

Abstract
The budding yeast mitotic exit network (MEN) is a signal transduction cascade that controls exit from mitosis by facilitating the release of the cell cycle phosphatase Cdc14 from the nucleolus. The G protein Tem1 regulates MEN activity. The Tem1 guanine nucleotide exchange factor (GEF) Lte1 associates with the cortex of the bud and activates the MEN upon the formation of an anaphase spindle. Thus, the cell cortex has an important but ill-defined role in MEN regulation. Here, we describe a network of conserved cortical cell polarity proteins that have key roles in mitotic exit. The Rho-like GTPase Cdc42, its GEF Cdc24 and its effector Cla4 [a member of the p21-activated kinases (PAKs)] control the initial binding and activation of Lte1 to the bud cortex. Moreover, Cdc24, Cdc42 and Ste20, another PAK, probably function parallel to Lte1 in facilitating mitotic exit. Finally, the cell polarity proteins Kel1 and Kel2 are present in complexes with both Lte1 and Tem1, and negatively regulate mitotic exit.
Keywords: Cdc24/Cdc42/Lte1/mitotic exit/PAK kinases
Introduction
In budding yeast, the cell cycle phosphatase Cdc14 forms a complex with the nucleolar protein Net1 during interphase and metaphase of mitosis (Shou et al., 1999). With anaphase onset, Cdc14 is released from the nucleolus and dephosphorylates key targets such as the anaphase promoting complex subunit Cdh1/Hct1 and the mitotic cyclin inhibitor Sic1. Together, these proteins then decrease the activity of mitotic cyclin-dependent kinase (Cdk-Clb). This decrease in Cdk-Clb activity is a pre requisite for mitotic exit (ME) and the initiation of the next cell cycle (Visintin et al., 1998).
The release of Cdc14 from the nucleolus is a two-step process. In early anaphase, separase (Esp1) and polo kinase (Cdc5) are required for the partial release of Cdc14 from the nucleolus (FEAR network). The full release of Cdc14 from the nucleolus is essential for ME and is regulated by a GTPase-driven signalling network, the mitotic exit network (MEN) (Pereira et al., 2002; Stegmeier et al., 2002).
A key protein of the MEN is the small Ras-like GTPase Tem1. The GTPase-activating protein (GAP) complex composed of Bfa1 and Bub2 inactivates Tem1 (Bardin et al., 2000; Pereira et al., 2000), while the guanine nucleotide exchange factor (GEF) Lte1 activates Tem1. Tem1 forms a complex with the Bfa1–Bub2 GAP at the budding yeast microtubule organizing centre, the spindle pole body (SPB) (Pereira et al., 2000). In contrast, the GEF, Lte1, is associated with the cortex of the bud, but not the mother cell (Bardin et al., 2000; Pereira et al., 2000). It has been proposed that the SPB-associated Bfa1–Bub2 GAP inactivates Tem1 until it becomes exposed to Lte1 with the migration of the nucleus into the bud during anaphase. This interdependency between MEN activation and nuclear migration into the bud prevents ME in cells where the anaphase spindle forms in the mother cell body.
How the selective binding of Lte1 to the bud cortex is achieved and how this binding is regulated are not understood. Moreover, at 30 or 37°C cells lacking LTE1 exit mitosis with nearly identical kinetics to wild-type cells, while at 10°C, Δlte1 cells arrest in the cell cycle at the end of mitosis (Adames et al., 2001; Pereira et al., 2002). In order to explain the non-essential role of Lte1 at 30°C, additional Lte1-independent mechanisms must activate the MEN. These processes may involve cell cortex-associated proteins, which ensure that, in the absence of Lte1, cells can still restrain ME until the spindle is correctly positioned along the mother-to-bud axis.
To understand how the MEN is activated in the absence of LTE1, we performed a screen for high-dosage suppressors of the cold-sensitive growth defect of Δlte1 cells. The conserved p21-activated kinase (PAK) STE20 was identified in this screen. Ste20 is associated with the bud cortex and functions in polarized growth and mating (Holly and Blumer, 1999). The cell polarity protein Cdc42 regulates Ste20 activity. This Rho-like GTPase is in turn activated by the GEF Cdc24 (Peter et al., 1996). Further data suggest that the Cdc24–Cdc42–Ste20 cascade has an overlapping function with Lte1. Moreover, we show that Cdc24, Cdc42 and the PAK kinase Cla4, which is also regulated by Cdc42 (Cvrckova et al., 1995; Benton et al., 1997), are required to localize Lte1 to the bud cortex and that phosphorylation and activation of Lte1 are dependent on Cla4. Finally, Kel1 and Kel2, two cell cortex proteins that regulate cell morphology (Philips and Herskowitz, 1998), negatively control MEN activity.
Results
A role for the PAK kinase Ste20 in ME
The fact that LTE1 is not essential at 30°C suggests that there are additional ways of activating the MEN. At 10°C, Δlte1 cells may arrest in the cell cycle because these alternative pathways are less active at lower temperatures. Increasing the activity of one of these pathways by overexpressing a gene functioning within it might rescue the cold sensitivity of Δlte1 cells. We therefore transformed Δlte1 cells with a high-copy-number yeast library and selected for growth at 10°C. About 95 out of 27 000 transformants were able to grow at 10°C. Of these, 55 plasmids carried TEM1 (Figure 1A, row 4), which has been described as a suppressor of Δlte1 cells, two plasmids harboured CDC15, which functions downstream of TEM1 (Jaspersen et al., 1998), 11 plasmids contained SPO12 (row 5), a common suppressor of MEN mutants (Jaspersen et al., 1998), and two plasmids carried SIC1, coding for a Cdk-Clb inhibitor (Schwob et al., 1994). In addition, 20 plasmids contained STE20 (Figure 1A, row 3), which encodes a conserved PAK involved in polarized growth and mating (Holly and Blumer, 1999). The remaining plasmids carried LTE1.

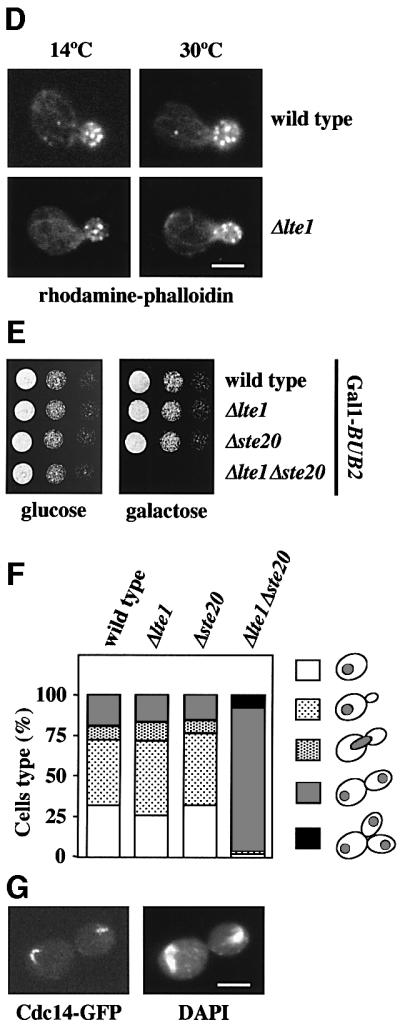
Fig. 1. A role of STE20 in ME. (A) Suppression of the Δlte1 growth defect by STE20. Serial dilutions (1:10) of cells of Δlte1 (row 2) with the 2 µm-based plasmids pRS426-STE20 (row 3), pRS426-TEM1 (row 4), pRS426-SPO12 (row 5), pRS426-LTE1 (row 6) and pRS426 (row 7) were grown for 10 days at 10°C on YPAD. Wild-type cells were used as control (row 1). Cells grew equally well at 30°C (not shown). (B) Overexpression of STE20 suppresses the ME defect of Δlte1 cells. Wild-type, Δlte1 and Δlte1 Gal1-STE20 cells with CDC14-GFP in YPRA were arrested in G1 with α-factor. Cells progressed synchronously through the cell cycle at 10°C upon removal of α-factor by washing with pre-cooled YPRA galactose medium, which also induced expression of Gal1-STE20. The number of cells with large buds and nucleolar Cdc14–GFP (n > 100) was determined over time. (C) Synthetic lethal phenotype of Δlte1 Δste20 cells is suppressed by Δbub2. Serial dilutions of cells of wild type (lane 1), Δlte1 (lane 2), Δste20 (lane 3), Δlte1 Δste20 pRS316-LTE1 with the additional plasmid pRS315 (lane 4), pRS315-LTE1 (lane 5) or pRS315-STE20 (lane 6) and Δlte1 Δste20 Δbub2 pRS316-LTE1 were grown on 5′-fluoroorotic acid (5-FOA) and YPAD plates at 30°C for 2 days. 5-FOA selects against URA3-based pRS316 derivatives. (D) F-actin of wild-type and Δlte1 cells grown at 14 and 30°C was stained with rhodamine–phalloidin. (E) Serial dilutions of wild-type, Δlte1, Δste20 and Δlte1 Δste20 cells with Gal1-BUB2 were grown for 2 days at 30°C. (F) Δlte1 Δste20 cells have a ME defect. Wild-type, Δlte1, Δste20 and Δlte1 Δste20 cells with Gal1-BUB2 CDC14-GFP grown in YPRA medium were washed and incubated for 3 h at 30°C in YPRA galactose medium to induce Gal1-BUB2 expression. Cells were fixed, stained with DAPI and analysed by fluorescence microscopy. The circles in the cartoon cells indicate the DAPI staining regions. n > 100. (G) An anaphase Gal1-BUB2 Δlte1 Δste20 CDC14-GFP cell of (F). Bars: 5 µm.
We then asked whether overexpression of STE20 rescued the ME defect of Δlte1 cells. Wild-type, Δlte1 and Δlte1 Gal1-STE20 cells with CDC14-GFP were arrested in G1 with α-factor and released into galactose medium, which induced expression of the Gal1 promoter of the Gal1-STE20 construct. We monitored the release of Cdc14–GFP from the nucleolus and the decrease in the proportion of cells, which had large buds, as markers of ME. At 10°C, wild-type cells released Cdc14–GFP from the nucleolus and exited mitosis after ∼7 h (Figure 1B). In contrast, most Δlte1 cells arrested in anaphase with separated DAPI staining regions (data not shown) and Cdc14–GFP trapped in the nucleolus (Figure 1B). Δlte1 cells in which Gal1-STE20 was induced did not arrest in anaphase and released Cdc14 from the nucleolus with the same kinetics as wild-type cells (Figure 1B). This suggested that Ste20 triggers ME of Δlte1 cells by facilitating the release of Cdc14 from the nucleolus.
An additional genetic interaction confirmed that STE20 shares a common function with LTE1. Deletion of either LTE1 or STE20 did not significantly affect growth of yeast cells at 23 and 30°C (Figure 1C, rows 2 and 3). However, cells lacking both Δlte1 and Δste20 were unable to grow at 23 and 30°C (Figure 1C, row 4, 5-FOA). The genetic interactions of LTE1 and STE20 indicate that the two genes share an overlapping function in mating, bud growth or ME. However, LTE1 did not show a synthetically lethal phenotype with STE11 (Table I), which functions downstream of Ste20. Thus, the overlapping function of LTE1 and STE20 is not dependent on the mating pheromone MAP kinase cascade.
Table I. Genetic interactions and localization of Lte1.
| Genotype | Synthetic lethality with Δlte1a | Suppression of the Δlte1 cold sensitivityb | Bud cortex association of GFP–Lte1c |
|---|---|---|---|
| Δbni1 | – | – | + |
| Δbub2 | – | + | + |
| Δbud6 | – | – | + |
| cdc24-1 | + | – | –d |
| cdc42-1 | + | – | –d |
| cdc42-118 | + | n.d. | +d |
| Δcla4 | – | – | – |
| Δgic1 | – | – | + |
| Δgic2 | – | – | + |
| Δgic1Δgic2 | – | – | n.d. |
| Δgin4 | – | n.d. | + |
| Δkel1 | – | + | + |
| Δkel2 | – | + | + |
| Δkel1Δkel2 | n.d. | n.d. | + |
| Δnum1 | n.d. | n.d. | + |
| Δpea2 | n.d. | n.d. | + |
| Δskm1 | – | n.d. | + |
| Δspa2 | n.d. | n.d. | + |
| Δste11 | – | n.d. | n.d. |
| Δste20 | + | – | + |
| Δzds1 | – | n.d. | + |
| Δzds2 | – | n.d. | + |
| Δyck1 | n.d. | n.d. | + |
n.d., not determined.
aDetermined at 23 and 30°C.
bCells were grown at 10°C.
cDetermined at 30°C, with the exception of cdc24-1, cdc42-1 and cdc42-118 cells.
dSee text for details.
We next tested whether LTE1 has a role in bud growth. Cells with defects in polarized growth often fail to organize the actin cytoskeleton (Chant, 1999). α-factor-synchronized wild-type and Δlte1 cells were grown at 10 and 30°C. At both temperatures, wild-type and Δlte1 cells grew buds with identical kinetics (data not shown) and formed polarized actin patches and cables (Figure 1D). These results exclude a major role of Lte1 in the organization of the actin cytoskeleton and polarized growth.
Δlte1 Δste20 cells may be unable to grow because the products of both genes facilitate ME. If this was the case, activation of the MEN should suppress the synthetically lethal phenotype of Δlte1 Δste20. Activation of the MEN would not be expected to suppress the lethality if the growth defect is caused by an overlapping function of both genes in cell polarity. Inactivation of the Bfa1–Bub2 GAP complex results in a hyperactive MEN (Pereira et al., 2002). Δbub2 Δlte1 Δste20 cells, but not Δlte1 Δste20 cells, grew at 30°C (Figure 1C, compare row 4 with 7). Similarly, the synthetically lethal phenotype of Δlte1 Δste20 cells was suppressed if the MEN was hyperactivated through overexpression of TEM1 or SPO12 (data not shown). Taken together, activation of the MEN suppressed the synthetic lethality of Δlte1 Δste20 cells, suggesting that these cells are defective in MEN activation. This, in turn, implies that STE20 has a role in ME.
To confirm a function of STE20 in the regulation of ME, we investigated the phenotype of Δlte1 Δste20 cells. To obtain conditional lethal Δlte1 Δste20 cells, we made use of the observation that Δbub2 Δlte1 Δste20 cells are viable (Figure 1C, row 7). Therefore, Gal1-BUB2 Δbub2 Δlte1 Δste20 cells in which BUB2 expression was repressed by glucose grew without defects (Figure 1E). However, induction of Gal1-BUB2 by the addition of galactose prevented growth of the Gal1-BUB2 Δbub2 Δlte1 Δste20 cells at 30°C. Controls established that Gal1-BUB2 expression did not inhibit growth of wild-type, Δlte1 or Δste20 cells.
We were now able to use this inducible BUB2 strain to determine the phenotypic consequences of the simultaneous loss of LTE1 and STE20 function. BUB2 was induced for 3 h in Gal1-BUB2 Δbub2 Δlte1 Δste20 CDC14-GFP cells at 30°C by the addition of galactose. Eighty-nine per cent of Gal1-BUB2 Δbub2 Δlte1 Δste20 cells grown in galactose arrested as large budded cells with separated DAPI staining regions (Figure 1F) and Cdc14–GFP in the nucleolus (Figure 1G), indicating a ME defect. In contrast, Δlte1 and Δste20 cells with Gal1-BUB2 behaved as Gal1-BUB2 cells. In these three cell types, only ∼25% of cells were in anaphase after growth in galactose medium (Figure 1F). Moreover, in contrast to the situation in Δlte1 Δste20 cells, Cdc14–GFP of wild-type, Δlte1 and Δste20 cells in anaphase was released from the nucleolus to trigger ME. The result of this experiment established that STE20 regulates ME.
Cdc24 and Cdc42 functionally overlap with Lte1
Ste20 interacts with the effector domain of GTP-bound Cdc42 (Peter et al., 1996). In turn, Cdc42 is converted into this active GTP-bound form by the GEF activity of Cdc24. These interdependencies raised the possibility that Cdc24 and Cdc42, like Ste20, functionally overlap with Lte1. To test this notion, we asked whether CDC24 and CDC42 show genetic interactions with LTE1. Δlte1, cdc24-1, cdc42-1 and cdc42-118 single mutants can all grow at 23 and 30°C. In contrast, deletion of LTE1 was lethal when combined with cdc24-1, cdc42-1 or cdc42-118 at either temperature (Figure 2A and B, row 4; Table I).
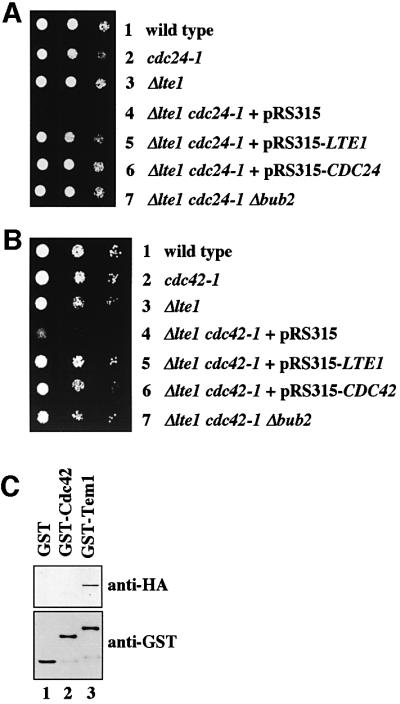
Fig. 2. CDC24 and CDC42 interact with LTE1. (A) Serial dilutions of cells of wild type (row 1), cdc24-1 (row 2), Δlte1 (row 3), Δlte1 cdc24-1 pRS316-LTE1 transformed with plasmid pRS315 (row 4), pRS315-LTE1 (row 5) or pRS315-CDC24 (row 6), and Δlte1 cdc24-1 Δbub2 (row 7) were grown on 5-FOA for 3 days at 23°C. (B) The indicated cell types were grown on 5-FOA for 3 days at 23°C. (C) Purified GST, GST–Cdc42 and GST–Tem1 bound to glutathione–Sepharose beads were incubated with a yeast lysate of 3HA-LTE1 cells. Eluted proteins were analysed by immunoblotting.
The genetic interaction of Δlte1 with cdc24-1 and cdc42 implied that Cdc24 and Cdc42 function in ME or that the GEF Lte1, like Cdc24, regulates the GTPase Cdc42. Two experiments were performed to discriminate between these possibilities. We first investigated whether the synthetically lethal phenotype of Δlte1 cdc24-1 and Δlte1 cdc42-1 cells was suppressed by the hyperactivation of the MEN through deletion of BUB2. Cells of Δlte1 cdc24-1 and Δlte1 cdc42-1 were able to grow when BUB2 was deleted (Figure 2A and B, row 7). Secondly, we tested whether Lte1 interacts with Tem1 but not Cdc42. For this approach, recombinant GST, GST–Tem1 and GST– Cdc42, purified from Escherichia coli, were bound to glutathione–Sepharose beads, which were incubated with a yeast extract of LTE1-3HA cells. Lte1-3HA interacted with GST–Tem1 (Figure 2C, lane 3), but not with GST (lane 1) or GST–Cdc42 (lane 2), indicating that Lte1 regulates Tem1 but not Cdc42. When taken together, these results are consistent with a function for Cdc24 and Cdc42 in ME.
Bud cortex binding of Lte1 is dependent on Cdc24 and Cdc42
Like Lte1, Cdc24, Cdc42 and Ste20 associate with the cortex of the bud (Peter et al., 1996; Toenjes et al., 1999; Bardin et al., 2000; Pereira et al., 2000). Cdc24, Cdc42 and Ste20 could therefore target Lte1 to the bud cortex in G1 of the cell cycle. These proteins could be required for either the initial binding of Lte1 to the bud cortex or to maintain the bud cortex interaction. To address the first possibility we determined the distribution of Lte1 in wild-type, cdc24-1 or cdc42 cells. For these localization studies, the chromosomal LTE1 was fused to GFP. The GFP-LTE1 gene fusion was under the control of the Gal1 promoter, which is induced by galactose but only weakly active when cells are grown in the presence of glucose. Gal1-GFP-LTE1 cells were able to grow at 10°C in galactose and glucose medium as wild-type cells. This demonstrated that GFP-LTE1 fulfilled the ME function of Lte1, even when only weakly expressed.
Logarithmically growing wild-type, cdc24-1, cdc42-1 and cdc42-118 cells were shifted to 37°C, and Gal1-GFP-LTE1 was simultaneously induced by the addition of galactose. Since Lte1 is not associated with the cell cortex of unbudded wild-type cells (Pereira et al., 2000), only cells with a bud were scored in the experiment. GFP–Lte1 of wild-type cells associated with the bud cortex (Figure 3A, 37°C and B). In contrast, GFP–Lte1 of budded cdc24-1 or cdc42-1 cells showed a diffuse staining at this restrictive temperature (Figure 3A and B, 37°C). In addition, for unknown reasons GFP–Lte1 of cdc42-1 cells often associated with tubular structures that were stained by DAPI and may represent mitochondria (data not shown). The plasmid-encoded wild-type genes complemented GFP–Lte1 mislocalization of cdc24-1 and cdc42-1 cells (data not shown). Mislocalization of GFP–Lte1 was not observed in cdc42-118 cells (Table I) (Kozminski et al., 2000), indicating that only a subset of Cdc42 defects affect Lte1 localization. GFP–Lte1 was expressed to similar levels in all cell types when Gal1-GFP-LTE1 was induced by the addition of galactose (Figure 3C). When cdc24-1 and cdc42-1 cells were shifted back to the permissive temperature, GFP–Lte1 associated with the bud cortex within 10 min, suggesting that the mislocalized GFP–Lte1 was able to bind to the bud cortex as soon as Cdc24 and Cdc42 became functional once again (Figure 3A and B, 23°C). A failure of GFP–Lte1 to associate with the bud cortex was also observed when cdc24-1 and cdc42-1 cells were first arrested in metaphase with nocodazole at 23°C, followed by a shift to 37°C and the simultaneous induction of Gal1-GFP-LTE1 by the addition of galactose (Figure 3D, 37°C). In contrast, GFP–Lte1 of wild-type cells bound under the same condition to the bud cortex, as did GFP–Lte1 when cdc24-1 and cdc42-1 were further incubated at 23°C (Figure 3D, 23°C). Finally, the role of Cdc42 in Lte1 cell cortex association was confirmed using the constitutively GTP-bound Cdc42G12A (Supplementary figure 9 available at The EMBO Journal Online). In conclusion, Cdc24 and Cdc42 were required for the initial binding of Lte1 to the bud cortex.

Fig. 3. Cdc24 and Cdc42 are required to target Lte1 to the bud cortex. (A) Wild-type, cdc24-1 and cdc42-1 cells with Gal1-GFP-LTE1 were shifted to 37°C simultaneously with the addition of galactose to induce Gal1-GFP-LTE1. GFP–Lte1 localization was determined by fluorescence microscopy 60 min after induction of the Gal1 promoter (panel 37°C). Cells were then shifted back to 23°C for 10 min and analysed (panel 23°C). (B) Quantification of (A). Cells with small and medium-sized buds were analysed for GFP–Lte1 bud cortex association after 60 min at 37°C and when cells were shifted back to 23°C. n > 100 (two experiments). (C) Cells of (A) grown at 23°C before the addition of galactose (lanes 1, 4 and 7), 60 min after the induction of Gal1-GFP-LTE1 at 37°C (lanes 2, 5 and 8) and when sifted back to 23°C (lanes 3, 6 and 9) were analysed by immunoblotting with anti-GFP antibodies. Tub2 was detected as loading control. (D) cdc24-1 and cdc42-1 cells with Gal1-GFP-LTE1 were arrested in YPRA in metaphase at 23°C with 15 µg/ml nocodazole. Gal1-GFP-LTE1 was either induced at 23°C by the addition of galactose or cells were shifted to 37°C simultaneously with the addition of galactose. Bud cortex localization of GFP–Lte1 is shown (n > 100). (E) GFP–Lte1 binds to the bud cortex in Δste20 cells. Gal1-GFP-LTE1 of Δste20 cells was induced for 60 min at 30°C. (F) Lte1 localization is not dependent on actin. Cells of Gal1-GFP-LTE1 in YPRA medium were incubated with (+) and without (–) Lat-A for 10 min at 30°C to depolymerize actin. Gal1-GFP-LTE1 was then expressed for 60 min by the addition of galactose. Cells were stained for F-actin by rhodamine–phalloidin and analysed by fluorescence microscopy. Bars: 5 µm.
To determine whether Cdc24 and Cdc42 are also required for maintaining Lte1 association with the bud cortex, we first expressed GFP–Lte1 in cdc24-1 and cdc42-1 cells incubated at the permissive temperature. GFP–Lte1 associated in 97 ± 2% of the cases with small and medium-sized buds of cdc24-1 and cdc42-1 cells. When cells were then shifted from 23 to 37°C for 60 min, a condition that was sufficient to inactivate cdc24-1 and cdc42-1 functions in the previous experiment, GFP–Lte1 remained associated with the bud cortex in 95 ± 4% (n = 125, two experiments) of the cells. This result indicated that Cdc24 and Cdc42 were not important to maintain the bud cortex association of GFP–Lte1.
We then investigated GFP–Lte1 localization in Δste20 cells. Upon the induction of the Gal1 promoter, GFP–Lte1 associated with the bud cortex of 97 ± 2% (n = 120, two experiments) of Δste20 Gal1-GFP-LTE1 cells with small and medium buds (Figure 3E). Thus, Ste20 does not play an essential role in Lte1 binding to the bud cortex.
Activated Cdc42 regulates polarized growth by rearranging the actin cytoskeleton (Li et al., 1995). Therefore, Cdc42-dependent localization of Lte1 might be mediated by actin. This possibility was tested by studying GFP–Lte1 localization in cells treated with the actin monomer sequestering drug latrunculin A (Lat-A) (Ayscough et al., 1997). Gal1-GFP-LTE1 cells were treated with Lat-A before or after the induction of the Gal1 promoter with galactose. In both cases, GFP–Lte1 associated with the bud cortex, even though the F-actin cytoskeleton was completely disrupted by Lat-A (Figure 3F; data not shown). We concluded that F-actin was not required to direct, or maintain, Lte1 at the bud cortex. The function of Cdc24 and Cdc42 in the initial binding of Lte1 to the bud cortex was, therefore, independent of their role in regulating the actin cytoskeleton.
Bud cortex association of Lte1 is dependent on the PAK kinase Cla4
Cdc24 and Cdc42 may directly localize Lte1 to the bud cortex or may do so indirectly by activating downstream effectors such as the formin Bni1, the PAKs Cla4 and Skm1, Gic1, Gic2, Zds1 or Zds2 (Gulli and Peter, 2001). To test whether these Cdc42 effectors were involved in the binding of Lte1 to the bud cortex, the localization of GFP–Lte1 was analysed in deletion mutants. GFP–Lte1 associated with the bud cortex of Δbni1, Δgic1, Δgic2, Δskm1, Δzds1 and Δzds2 cells (Table I) as in wild-type cells. In contrast, in Δcla4 cells with small and medium-sized buds, the GFP–Lte1 signal was dispersed in the cytoplasm (Figure 4A and B). This defect in GFP–Lte1 localization of Δcla4 cells was complemented by an episomal plasmid bearing CLA4, but not when the plasmid carried the kinase-dead CLA4K594R (Figure 4A and B). As shown by immunoblotting, the levels of GFP–Lte1 were similar in the three cell types (data not shown). The role of CLA4 in localizing Lte1 to the bud cortex was confirmed by demonstrating that the Cdc42G12V-induced cell cortex targeting of GFP–Lte1 in unbudded G1 cells was dependent on CLA4 (Supplementary figure 9). Thus, localization of Lte1 with the cortex of the bud was dependent on Cla4 kinase activity.

Fig. 4. Lte1 phosphorylation is CLA4 dependent. (A) GFP–Lte1 localization was determined as described in Figure 3E. (B) Quantification of (A). n > 100 (average of two experiments). Small and medium budded cells were analysed. (C) GFP–Lte1 of wild-type (THY279) and cdc12-6 cells (THY278) was induced for 1.5 h at 23°C. Then cells were incubated for 20 min at 37°C. GFP–Lte1 was analysed by fluorescence microscopy. (D) GFP–Lte1 localization of Δgin4 cells was determined as in Figure 3E. (E) Sec3–GFP of CLA4 and Δcla4 cells was determined as in Figure 3E. (F) Quantification of (E). n > 100 (two experiments). Bars: 5 µm.
The septin ring is required for the compartmentalization of cell cortex proteins. In cdc12-6 cells, which at the restrictive temperature completely disassemble the septin ring, bud cortex proteins such as Sec3, Sec5, Spa2 and Myo4p are mislocalized (Barral et al., 2000). Similarly, GFP–Lte1 became mislocalized when cdc12-6 cells were incubated for 20 min at 37°C (Figure 4C). A mild defect in septin ring morphology (but not assembly) has been reported for Δcla4 cells (Longtine et al., 2000). We investigated whether this septin defect is responsible for the failure of Lte1 to bind to the bud cortex in Δcla4 cells. Cells deleted in the Nim1-related kinase GIN4 have a similar septin morphology defect as Δcla4 cells (Longtine et al., 2000). However, in contrast to Δcla4 cells, GFP–Lte1 was still associated with the bud cortex of Δgin4 cells (Figure 4D; Table I). Moreover, the bud cortex localization of Sec3–GFP, which is disturbed in cdc12-6 cells (Barral et al., 2000) (data not shown), was not affected in Δcla4 cells (Figure 4E and F). When round, medium to large, budded CLA4 and Δcla4 cells were scored, ∼40% of cells showed Sec3–GFP at the bud cortex (Figure 4F). In addition, Δcla4 cells displayed elongated buds due to a delay in G2/M transition (Longtine et al., 2000). In ∼55% of these Δcla4 cells, Sec3–GFP was at the bud tip. Thus, the septin defect of Δcla4 cells does not cause mislocalization of Sec3. This result suggests that mislocalization of Lte1 in Δcla4 cells is not caused by the mild septin defect; rather, targeting Lte1 to the cortex is a specific function of Cla4.
Phosphorylation of Lte1 requires Cla4
Lte1 is a phosphoprotein (Bardin et al., 2000). We asked whether Lte1 phosphorylation could depend on Cla4. Lte1 phosphorylation was studied in α-factor-synchronized wild-type and Δcla4 cells in which LTE1 was fused to protein A (LTE1-ProA) to make it easily detectable by immunoblotting. In wild-type cells, Lte1–ProA became phosphorylated at multiple sites with the emergence of buds in late G1 of the cell cycle (Bardin et al., 2000) (Figure 5A, t = 30–40) as indicated by the appearance of Lte1–ProA phospho-forms that migrated more slowly than the non-phosphorylated Lte1–ProA of α-factor-arrested cells (Figure 5A, t = 0). Most of Lte1–ProA became dephosphorylated when cells had grown sufficiently to accumulate large buds (Figure 5A, t = 90–100). In contrast, no Lte1–ProA phosphorylation was seen at any cell cycle stage in Δcla4 cells.

Fig. 5. Phosphorylation of Lte1 is CLA4 dependent. (A) Phos phorylation of Lte1 is disturbed in Δcla4 cells. α-factor-synchronized wild-type and Δcla4 cells were analysed for budding (top) and Lte1–ProA phosphorylation (bottom). (B) Lte1–ProA of CLA4 and Δcla4 cells of (A) (t = 60–80) was enriched with IgG beads and treated with buffer (lanes 1 and 4), alkaline phosphatase (lanes 2 and 5) and alkaline phosphatase with inhibitor (lanes 3 and 6). Samples were analysed by immunoblotting. (C) Wild-type (lane 1), Δcla4 (lane 2) and Δste20 cells (lane 3) with LTE1-ProA were arrested in metaphase with 15 µg/ml nocodazole at 30°C. Cell extracts were analysed for Lte1 phosphorylation.
To ensure that Lte1 was not phosphorylated in Δcla4 cells, Lte1–ProA of small budded wild-type and Δcla4 cells (Figure 5A, t = 60–80) was enriched with IgG beads. The purified Lte1–ProA was incubated with buffer, phosphatase or phosphatase in the presence of inhibitors. While the mobility of Lte1–ProA of CLA4 cells was changed when incubated with phosphatase (Figure 5B, lane 2) due to dephosphorylation of Lte1, such a phosphatase-induced mobility shift was not observed in Δcla4 cells (lane 5). This result confirmed that all phosphorylation events of Lte1 that contribute to the mobility shift were dependent on Cla4.
Given the MEN function of Ste20, it was possible that Ste20 could also be participating in Lte1 phosphorylation. We arrested wild-type, Δcla4 and Δste20 cells with LTE1-ProA in metaphase using the microtubule-depolymerizing drug nocodazole to assess this possibility. Lte1–ProA was hyper-phosphorylated in wild-type and Δste20 cells, but not in Δcla4 cells (Figure 5C). In summary, these results suggest a role for Cla4, but not Ste20, in Lte1 phosphorylation.
A role of Cla4 in ME
If Cla4-mediated binding and phosphorylation of Lte1 are essential for the activation of Lte1, we would expect Δcla4 cells to show the same ME defect at 10°C as is seen in Δlte1 cells. Moreover, as is the case for Δlte1 cells, this ME defect should be suppressed by hyperactivation of the MEN through overexpression of TEM1 or deletion of BUB2. Indeed, Δcla4 cells failed to grow at 10°C (Figure 6A, row 2) and this growth defect was suppressed by high gene dosage of TEM1 and by Δbub2 (rows 5 and 7).

Fig. 6. Cla4 functions in ME. (A) Cells lacking CLA4 are cold sensitive. Serial dilutions of the indicated cell types were grown for 10 days at 10°C on YPAD plates. All cell types grew equally well at 30°C (data not shown). (B) Cells of Δcla4 arrest at the end of anaphase. α-factor-synchronized wild-type and Δcla4 cells with CDC14-GFP were analysed for cell cycle progression at 10°C. Large buds and nucleolar Cdc14–GFP were determined (n > 100). (C) Δcla4 cells of (B) incubated for 6 h at 10°C were analysed by indirect immunofluorescence. DNA was stained with DAPI. The bud is elongated due to a function of Cla4 in polarized growth. (D) Cells of (B) were analysed after 8 h at 10°C. Cartoons are as in Figure 1F. (E) The indicated cell types were incubated for 9 h at 10°C, fixed and DNA stained with DAPI. Phase contrast with DAPI immunofluorescence. (F) Quantification of (E). n > 100. Bars: 5 µm.
To ensure that the failure of Δcla4 cells to grow at 10°C was indeed due to a defect in ME, we monitored cell cycle progression of α-factor-synchronized wild-type and Δcla4 cells with CDC14-GFP at 10°C. As expected, wild-type cells released Cdc14–GFP at the beginning of anaphase and exited mitosis to start a new cell cycle (as indicated by the decrease of cells with large buds after 6 h, Figure 6B). Δcla4 cells progressed through interphase as wild-type cells (Figure 6B), but then most Δcla4 cells stopped cell cycle progression in late anaphase. The late anaphase arrest is indicated by the separated DAPI staining regions (Figure 6D, cells of B after 8 h), the long anaphase spindle and Cdc14–GFP entrapped in the nucleolus (Figure 6B and C). Thus, Δcla4 cells are defective in ME at 10°C.
The cold-sensitive ME defect of Δcla4 cells may be a direct reflection of the failure to localize and activate Lte1. In this case, cells from which both Δcla4 and Δlte1 have been deleted should have the same phenotype as cells with the singly deleted CLA4 or LTE1, because the ME defect in all three mutants resulted from lack of Lte1 function. Indeed, Δcla4 Δlte1 cells were similarly defective in ME as Δlte1 or Δcla4 cells. In addition, overexpression of CLA4 did not suppress the cold-sensitive growth defect of Δlte1 cells (data not shown). This supports the notion that the role played by Cla4 in MEN activation is not independent of Lte1, as is the case for Ste20, rather, it indicates a function of Cla4 upstream of, or alongside, Lte1.
Some of the phenotypes of Δcla4 cells, like the elongated bud, are dependent on the Wee1-like kinase Swe1, which delays entry into mitosis through Cdk1 phosphorylation as part of the morphogenesis checkpoint (Longtine et al., 2000). We investigated whether the late anaphase arrest of Δcla4 cells required Swe1. Δcla4 Δswe1 cells resembled Δcla4 cells in being cold sensitive for growth and arresting in late anaphase with separated DAPI staining regions when incubated for 9 h at 10°C (Figure 6E and F). About 20% of the cell cycle-arrested Δcla4 cells showed elongated buds due to the Swe1-dependent delay in mitotic entry. Consistently, cells with elongated buds were not observed in Δcla4 Δswe1 cells (Figure 6E). Together, SWE1 is not involved in the late anaphase arrest of Δcla4 cells.
The kelch domain proteins Kel1 and Kel2 negatively regulate the MEN
Cdc24 and Cdc42 were only required for the initial binding of Lte1 to the bud cortex, but not for the subsequent anchorage of Lte1. Therefore, it was likely that Lte1 interacted with additional proteins at the cell cortex. We tested mutants deleted in BUD6, KEL1, KEL2, NUM1, PEA2, SPA2 and YCK1, which all encode cortical proteins (Chant, 1999), for their involvement in Lte1 localization and genetic interaction with Δlte1. In all mutant cells, GFP–Lte1 localized with the bud cortex (Table I). We noticed that deletion of KEL1 or KEL2 suppressed the cold-sensitive growth defect of Δlte1 cells (Figure 7A, rows 5 and 6). A similar suppression of the Δlte1 phenotype was observed when the inhibitory Bfa1–Bub2 GAP was inactivated by the deletion of BUB2 (Figure 7B). This indicated that, like the Bfa1–Bub2 GAP complex, Kel1 and Kel2 have an inhibitory influence on the MEN.


Fig. 7. Kel1 and Kel2 are present in complexes with Lte1 and Tem1. (A) Deletion of KEL1 or KEL2 suppresses the cold-sensitive growth defect of Δlte1 cells. Serial dilutions of the indicated cell types were grown on YPDA plates for 10 days at 10°C. (B) Deletion of KEL1 suppresses the ME defect of Δlte1 cells. The indicated cells were analysed as in Figure 6B. (C) Co-immunoprecipitation of Tem1 and Lte1 with Kel1 and Kel2. Cells of KEL1-3Myc (lanes 1–4) or KEL2-3Myc (lanes 5–8) without additional tag (lanes 1 and 5) or with additional LTE1-3HA (lanes 2 and 6), TEM1-3HA (lanes 3 and 7) and TEM1-3HA Δlte1 (lanes 4 and 8) were grown at 30°C. Anti-HA immunoprecipitates were analysed by immunoblotting. The arrowhead indicates a protein band that was non-specifically precipitated with the anti-HA beads and migrated slightly faster than Lte1-3HA (arrow).
We asked whether the deletion of KEL1 suppressed the ME defect of Δlte1 cells at 10°C. For this experiment, the formation of large buds and release of the nucleolar Cdc14 were used as markers for cell cycle progression and ME of α-factor-synchronized wild-type, Δlte1, Δkel1, Δbub2, Δkel1 Δlte1 and Δbub2 Δlte1 cells that contained CDC14-GFP. As expected, Δlte1 cells arrested in late anaphase as large budded cells with Cdc14–GFP in the nucleolus (Figure 7B) and separated DAPI staining regions (data not shown). In contrast, Δkel1, Δbub2, Δkel1 Δlte1 and Δbub2 Δlte1 cells progressed though the cell cycle and exited mitosis with similar kinetics to wild type (Figure 7B). The outcome of this experiment suggested that, like the Bfa1–Bub2 GAP, Kel1 and Kel2 oppose the activating function of Lte1.
Kel1 and Kel2 may regulate ME through binding to Lte1 and Tem1. Immunoprecipitation experiments were performed with strains in which Lte1 and Tem1 were tagged with the haemagglutinin (HA) and KEL1 and KEL2 with the Myc epitope. Co-immunoprecipitation of Kel1-3Myc and Kel2-3Myc, but not of Tem1 with Lte1-3HA, was observed (Figure 7C, lanes 2 and 6), indicating complexes containing Kel1, Kel2 and Lte1. Moreover, Kel1-3Myc and Kel2-3Myc were detected in the Tem1-3HA immunoprecipitation (lanes 3 and 7). These anti-HA immunoprecipitations were specific since they were not observed in cells without an HA-tagged protein but with KEL1-3Myc (lane 1) or KEL2-3Myc (lane 5). The failure to detect Tem1 in the Lte1-3HA immunoprecipitation indicates that the interaction of Lte1 and Tem1, which was detected by in vitro binding (Figure 2C), is either relatively weak or only transient.
Tem1 may bind to Kel1 and Kel2 independently of Lte1. If this is the case, deletion of LTE1 should not affect the ability to co-immunoprecipitate Kel1 or Kel2 with Tem1. Consistently, Kel1-3Myc and Kel2-3Myc were still detected in the Tem1-3HA immunoprecipitations from Δlte1 KEL1-3Myc TEM1-3HA and Δlte1 KEL2-3Myc TEM1-3HA cells (Figure 7C, lanes 4 and 8). Thus, the interaction between Tem1 and Kel1 or Kel2 was independent of Lte1. When taken together, these data establish that Kel1 and Kel2 interact with both Lte1 and Tem1, and have an inhibitory effect on the MEN.
Discussion
Most MEN components are associated with the SPB. In contrast, Lte1, which acts in the early stages of MEN activation, is localized at the bud cortex (Bardin et al., 2000; Pereira et al., 2000). This raised the possibility that proteins that regulate and target Lte1 to the bud cortex may also be cortical. In addition, cell cortex proteins may regulate ME independently of Lte1, ensuring that in the absence of Lte1, cells delay ME until the spindle is correctly positioned along the mother to bud axis. In this study, we report that the cell cortex proteins Cdc24, Cdc42, Cla4, Ste20, Kel1 and Kel2 regulate ME in at least three different ways (Figure 8).

Fig. 8. Model for how the bud cortex regulates ME. See Discussion for details.
Cdc24, Cdc42 and Cla4 target Lte1 to the bud cortex
The Rho-type GTPase Cdc42 is a key regulator of cell polarity in both yeast and higher eukaryotes. In yeast, Cdc42 localizes to the plasma membrane around the entire cell periphery and at internal membranes throughout the cell cycle. However, Cdc42 clusters at the incipient bud site prior to bud emergence in G1 and at the mother-bud neck region in late anaphase (Richman et al., 2002). Central to the initiation of actin polymerization is the local activation of Cdc42 through the GEF Cdc24. In the active GTP-bound state, Cdc42 then interacts with its downstream effectors such as the PAKs Cla4 and Ste20, which in turn control the assembly of actin filaments and their organization into complex structures (Gulli and Peter, 2001).
We show that bud cortex binding of the MEN component Lte1 is dependent upon the function of Cdc24, Cdc42 and the downstream effector Cla4 (Figures 3 and 4). Moreover, a constitutively active Cdc42G12V was able to target Lte1 prematurely to the cell cortex of unbudded G1 cells in dependence of Cla4 (Supplementary figure 9). Although GFP–Lte1 bud cortex localization is dependent on a functional septin ring (Figure 8), the mild septin defect of Δcla4 cells (Longtine et al., 2000) is not the cause for the mislocalization of GFP–Lte1. First, GFP–Lte1 is still associated with the bud cortex of Δgin4 cells, which have a similar septin defect to Δcla4 cells. Secondly, Sec3 associates with the bud cortex in dependence of the septin ring (Barral et al., 2000). We show that Sec3 is still attached to the bud cortex of Δcla4 cells. Therefore, we suggest that the Cdc24–Cdc42–Cla4 cascade has a direct role in localizing Lte1 to the bud cortex.
Neither actin nor microtubules were necessary for the association of Lte1 with the bud cortex (Figure 3F; our unpublished data). It is, therefore, likely that Lte1 diffuses to the plasma membrane of the emerging bud, where it becomes targeted to the cortex. A likely interpretation of our results is that the Cdc24–Cdc42–Cla4 cascade is involved in the initial targeting event of Lte1 to the bud cortex. The molecular mechanisms by which Cdc24, Cdc42 and Cla4 target Lte1 to the bud cortex remain to be established. One possibility is that Cla4-mediated phosphorylation of Lte1 may trigger a conformational change in Lte1 that allows bud cortex binding. Our findings further indicate that Cdc24 and Cdc42 are only required for the initial binding of Lte1 to the bud cortex. This suggests that after the initial targeting event, Cdc24, Cdc42 and Cla4 do not seem to exercise any role as adaptor proteins linking Lte1 to the cortex.
Several observations suggest that Cla4 participates in the activation of Lte1 and that this is the primary function of Cla4 in ME. Like Δlte1 cells, Δcla4 cells showed a ME defect at 10°C that was suppressed by high gene dosage of TEM1 or deletion of BUB2 (this study). The lack of genetic interactions between CLA4 and LTE1, and the observation that the ME defect arising from loss of either gene were not additive, suggest that the main function of Cla4 in MEN regulation is the activation of Lte1. Thus, it is likely that the Lte1 protein of Δcla4 cells, which is mislocalized in a non-phosphorylated form, is inactive and accounts for the ME defect of Δcla4 cells. Considering the role of protein phosphorylation in the activation of some GEFs (Gulli and Peter, 2001), it is possible that Cla4-mediated phosphorylation of Lte1 leads to its activation.
Lee et al. (2001) reported that Lte1 phosphorylation is dependent on polo kinase Cdc5. However, in our hands, Lte1 phosphorylation was not affected in synchronized cultures of cdc5-1 or Cdc5-depleted Gal1-CDC5 cells. This indicates that phosphorylation of Lte1 is predominantly Cla4 dependent.
Evidence for an Lte1-independent role for Cdc24, Cdc42 and Ste20 in ME
Ste20 and Cla4 belong to the PAK-like kinases that become activated through Cdc42. These closely related kinases have overlapping functions in actin polarization (Cvrckova et al., 1995; Peter et al., 1996; Benton et al., 1997; Tjandra et al., 1998; Holly and Blumer, 1999). However, in contrast to Cla4, Ste20 was not required to localize Lte1 to the bud cortex, nor was it essential for Lte1 phosphorylation and activation (Δste20 cells are not cold sensitive). In addition, only STE20 but not CLA4 interacted with LTE1 in genetic assays. The cdc24-1 and cdc42-1 alleles, like Δste20, showed genetic interactions with Δlte1 and, like Δcla4, were both defective for the initial binding of Lte1 to the bud cortex. The dual behaviour of Cla4, Cdc24, Cdc42 and Ste20 suggests that these proteins function in at least two ways. First, Cdc24 and Cdc42 use the effector Cla4 to target Lte1 to the bud cortex and activate Lte1. Secondly, Cdc24 and Cdc42 activate Ste20 to fulfil an essential function that overlaps with Lte1. If this is the case, we would expect to find cdc42 alleles, which are only defective for one of the two functions. Indeed, in cdc42-118 cells Lte1 was targeted to the bud cortex in a wild-type manner, but these cells required STE20 for survival (Table I).
Our results are consistent with a role of CDC24, CDC42 and STE20 in ME (Figure 8). Δlte1 was only synthetically lethal when combined with cdc24-1, cdc42 and Δste20, but not with other cell polarity mutants that result in defects in actin organization, such as Δcla4, Δbni1, Δbud6 and Δgic1 Δgic2 (Figures 1 and 2; Table I). This emphasizes that the Δlte1 Δste20 lethality is due to a specific defect in the regulation of ME rather than a secondary consequence of defects in actin polarization. Δlte1 also shows a synthetically lethal phenotype with genes involved in MEN activation, such as separase ESP1 (Stegmeier et al., 2002), polo kinase CDC5 (our unpublished data) and the MEN component MOB1 (Luca and Winey, 1998). A function of Cdc24, Cdc42 and Ste20 in ME is further supported by the suppression of lethality in cdc24-1 Δlte1, cdc42-1 Δlte1 and Δste20 Δlte1 cells by the hyperactivation of the MEN either through Bfa1–Bub2 complex inactivation or TEM1 or SPO12 overexpression. In addition, overexpression of STE20 not only suppressed the cold-sensitive growth defect of Δlte1 cells, but also the failure of Δlte1 cells to release Cdc14 from the nucleolus at 10°C (Figure 1B). The latter phenotype suggests that Ste20 triggers Cdc14 release independently of Lte1. Moreover, Δlte1 Δste20 cells displayed a ME defect under conditions in which neither mutant on its own had any defect (Figure 1E–G). Finally, it is important to note that the fission yeast PAK-like kinase Sid1 is part of the septum initiation network (SIN), which is similar in composition and function to the MEN (Balasubramanian et al., 2000).
At present, it is unclear how Cdc24, Cdc42 and Ste20 regulate ME. Overexpression of STE20 in cells arrested in metaphase with nocodazole, when the MEN is inactive, did not trigger the release of Cdc14 from the nucleolus (our unpublished data). This contrasts with the MEN-independent release of Cdc14 from the nucleolus upon overproduction of separase Esp1 (Stegmeier et al., 2002). These results are consistent with a model in which Ste20 requires an active MEN to trigger Cdc14 release. One attractive possibility is that Ste20 regulates the polo kinase Cdc5, which in turn controls the MEN through inactivation of the inhibitory Bfa1–Bub2 complex. PAK kinase has been shown to activate polo kinase in other systems (Ellinger-Ziegelbauer et al., 2000) and the role of Cdc5 in regulating the Bfa1–Bub2 complex is well established (Lee et al., 2001).
Kel1 and Kel2 negatively regulate the MEN
Kel1 and Kel2 are conserved kelch domain proteins associated with the bud cortex that are involved in cell fusion and morphology (Philips and Herskowitz, 1998). Co-immunoprecipitation revealed that both Kel1 and Kel2 form complexes with Lte1 (this study). Consistent with our data, a complex containing Kel1 and Lte1 has been purified, indicating a direct interaction between the two proteins (Gavin et al., 2002). Kel1 and Kel2 may act as adaptor proteins that target Lte1 to the bud cortex. However, binding of Lte1 to the bud cortex in mutants where both KEL1 and KEL2 have been deleted make such a model unlikely. Instead, the observation that deletion of KEL1 or KEL2 mimicked the inactivation of the inhibitory Bfa1–Bub2 complex in suppressing the cold-sensitive ME defect of Δlte1 cells supports the notion that Kel1 and Kel2 are negative regulators of ME (Figure 8). In this respect, it is interesting that Kel1 and Kel2 co-immunoprecipitated with Tem1 and that this interaction was independent of Lte1. Kel1 and Kel2 therefore bind the GTPase Tem1 and the GEF Lte1 independently, possibly preventing their association. This separation and the sequestration of Tem1 by Kel1 and Kel2 may prevent premature activation of Tem1 by Lte1 or by another mechanism.
Do Cdc24, Cdc42, PAKs and Kel homologues have a role in regulating ME in other organisms?
Kel1 and Kel2 are related to the fission yeast Tea1, which interacts with the plus end of microtubules and also functions in polarized growth (Mata and Nurse, 1997). It will be important to test whether Tea1 regulates the SIN network in fission yeast (Balasubramanian et al., 2000).
Cdc24, Cdc42 and PAK kinases are conserved from yeast to mammalian cells. Human and yeast CDC42 are 80% identical and functionally interchangeable, suggesting that the key functions of Cdc42 are highly conserved (Shinjo et al., 1990). It is important to note that in higher eukaryotes Rho GTPases have been implicated in the regulation of late events in the cell cycle around the time of mitotic exit, such as central spindle formation and cytokinesis (Tatsumoto et al., 1999; Jantsch-Plunger et al., 2000).
Materials and methods
Growth conditions and yeast strains
Yeast strains were grown in yeast extract, peptone, dextrose medium containing 100 mg/l adenine (YPAD medium). For the induction of the Gal1 promoter, yeast cells were grown in yeast extract, peptone, 3% raffinose, 100 mg/ml adenine medium (YPRA). Galactose (2%) was added to induce the Gal1 promoter. Yeast strains were derivatives of S288c (Sikorski and Hieter, 1989) (Table II) with the exception of THY278 and THY279, which were derived from YEF473A (gift of Dr D.Lew). Yeast strains were constructed using PCR-amplified cassettes (Longtine et al., 1998).
Table II. Yeast strains and plasmids.
| Name | Genotype—construction | Source or reference |
|---|---|---|
| Yeast strains | ||
| CRY7 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 LTE1-ProA-KanMX6 Δste20::klTRP1a | This study |
| CRY9 | MATa ura3-52 his3Δ200 leu2Δ1 Δlte1::KanMX6 Δbub2::His3MX6 cdc24-1 | This study |
| ESM356 | MATa ura3-52 trp1Δ63 his3Δ200 leu2Δ1 | This study |
| ESM1192 | MATa ura3-52 his3Δ200 leu2Δ1 Δlte1::KanMX6 | This study |
| ESM1362 | MATa ura3-52 trp1Δ63 his3Δ200 leu2Δ1 CDC14-GFP-klTRP1 | This study |
| GPY104 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 LTE1-ProA-KanMX6 | This study |
| GPY405 | MATa ura3-52 trp1Δ63 his3Δ200 leu2Δ1 Δbub2::klTRP1 CDC14-GFP-His3MX6 | This study |
| GPY406 | MATa ura3-52 trp1Δ63 his3Δ200 leu2Δ1 Δlte1::KanMX6 Δbub2::klTRP1 CDC14-GFP-His3MX6 | This study |
| GPY413 | MATa ura3-52 his3Δ200 leu2Δ1 Δlte1::KanMX6 CDC14-GFP-His3MX6 | This study |
| THY5 | MATa ura3-52 trp1Δ63 his3Δ200 leu2Δ1 KanMX6-Gal1-GFP-LTE1 | Pereira et al. (2000) |
| THY72 | MATa ura3-52 his3Δ200 leu2Δ1 Δlte1::KanMX6 cdc24-1 pSM903 | This study |
| THY87 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 Δlte1::KanMX6 Δste20::klTRP1 pSM903 | This study |
| THY91 | MATa ura3-52 trp1Δ63 his3Δ200 leu2Δ1 Δste20::klTRP1 KanMX6-Gal1-GFP-LTE1 | This study |
| THY94 | MATa ura3-52 trp1Δ63 his3Δ200 leu2Δ1 Δste20::klTRP1 | This study |
| THY95 | MATa ura3-52 his3Δ200 leu2Δ1 KanMX6-Gal1-GFP-LTE1 cdc24-1 | This study |
| THY106 | MATa ura3-52 his3Δ200 leu2Δ1 KanMX6-Gal1-GFP-LTE1 cdc42-1 | This study |
| THY121 | MATa ura3-52 his3Δ200 leu2Δ1 cdc42-1 | This study |
| THY122 | MATa ura3-52 his3Δ200 leu2Δ1 cdc24-1 | This study |
| THY136 | MATa ura3-52 trp1Δ63 his3Δ200 leu2Δ1 Δkel1::klTRP1 | This study |
| THY137 | MATa ura3-52 trp1Δ63 his3Δ200 leu2Δ1 Δkel2::klTRP1 | This study |
| THY142 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 KEL1-3Myc-His3MX6 TEM1-3HA-KanMX6 | This study |
| THY145 | MATa ura3-52 his3Δ200 leu2Δ1 Δlte1::KanMX6 cdc42-1 pSM903 | This study |
| THY147 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 KEL1-3Myc-His3MX6 | This study |
| THY148 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 KEL2-3Myc-His3MX6 | This study |
| THY149 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 KEL2-3Myc-His3MX6 TEM1-3HA-KanMX6 | This study |
| THY150 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 KEL2-3Myc-His3MX6 LTE1-3HA-KanMX6 | This study |
| THY157 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 KEL1-3Myc-His3MX6 LTE1-3HA-KanMX6 | This study |
| THY163 | MATa ura3-52 trp1Δ63 his3Δ200 leu2Δ1 Δkel1::klTRP1 Δlte1::KanMX6 | This study |
| THY164 | MATa ura3-52 trp1Δ63 his3Δ200 leu2Δ1 Δkel2::klTRP1 Δlte1::KanMX6 | This study |
| THY170 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 Δcla4::klTRP1 KanMX6-Gal1-GFP-LTE1 | This study |
| THY172 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 Δcla4::klTRP1 LTE1-ProA-KanMX6 | This study |
| THY175 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 KEL1-3Myc-His3MX6 | This study |
| TEM1-3HA-KanMX6 Δlte1::klTRP1 | ||
| THY176 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 KEL2-3Myc-His3MX6 | This study |
| TEM1-3HA-KanMX6 Δlte1::klTRP1 | ||
| THY190 | MATa ura3-52 trp1Δ63 his3Δ200 leu2Δ1 Δkel1::klTRP1 CDC14-GFP-His3MX6 | This study |
| THY191 | MATa ura3-52 trp1Δ63 his3Δ200 leu2Δ1 Δkel1::klTRP1 Δlte1::KanMX6 CDC14-GFP-His3MX6 | This study |
| THY192 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 Δcla4::klTRP1 | This study |
| THY196 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 Δbub2::His3MX6 | |
| Δlte1::KanMX6 Δste20::klTRP1 | This study | |
| THY204 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 Δcla4::klTRP1 CDC14-GFP-His3MX6 | This study |
| THY205 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 Δcla4::klTRP1 Δbub2::His3MX6 | This study |
| THY206 | MATa ura3-52 his3Δ200 leu2Δ1 Δbub2::His3MX6 Δlte1::KanMX6 cdc42-1 | This study |
| THY216 | MATa ura3-52 his3Δ200 leu2Δ1-Gal1-STE20-LEU2 Δlte1::KanMX6 CDC14-GFP-His3MX6 | This study |
| THY217 | MATa ura3-52 lys2-801 ade2-101 his3Δ200 leu2Δ1 CDC14-GFP-His3MX6 | This study |
| THY230 | MATa ura3-52 trp1Δ63 his3Δ200 leu2Δ1 Δgin4::klTRP1 KanMX6-Gal1-GFP-LTE1 | This study |
| THY234 | MATa ura3-52-Gal1-BUB2-URA3 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 Δlte1::KanMX6 | This study |
| Δste20::klTRP1 Δbub2::His3MX6 | ||
| THY235 | MATa ura3-52-Gal1-BUB2-URA3 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 Δlte1::KanMX6 | This study |
| Δste20::klTRP1 Δbub2::His3MX6 CDC14-GFP-LEU2 | ||
| THY236 | MATa ura3-52-Gal1-BUB2-URA3 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 | This study |
| THY237 | MATa ura3-52-Gal1-BUB2-URA3 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 Δlte1::KanMX6 | This study |
| THY238 | MATa ura3-52-Gal1-BUB2-URA3 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 Δste20::klTRP1 | This study |
| THY278 | MATa ura3-52 lys2 trp1 his3 leu2 KanMX6-Gal1-GFP-LTE1 cdc12-6 | This study |
| THY279 | MATa ura3-52 lys2 trp1 his3 leu2 KanMX6-Gal1-GFP-LTE1 | This study |
| THY285 | MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 Δcla4::klTRP1 Δswe1::His3MX6 | This study |
| YPH499 |
MATa ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 |
Sikorski and Hieter (1989) |
| Plasmids | ||
| pKA112 | pRS315 carrying CDC42 | K.Ayscough |
| pBB1 | pRS315 carrying Gal1-CDC42 | Benton et al. (1997) |
| pBB2 | pRS315 carrying Gal1-CDC42G12V | Benton et al. (1997) |
| pBB3 | pRS315 carrying Gal1-CDC42D118A | Benton et al. (1997) |
| pSM771 | pRS426 carrying TEM1 | This study |
| pSM903 | pRS316 carrying LTE1 | This study |
| pSM919 | pRS315 carrying LTE1 | This study |
| pSM923 | pRS426 carrying LTE1 | This study |
| pSM1033 | pRS316 carrying CLA4K594R-9Myc | This study |
| pTH20 | pRS426 carrying SPO12 | This study |
| pTH31 | pRS426 carrying STE20 | This study |
| pTH35 | pRS315 carrying STE20 | This study |
| pTH37 | pRS315 carrying CDC24 | This study |
| pTH102 | pRS316 carrying CLA4 | This study |
| pTH127 | pRS316 carrying SEC3-GFP | This study |
aklTRP1 encodes the Kluyveromyces lactis TRP1 gene.
In vitro binding experiments
GST, GST–Cdc42 and GST–Tem1 were expressed in E.coli BL21 and purified using glutathione–Sepharose (Amersham Pharmacia). The GST proteins bound to glutathione–Sepharose were presented to a yeast lysate of 3HA-LTE1 cells for 60 min at 4°C in UB buffer (0.05 M HEPES pH 7.5, 0.1 M KCl, 3 mM MgCl2, 1 mM EGTA, 1 mM DTT, 1 mM PMSF, 0.2% Triton X-100, 1 mM GTP). After three washes with UB buffer, the associated proteins were eluted with sample buffer and analysed by immunoblotting.
Antibodies, immunoprecipitations, indirect immunofluorescence, GFP–Lte1 localization and actin staining with phalloidin
Monoclonal mouse anti-HA (12CA5), mouse anti-Myc (9E10) or rabbit anti-GFP antibodies were obtained from Boehringer Ingelheim. Mouse monoclonal anti-α-tubulin antibody WA3, rabbit anti-Tem1 and anti-Tub2p antibodies have been described previously (Pereira et al., 2000). Secondary antibodies were from Jackson Research Laboratories. Lte1-3HA and Tem1-3HA were immunoprecipitated from yeast cell extracts using anti-HA antibodies covalently coupled to ProA–Sepharose beads. Indirect immunofluorescence was performed using a standard protocol. GFP-labelled cells were analysed by fluorescence microscopy after fixing the cells with paraformaldehyde (Pereira et al., 2000). DNA was stained with 4′,6-diamidino-2-phenylindole (DAPI). F-actin of yeast cells was stained with rhodamine–phalloidin (Ayscough et al., 1997).
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We thank Drs K.Ayscough, F.Cross, D.Drubin, D.Lew and K.Nasmyth for plasmids, yeast strains and Lat-A. We are grateful to Dr I.Hagan for comments on the manuscript. The work of E.S. is supported by Cancer Research UK.
References
- Adames N.R., Oberle,J.R. and Cooper,J.A. (2001) The surveillance mechanism of the spindle position checkpoint in yeast. J. Cell Biol., 153, 159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayscough K.R., Stryker,J., Pokala,N., Sanders,N., Crews,P. and Drubin,D.G. (1997) High rates of actin filament turnover in budding yeast and roles for actin in establishment and maintenance of cell polarity revealed using the actin inhibitor latrunculin-A. J. Cell Biol., 137, 399–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian M.K., McCollum,D. and Surana,U. (2000) Tying the knot: linking cytokinesis to the nuclear cycle. J. Cell Sci., 113, 1503–1513. [DOI] [PubMed] [Google Scholar]
- Bardin A.J., Visintin,R. and Amon,A. (2000) A mechanism for coupling exit from mitosis to partitioning of the nucleus. Cell, 102, 21–31. [DOI] [PubMed] [Google Scholar]
- Barral Y., Mermall,V., Mooseker,M.S. and Snyder,M. (2000) Compartmentalization of the cell cortex by septins is required for maintenance of cell polarity in yeast. Mol. Cell, 5, 841–851. [DOI] [PubMed] [Google Scholar]
- Benton B.K., Tinkelenberg,A., Gonzalez,I. and Cross,F.R. (1997) Cla4p, a Saccharomyces cerevisiae Cdc42p-activated kinase involved in cytokinesis, is activated at mitosis. Mol. Cell. Biol., 17, 5067–5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chant J. (1999) Cell polarity in yeast. Annu. Rev. Cell Dev. Biol., 15, 365–391. [DOI] [PubMed] [Google Scholar]
- Cvrckova F., Devirgilio,C., Manser,E., Pringle,J.R. and Nasmyth,K. (1995) Ste20-like protein-kinases are required for normal localization of cell growth and for cytokinesis in budding yeast. Genes Dev., 9, 1817–1830. [DOI] [PubMed] [Google Scholar]
- Ellinger-Ziegelbauer H., Karasuyama,H., Yamada,E., Tsujikawa,K., Todokoro,K. and Nishida,E. (2000) Ste20-like kinase (SLK), a regulatory kinase for polo-like kinase (Plk) during the G2/M transition in somatic cells. Genes Cells, 5, 491–498. [DOI] [PubMed] [Google Scholar]
- Gavin A.C. et al. (2002) Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature, 415, 141–147. [DOI] [PubMed] [Google Scholar]
- Gulli M.-P. and Peter,M. (2001) Temporal and spatial regulation of Rho-type guanine-nucleotide exchange factors: the yeast perspective. Genes Dev., 15, 365–379. [DOI] [PubMed] [Google Scholar]
- Holly S.P. and Blumer,K.J. (1999) PAK-family kinases regulate cell and actin polarization throughout the cell cycle of Saccharomyces cerevisiae.J. Cell Biol., 147, 845–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jantsch-Plunger V., Gönczy,P., Romano,A., Schnabel,H., Hamill,D., Schnabel,R., Hyman,A.A. and Glotzer,M. (2000) CYK-4: a Rho family GTPase activating protein (GAP) required for central spindle formation and cytokinesis. J. Cell Biol., 149, 1391–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspersen S.L., Charles,J.F., Tinker-Kulberg,R.L. and Morgan,D.O. (1998) A late mitotic regulatory network controlling cyclin destruction in Saccharomyces cerevisiae. Mol. Biol. Cell, 9, 2803–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozminski K.G., Chen,A.J., Rodal,A.A. and Drubin,G.D. (2000) Functions and functional domains of the GTPase Cdc42p. Mol. Biol. Cell, 11, 339–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.E., Jensen,S., Frenz,L.M., Johnson,A.L., Fresquet,D. and Johnston,L.H. (2001) The Bub2-dependent mitotic pathway in yeast acts every cell cycle and regulates cytokinesis. J. Cell Sci., 114, 2345–2354. [DOI] [PubMed] [Google Scholar]
- Li R., Zheng,Y. and Drubin,D.G. (1995) Regulation of cortical actin cytoskeleton assembly during polarized cell growth in budding yeast. J. Cell Biol., 128, 599–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine M.S., McKenzie,A., Demarini,D.J., Shah,N.G., Wach,A., Brachat,A., Philippsen,P. and Pringle,J.P. (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast, 14, 953–961. [DOI] [PubMed] [Google Scholar]
- Longtine M.S., Threesfeld,C.L., McMillan,J.N., Weaver,E., Pringle,J.R. and Lew,D.J. (2000) Septin-dependent assembly of a cell cycle-regulatory module in Saccharomyces cerevisiae. Mol. Cell. Biol., 20, 4049–4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luca F.C. and Winey,M. (1998) MOB1, an essential yeast gene required for completion of mitosis and maintenance of ploidy. Mol. Biol. Cell, 9, 29–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata J. and Nurse,P. (1997) tea1 and the microtubular cytoskeleton are important for generating global spatial order within the fission yeast cell. Cell, 89, 939–949. [DOI] [PubMed] [Google Scholar]
- Pereira G., Höfken,T., Grindlay,J., Manson,C. and Schiebel,E. (2000) The Bub2p spindle checkpoint links nuclear migration with mitotic exit. Mol. Cell, 6, 1–10. [PubMed] [Google Scholar]
- Pereira G., Manson,C., Grindlay,J. and Schiebel,E. (2002) Regulation of the Bfa1p–Bub2p complex at spindle pole bodies by the cell cycle phosphatase Cdc14p. J. Cell Biol., 157, 367–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter M., Neiman,A.M., Park,H.O., van Lohuizen,M. and Herskowitz,I. (1996) Functional analysis of the interaction between the small GTP binding protein Cdc42 and the Ste20 protein kinase in yeast. EMBO J., 15, 7046–7059. [PMC free article] [PubMed] [Google Scholar]
- Philips J. and Herskowitz,I. (1998) Identification of Kel1p, a kelch domain-containing protein involved in cell fusion and morphology in Saccharomyces cerevisiae. J. Cell Biol., 143, 375–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richman T.J., Sawyer,M.M. and Johnson,D.I. (2002) Saccharomyces cerevisiae Cdc42p localizes to cellular membranes and clusters at sites of polarized growth. Eukary. Cell, 1, 458–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwob E., Bohm,T., Mendenhall,M.D. and Nasmyth,K. (1994) The B-type cyclin kinase inhibitor p40 (Sic1) controls the G1 to S transition in Saccharomyces cerevisiae. Cell, 79, 233–244. [DOI] [PubMed] [Google Scholar]
- Shinjo K., Koland,J.G., Hart,M.J., Narasimhan,V., Johnson,D.I., Evans,Y. and Cerione,R.A. (1990) Molecular cloning of the gene for the human placental GTP-binding protein Gp (G25K): identification of this GTP-binding protein as the human homolog of the yeast cell-division-cycle protein CDC42. Proc. Natl Acad. Sci. USA, 87, 9853–9857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shou W. et al. (1999) Exit from mitosis is triggered by Tem1-dependent release of the protein phosphatase Cdc14 from nucleolar RENT complex. Cell, 97, 233–244. [DOI] [PubMed] [Google Scholar]
- Sikorski R.S. and Hieter,P. (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae.Genetics, 122, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegmeier F., Visintin,R. and Amon,A. (2002) Separase, polo kinase, the kinetochore protein Slk19 and Spo12 function in a network that controls Cdc14 localization during early anaphase. Cell, 108, 207–220. [DOI] [PubMed] [Google Scholar]
- Tatsumoto T., Xie,X., Blumenthal,R., Okamoto,I. and Miki,T. (1999) Human ECT2 is an exchange factor for Rho GTPases, phosphorylated in G2/M phases and involved in cytokinesis. J. Cell Biol., 147, 921–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjandra H., Compton,J. and Kellogg,D. (1998) Control of mitotic events by the Cdc42 GTPase, the Clb2 cyclin and a member of the PAK kinase family. Curr. Biol., 8, 991–1000. [DOI] [PubMed] [Google Scholar]
- Toenjes K., Sawyer,M.M. and Johnson,D.I. (1999) The guanine-nucleotide-exchange factor Cdc24p is targeted to the nucleus and polarized growth sites. Curr. Biol., 9, 1183–1186. [DOI] [PubMed] [Google Scholar]
- Visintin R., Craig,K., Hwang,E.S., Prinz,S., Tyers,M. and Amon,A. (1998) The phosphatase Cdc14 triggers mitotic exit by reversal of Cdk-dependent phosphorylation. Mol. Cell, 2, 709–718. [DOI] [PubMed] [Google Scholar]