate of naked DNA* (Fig. 1). Direct comparison of DNA* and NUC* in parallel NER reactions shows only about a 2-fold reduction in repair activity because of the presence of the nucleosome.
The 32P end-labeled substrates (DNA* and NUC*) were incubated with nuclear extracts for increasing times in the presence of a DNA repair mix (see Materials and Methods). At different times, samples were removed, and the DNA was extracted, isolated, digested with T4 endonuclease V, and separated by denaturing gel electrophoresis. Results in Fig. 1A demonstrate the removal of CTDs over time by the increased resistance of the intact fragment (u) to T4 endonuclease V cleavage, which results in the appearance of a shorter fragment (d) if the fragment contains a CTD. Both DNA* and NUC* are effectively repaired, although the nucleosome is repaired at about half the rate of the naked DNA (Fig. 1B). The large standard deviations in Fig. 1B reflect the variations obtained in different extract preparations. Indeed, multiple repeats of the experiment conducted with the same extract preparation showed significantly lower standard deviations (≤5%).
Activities of Repair Enzymes on Damaged DNA Assembled into Nucleosomes.
The effect of nucleosome formation on the repair-associated activities of T4 endonuclease V, Escherichia coli UV photolyase, and NER in Xenopus nuclear extracts was compared. The naked DNA repair rates were normalized in time units, so that nucleosome repair rates could be directly compared graphically. As observed (15), there is a significant inhibition of the noneukaryotic repair enzymes when damaged DNA is assembled into nucleosomes (NUC*) (Fig. 2; compare open symbols, dashed lines). On the other hand, the extract effectively repairs the damaged NUC* (Fig. 2; compare closed symbols, solid lines), suffering only a 2-fold reduction in the time course of CTD removal. Thus, it appears that factors responsible for manipulating nucleosome structure may be coupled to NER in the Xenopus nuclear extracts (7, 29).
Figure 2.

Comparison of NER, UV photolyase, and T4 endonuclease V activities with nucleosomes. Naked DNA data for each reaction have been normalized to achieve 90% repair in one “relative time unit” to graphically compare nucleosome repair rates. T4 endonuclease V and UV photolyase data are represented by open symbols (– – –), and NER data are represented with closed symbols (——). Each data point represents the average of triplicate experiments performed with three different preparations of the respective repair systems.
Nucleosome Assembly After Repair.
We then examined DNA* for the presence of nucleosomes after a 1-h incubation with the extract, because nucleosome assembly of UV-damaged circular DNA occurs concomitantly with NER synthesis in Xenopus egg extracts (30, 31). Radiolabeled samples (DNA, NUC, DNA*, and NUC*) were loaded directly onto native polyacrylamide gels after the repair incubation (Fig. 3). (The presence of glycerol in the repair reaction buffer allowed direct loading of samples without further manipulation.) After 1 h of repair reactions, almost 50% of the damaged fragments (DNA*) are assembled into nucleosomes (Fig. 3, lane 5). In contrast, none of the undamaged fragments (DNA) are present as nucleosomes after the hour-long incubation (Fig. 3, lane 3), indicating that nucleosome loading by the extract occurs much more rapidly on the damaged and/or nascent repaired DNA. Kinetic experiments demonstrated that nucleosome formation occurs more slowly than the repair reaction (i.e., CTD removal) after an initial lag phase (data not shown). This slower rate of nucleosome formation indicates that nucleosome assembly occurs after repair is complete. The remaining lanes in Fig. 3 (lanes 6–9) show that no additional bands appear below the nucleosome band during NER. The absence of signal below this band (especially in lane 9) excludes the possibility of complete histone octamer displacement as a mechanism for manipulating nucleosome structure during repair (discussed in refs. 32 and 33).
Figure 3.

Nucleosome formation during DNA repair in Xenopus extracts. The picture is a PhosphorImage of a native polyacrylamide gel showing DNA and nucleosome complexes before and after repair. The samples are molecular weight markers (lane 1), undamaged naked DNA before (lane 2) and after (lane 3) 1 h of repair, damaged naked DNA* before (lane 4) and after (lane 5) 1 h of repair, undamaged DNA assembled into a nucleosome (NUC) before (lane 6) and after (lane 7) 1 h of repair, and damaged DNA* assembled into a nucleosome (NUC*) before (lane 8) and after (lane 9) 1 h of repair. Migration of naked DNA (DNA) and nucleosomes (NUC) is indicated on the right.
To examine the newly formed nucleosomes for the presence of CTDs, the upper and lower bands observed on native gels (e.g., Fig. 3, lane 5) after 1 h of repair were extracted and purified. The DNA samples were isolated and digested with T4 endonuclease V and separated on denaturing gels (Fig. 4). The upper band formed during repair of DNA* (see Fig. 3, lane 5) contains almost no CTDs (Fig. 4, lane 5), whereas the lower band has both newly repaired and damaged DNA fragments (Fig. 4, lane 6). This difference in the upper and lower bands further supports the notion that NER precedes nucleosome formation in the damaged fragments (DNA*).
Figure 4.

CTD content of newly formed nucleosomes: PhosphorImage of a denaturing gel after T4 endonuclease V digestion of the repair samples. Samples are molecular weight markers (lane 1); DNA* (lane 2); DNA* treated with T4 endonuclease V (lane 3); DNA* treated with T4 endonuclease V after 1 h of repair (lane 4); DNA* subjected to repair for 1 h, followed by isolation on native gels as nucleosomes with subsequent T4 endonuclease V digest (lane 5); and DNA* subjected to repair for 1 h, followed by isolation on native gels as naked DNA with subsequent T4 endonuclease V digest (lane 6). Migration of undamaged (u) and damaged (d) DNA is indicated on the right.
The structural integrity of newly formed nucleosomes was also examined by DNase 1 footprinting (28). Hydroxyl radical footprinting techniques, such as those described by Hayes et al. (34), could not be used because of the presence of glycerol in the NER reaction buffer. Nonetheless, we adopted their strategy of isolating digested nucleosomes by native gel electrophoresis before observing the footprint under denaturing electrophoresis conditions (34). Once again, DNA* was incubated with the Xenopus nuclear extract for 1 h. A small aliquot was removed and assayed for repair as a control. DNase I was then directly added to the reaction, and the digestion was optimized for single hit kinetics to minimize disruption of the DNA-protein complexes (28). As before, the samples were directly loaded onto a native gel, and the upper and lower bands were isolated. Sequencing gels of the radiolabeled material from the upper band and appropriate controls are presented in Fig. 5. The results indicate that the newly formed nucleosomes have structures very similar to those reconstituted by histone octamer exchange. Indeed, the footprints for newly formed nucleosomes are nearly identical to those found with the undamaged NUC controls (Fig. 5, compare lane 5 to lanes 10 and 11). Very subtle differences do exist, however, between DNase 1 footprints of NUC* (lane 9) and NUC (lanes 5, 10, and 11). Because nucleosomes formed during repair have a footprint similar to undamaged nucleosomes, these data support the notion that CTDs are repaired before nucleosome assembly. Thus, the newly formed complex observed on native gels after repair of DNA* appears to be a bona fide (or canonical) nucleosome core.
Figure 5.
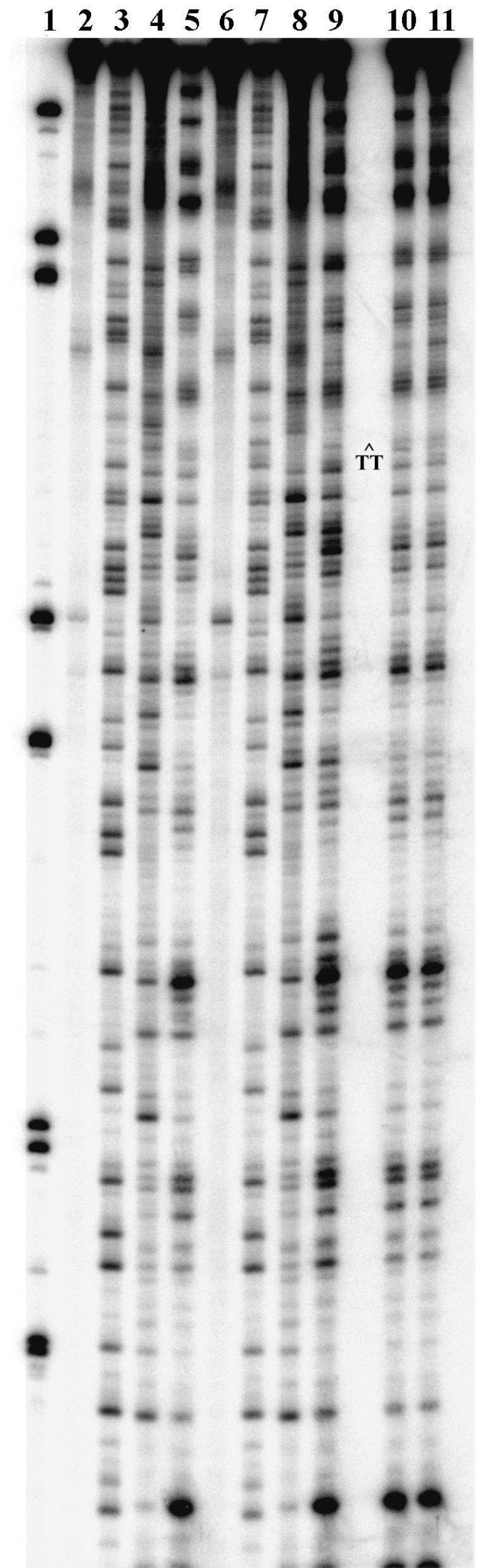
DNase 1 footprints of nucleosomes: PhosphorImage of a sequencing gel of naked and nucleosome DNA after DNase 1 digestion. Samples are molecular weight markers (lane 1), undamaged DNA (lane 2), G sequencing of undamaged DNA (lane 3), DNase 1 digest of undamaged DNA (lane 4) and NUC (lane 5), damaged DNA* (lane 6), G sequencing of damaged DNA* (lane 7), and DNase 1 digest of damaged DNA* (lane 8) and NUC* (lane 9). Lanes 10 and 11 are for 2-min (lane 11) or 3-min (lane 12) DNase 1 digests of newly formed nucleosomes from the repair of damaged DNA* after 1 h of extract repair and isolation on a native gel.
Discussion
Despite the degree of compaction of DNA in the chromatin hierarchy, most DNA repair in the eukaryotic genome occurs within nucleosomes. In this study, we show that the rate of repair in Xenopus nuclear extracts of a single CTD near the center of a DNA fragment assembled into a nucleosome decreases by only about 2-fold compared with that of naked DNA* (Fig. 1). In contrast, the activities of T4 endonuclease V and E. coli UV photolyase on nucleosome DNA are between 100- and 1,000-fold slower than on naked DNA* (Fig. 2). A dynamic nucleosome model has been proposed for access of DNA in nucleosomes (35, 36) and predicts that CPDs near the edges of nucleosomes (or in free DNA) should be far more accessible (by 100- to 1,000-fold) than CPDs near the center. As noneukaryotic enzymes also experience a dynamic “nucleosome-like” structure, with DNA bound by HU family proteins (37), this mechanism may account for the significant inhibition of these enzymes by nucleosome assembly. However, the variation in repair rates of CPDs in the interior of the 5S rDNA nucleosome indicates that repair proteins in the Xenopus extracts do not follow the dynamic nucleosome model, at least for mononucleosomes (25). Therefore, NER may be accompanied by a transient disruption of the dynamic nucleosome structure that is sufficient to allow repair enzymes access to a DNA lesion.
Recently, we showed that NER of CPDs in 5S rDNA by Xenopus oocyte extracts is quite efficient at several sites within the nucleosome, including −7 and −8 (relative to the transcription start) (25). The two predominant nucleosome dyad positions in the 175-bp 5S fragment are −3 and +7, or 4–5 nt and 14–15 nt from these two sites, respectively. In the present report, the single CPD is located 5 nt from the (presumed) dyad axis of the TG motif-bracketed oligos (15). The difference between the rate of NER in naked 5S rDNA and that of 5S rDNA assembled into nucleosomes was 2-fold (25), or the same value we find in the present report for the TG motif-bracketed oligos (Fig. 1). This difference is surprising, considering the fact that the TG motif-bracketed oligos yield stronger positioned nucleosomes than the 5S rDNA sequence (16, 38).
The crystal structure of the 5S rDNA nucleosome indicates that a majority of the DNA–histone ion pair interactions within the nucleosome are bridged by water molecules (1, 3, 39). It is possible that only some of these sites are disrupted during repair and the remaining histone-DNA interactions are sufficient to maintain an “intermediate” nucleosome structure. Such a mechanism would not necessarily depended on additional proteins to assist in repair, and NER may have evolved to take advantage of the dynamic state of nucleosomes (35, 36). On the other hand, transient modification of histones, such as the reversible acetylation of ɛ-amino groups of lysines (40), may “prepare” nucleosomes for disruption during repair (e.g., see ref. 9). Indeed, binding of a transcriptional activator to a high-affinity site incorporated into an acetylated nucleosome results in complete disruption (but not displacement) of nucleosomes, sufficient for the binding of a second transcription factor (41).
The assembly of nucleosomes after NER has been extensively investigated in both intact cells and cell extracts (reviewed in refs. 9 and 29). For example, immediately after NER in human cells, there is an initial rapid association of newly synthesized repair patches with nucleosomes that may represent nucleosome refolding at nascent repair sites (42, 43). Furthermore, in Xenopus egg and human cell extracts, chromatin assembly factor 1 facilitates nucleosome assembly on UV-irradiated circular DNA templates after NER synthesis (29, 31). In the present study, we find that nucleosome formation occurs after a short time lag on the newly repaired DNA* (Fig. 3), and the DNase I footprint of these nucleosomes is very similar to the undamaged (NUC) control (Fig. 5). Moreover, nucleosome formation during the 1-h incubation is limited to the damaged DNA fragments, as the undamaged fragments remain free of nucleosomes during the incubation. It should be emphasized that the nucleosome loading activity we observe occurs in the presence of a huge excess of undamaged DNA. Therefore, nucleosome assembly activity is very specific for the UV-damaged/repaired DNA and may be linked (or coupled) to NER of these short linear DNA substrates. This result is analogous to that found with DNA plasmids (30, 31) in Xenopus nuclear extracts.
After submission of this paper, Ura et al. (44) reported that ATP-dependent chromatin remodeling facilitates excision of UV-induced (6–4) photoproducts in linker DNA of synthetic dinucleosomes. These authors observed an almost 4-fold enhancement of (6–4) photoproduct excision from the linker, yielding ≈27% the repair efficiency of naked DNA, when the dinucleosomes are treated with a chromatin assembly and remodeling factor. On the other hand, no enhancement of excision was observed with chromatin assembly and remodeling factor treatment when the (6–4) photoproduct was located near the dyad center of each nucleosome core (44). In the present report, we find efficient CPD removal (≈50% that of naked DNA) with Xenopus oocyte extracts, compared with access to the CPD by enzymes in vitro, when the CPD is near the nucleosome dyad (5 nt away). Therefore, the oocyte extracts may contain ATP-dependent chromatin remodeling proteins that allow access of NER proteins to the center of nucleosome cores.
In conclusion, we found that NER in Xenopus oocyte nuclear extracts effectively repairs a single UV lesion located near the center of a well-positioned nucleosome and is much more efficient than the activities of two noneukaryotic repair enzymes. In addition, nucleosome assembly occurs after a short time lag after NER in these extracts, and this assembly activity is specific for nascent repaired DNA. Finally, NER can repair lesions within nucleosomes without long-lasting disruption of preformed nucleosome structure.
Acknowledgments
We dedicate this paper to the memory of our colleague, Alan P. Wolffe, Senior Vice President and Chief Science Officer, Sangamo Biosciences, Inc., who was a beacon for the interplay of chromatin structure and DNA repair mechanisms. We thank R. Stephen Lloyd for supplying purified T4 endonuclease V and Antonio Conconi for sharing his expertise with the nuclear extract. This study was supported by National Institutes of Health Grant ES04106 (to M.J.S.) from the National Institute of Environmental Health Sciences and by Pacific Northwest National Laboratory Directed Research and Development funds (to E.J.A.).
Abbreviations
- CPD
cis-syn cyclobutane pyrimidine dimer
- NER
nucleotide excision repair
- CTD
cis-syn cyclobutane thymine dimer
- DNA*
damaged DNA
- NUC
nucleosome
- NUC*
nucleosome with damaged DNA
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Luger K, Mader A, Richmond R, Sargent D, Richmond T J. Nature (London) 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 2.van Holde K. Chromatin. New York: Springer; 1988. [Google Scholar]
- 3.Luger K, Richmond T J. Curr Opin Struct Biol. 1998;8:33–40. doi: 10.1016/s0959-440x(98)80007-9. [DOI] [PubMed] [Google Scholar]
- 4.Wolffe A. Chromatin: Structure and Function. 3rd Ed. San Diego: Academic; 1998. [Google Scholar]
- 5.Lewin B. Genes VII. London: Oxford Univ. Press; 2000. [Google Scholar]
- 6.Friedberg E, Walker G, Siede W. DNA Repair and Mutagenesis. Washington, DC: Am. Soc. Microbiol.; 1995. [Google Scholar]
- 7.Meijer M, Smerdon M J. BioEssays. 1999;21:596–603. doi: 10.1002/(SICI)1521-1878(199907)21:7<596::AID-BIES8>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 8.Pfeifer G P. Photochem Photobiol. 1997;65:270–283. doi: 10.1111/j.1751-1097.1997.tb08560.x. [DOI] [PubMed] [Google Scholar]
- 9.Smerdon M J, Conconi A. Prog Nucleic Acid Res Mol Biol. 1999;62:227–255. doi: 10.1016/s0079-6603(08)60509-7. [DOI] [PubMed] [Google Scholar]
- 10.Cadet J, Anselmino C, Douki T, Voituriez L. J Photochem Photobiol B. 1992;15:277–298. doi: 10.1016/1011-1344(92)85135-h. [DOI] [PubMed] [Google Scholar]
- 11.Mann D B, Springer D L, Smerdon M J. Proc Natl Acad Sci USA. 1997;94:2215–2220. doi: 10.1073/pnas.94.6.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gale J M, Nissen K A, Smerdon M J. Proc Natl Acad Sci USA. 1987;84:6644–6648. doi: 10.1073/pnas.84.19.6644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gale J M, Smerdon M J. J Mol Biol. 1988;204:949–958. doi: 10.1016/0022-2836(88)90054-x. [DOI] [PubMed] [Google Scholar]
- 14.Polach K, Lowary P, Widom J. J Mol Biol. 2000;298:211–223. doi: 10.1006/jmbi.2000.3644. [DOI] [PubMed] [Google Scholar]
- 15.Kosmoski J, Smerdon M J. Biochemistry. 1999;38:9485–9494. doi: 10.1021/bi990297h. [DOI] [PubMed] [Google Scholar]
- 16.Shrader T, Crothers D. J Mol Biol. 1990;216:69–84. doi: 10.1016/S0022-2836(05)80061-0. [DOI] [PubMed] [Google Scholar]
- 17.Hara R, Mo J, Sancar A. Mol Cell Biol. 2000;20:9173–9181. doi: 10.1128/mcb.20.24.9173-9181.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suquet C, Mitchell D L, Smerdon M J. J Biol Chem. 1995;270:16507–16509. doi: 10.1074/jbc.270.28.16507. [DOI] [PubMed] [Google Scholar]
- 19.Mitchell D L, Nguyen T D, Cleaver J E. J Biol Chem. 1990;265:5353–5356. [PubMed] [Google Scholar]
- 20.Kim J K, Patel D, Choi B S. Photochem Photobiol. 1995;62:44–50. doi: 10.1111/j.1751-1097.1995.tb05236.x. [DOI] [PubMed] [Google Scholar]
- 21.Lee J H, Hwang G S, Choi B S. Proc Natl Acad Sci USA. 1999;96:6632–6636. doi: 10.1073/pnas.96.12.6632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wood R. Annu Rev Biochem. 1996;65:135–167. doi: 10.1146/annurev.bi.65.070196.001031. [DOI] [PubMed] [Google Scholar]
- 23.Conconi A, Liu X, Koriazova L, Ackerman E, Smerdon M J. EMBO J. 1999;18:1387–1396. doi: 10.1093/emboj/18.5.1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oda N, Saxena J, Jenkins T, Prasad R, Wilson S, Ackerman E. J Biol Chem. 1996;271:13816–13820. doi: 10.1074/jbc.271.23.13816. [DOI] [PubMed] [Google Scholar]
- 25.Liu X, Smerdon M J. J Biol Chem. 2000;275:23729–23735. doi: 10.1074/jbc.M002206200. [DOI] [PubMed] [Google Scholar]
- 26.Ackerman E, Koriazova L, Saxena J, Spoonde A. Methods Mol Biol. 1999;113:347–355. doi: 10.1385/1-59259-675-4:347. [DOI] [PubMed] [Google Scholar]
- 27.Dodson M, Lloyd R. Mutat Res. 1989;218:49–65. doi: 10.1016/0921-8777(89)90011-6. [DOI] [PubMed] [Google Scholar]
- 28.Revzin A. Footprinting of Nucleic Acid Complexes. New York: Academic; 1993. [Google Scholar]
- 29.Moggs J, Almouzni G. Biochimie. 1999;81:45–52. doi: 10.1016/s0300-9084(99)80037-6. [DOI] [PubMed] [Google Scholar]
- 30.Gaillard P, Martini E, Kaufman P, Stillman B, Moustacchi E, Almouzni G. Cell. 1996;86:887–898. doi: 10.1016/s0092-8674(00)80164-6. [DOI] [PubMed] [Google Scholar]
- 31.Gaillard P, Moggs J, Roche D, Quivy J, Becker P, Wood R, Almouzni G. EMBO J. 1997;16:6281–6289. doi: 10.1093/emboj/16.20.6281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kornberg R, Lorch Y. Curr Opin Genet Dev. 1999;9:148–151. doi: 10.1016/S0959-437X(99)80022-7. [DOI] [PubMed] [Google Scholar]
- 33.Owen-Hughes T, Workman J L. EMBO J. 1996;15:4702–4712. [PMC free article] [PubMed] [Google Scholar]
- 34.Hayes J, Tullius T, Wolffe A. Proc Natl Acad Sci USA. 1990;87:7405–7409. doi: 10.1073/pnas.87.19.7405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Polach K J, Widom J. J Mol Biol. 1995;254:130–149. doi: 10.1006/jmbi.1995.0606. [DOI] [PubMed] [Google Scholar]
- 36.Polach K J, Widom J. J Mol Biol. 1996;258:800–812. doi: 10.1006/jmbi.1996.0288. [DOI] [PubMed] [Google Scholar]
- 37.Sandman K, Pereira S L, Reeve J N. Cell Mol Life Sci. 1998;54:1350–1364. doi: 10.1007/s000180050259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thastrom A, Lowary P T, Widlund H R, Cao H, Kubista M, Widom J. J Mol Biol. 1999;288:213–229. doi: 10.1006/jmbi.1999.2686. [DOI] [PubMed] [Google Scholar]
- 39.Luger K, Richmond T J. Opin Genet Dev. 1998;8:140–146. doi: 10.1016/s0959-437x(98)80134-2. [DOI] [PubMed] [Google Scholar]
- 40.Cheung W L, Briggs S D, Allis C D. Curr Opin Cell Biol. 2000;12:326–333. doi: 10.1016/s0955-0674(00)00096-x. [DOI] [PubMed] [Google Scholar]
- 41.Ng K W, Ridgway P, Cohen D R, Tremethick D J. EMBO J. 1997;16:2072–2085. doi: 10.1093/emboj/16.8.2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smerdon M J, Lieberman M W. Proc Natl Acad Sci USA. 1978;75:4238–4241. doi: 10.1073/pnas.75.9.4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baxter B K, Smerdon M J. J Biol Chem. 1998;273:17517–17524. doi: 10.1074/jbc.273.28.17517. [DOI] [PubMed] [Google Scholar]
- 44.Ura K, Araki M, Saeki H, Masutani C, Ito T, Iwai S, Mizukoshi T, Kaneda Y, Hanaoka F. EMBO J. 2001;20:2004–2014. doi: 10.1093/emboj/20.8.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]