

Abstract
CTLA-4 and PD-1 are receptors that negatively regulate T-cell activation. Ligation of both CTLA-4 and PD-1 blocked CD3/CD28-mediated upregulation of glucose metabolism and Akt activity, but each accomplished this regulation using separate mechanisms. CTLA-4-mediated inhibition of Akt phosphorylation is sensitive to okadaic acid, providing direct evidence that PP2A plays a prominent role in mediating CTLA-4 suppression of T-cell activation. In contrast, PD-1 signaling inhibits Akt phosphorylation by preventing CD28-mediated activation of phosphatidylinositol 3-kinase (PI3K). The ability of PD-1 to suppress PI3K/AKT activation was dependent upon the immunoreceptor tyrosine-based switch motif located in its cytoplasmic tail, adding further importance to this domain in mediating PD-1 signal transduction. Lastly, PD-1 ligation is more effective in suppressing CD3/CD28-induced changes in the T-cell transcriptional profile, suggesting that differential regulation of PI3K activation by PD-1 and CTLA-4 ligation results in distinct cellular phenotypes. Together, these data suggest that CTLA-4 and PD-1 inhibit T-cell activation through distinct and potentially synergistic mechanisms.
Precise regulation of T-lymphocyte proliferation is critical to the acquired immune response and hematopoietic homeostasis. Activation of resting T cells is a complex process, dependent upon intracellular signals initiated through the T-cell antigen receptor (TCR) and modulated by costimulatory and negative receptor signals. Receptors such as CD28 and ICOS transduce signals necessary to fully activate T cells. In contrast, receptors like CTLA-4 and PD-1 transduce signals that are inhibitory to lymphocyte activation (27, 55, 55, 57). The balance between “positive” and “negative” signaling is thought to enable effective immune responses while maintaining immunological tolerance and preventing autoimmunity (10, 63).
CD3/CD28 costimulation initiates a series of intracellular signals that increase cellular metabolism, oppose cell death, and drive progression through the cell division cycle (2). Ligation-dependent tyrosine phosphorylation of CD28 enables the recruitment and activation of phosphatidylinositol 3-kinase (PI3K), resulting in the intracellular accumulation of 3-phosphorylated lipids (49, 67). The lipid products of PI3K bind pleckstrin homology domain-containing proteins, including the serine/threonine kinase Akt, which is thought to play an important role in diverse cellular processes, including cytokine synthesis (34, 54) and survival (24, 33). Additionally, Akt plays a regulatory role in glucose metabolism by increasing Glut1 expression (4) and glycolysis (26, 54). Potentially, Akt catalytic activity can be modulated either by decreasing the intracellular concentration of 3-phosphorylated phosphatidylinositol lipids or direct dephosphorylation by serine/threonine phosphatases.
Antibody-mediated blockade of CTLA-4 prevents development of tolerance, augments anti-tumor responses, and exacerbates autoimmune disease (52). Despite much attention, the mechanism underlying CTLA-4-mediated inhibition of T lymphocytes remains obscure. CTLA-4 shares the B7 ligand with CD28 but binds with a higher avidity (20); thus, ligand sequestration may be significant in vivo (9). The finding that the CTLA-4 extracellular domain alone is insufficient to prevent lymphadenopathy in mice suggested that CTLA-4 may also propagate signals that actively counter TCR/CD28 signaling (25, 41). To this end the cytoplasmic domain of CTLA-4 has been reported to interact with the tyrosine phosphatase SHP-2 (40) and serine/threonine phosphatase PP2A (17). Whether the CTLA-4 cytoplasmic tail requires phosphorylation to recruit effector molecules remains controversial (18, 23, 31, 40, 71). In one study, however, SHP-2 recruitment was thought to dephosphorylate the TCR zeta chains (36). The significance of PP2A to CTLA-4 function is unclear, since PP2A also binds to CD28 (17). Moreover, mutations in CTLA-4 that alter regulatory subunit association with bound PP2A affect the ability of CTLA-4 to inhibit interleukin-2 (IL-2) transcription in T cells (3).
PD-1 is expressed on activated T lymphocytes but also on B cells, suggesting involvement in a broader spectrum of immune regulation than CTLA-4 (1). To date, all reports that specifically target PD-1 using PD-1 antibody (Ab) have shown that PD-1 acts as a negative regulator of T-cell activation (5, 47). Consistent with this finding, PD-1-deficient mice show splenomegaly and increased susceptibility to autoimmune disease (46, 58). Despite the shared ability to block T-cell activation, the cytoplasmic domain of PD-1 shows significant differences from CTLA-4 by encoding both an immunoreceptor tyrosine-based inhibitory motif (ITIM) and an immunoreceptor tyrosine-based switch motif (ITSM), of which the ITSM appears to be the most important for mediating PD-1 suppression of lymphocyte activation (16, 47). Understanding to what extent PD-1 and CTLA-4 employ similar or distinct mechanisms to block T-cell activation will have important implications for the design of immunotherapies that aim to alter the immune response. Here, we show that CTLA-4 and PD-1 propagate biochemically separable, inhibitory signals that converge at Akt to limit cellular metabolism. While PD-1 disrupts the intracellular accumulation of 3-phosphorylated phosphatidylinositol lipids, CTLA-4 targets downstream effectors of PI3K through activation of the serine/threonine phosphatase PP2A.
MATERIALS AND METHODS
Lymphocyte isolation and stimulation.
Peripheral blood lymphocytes were isolated from normal volunteer donors following apheresis and elutriation under a protocol approved by The University of Pennsylvania Institutional Review Board. CD4+ T cells were purified by negative selection using magnetic beads (Dynal, Lake Success, NY) as previously described and were routinely more than 95% CD3+, more than 98% CD28+, and less than 3% CD8+ as determined by flow cytometry (56). Cells were stimulated at 37°C by mixing with beads at a ratio of 1:3 (cells to beads). Anti-CD3 (OKT3), anti-hCD28 (9.3), anti-major histocompatibility complex (anti-MHC) class I (W6/32), anti-mCD28 (37.51; BD Biosciences), and anti-PD-1 (#17; a kind gift of Beatriz Carreno, Inflammation, Wyeth Research, Cambridge, MA) (5) were covalently attached to polyurethane-coated tosyl-activated Dynal beads according to the manufacturer's instructions. Anti-CD3 Ab represents 5% of the total protein added to the beads, anti-CD28 Ab represents 10% of the remaining 95%, and the remaining Ab was composed of the antibody indicated.
Glucose uptake and glycolysis.
Glucose uptake was measured using a modified method of Whetton et al. (69). Briefly, stimulated T cells were incubated for 15 min at 37°C in glucose uptake buffer (8.1 mM Na2HPO4, 1.4 mM KH2PO4, 2.4 mM KCl, 136 mM NaCl, 0.5 mM MgCl2, 0.9 mM CaCl2 [pH 7.4]) to deplete intracellular glucose stores. Triplicate samples of 1 × 106 cells were incubated with 1 μCi of [3H]2-deoxyglucose (NEN, Boston, MA) in glucose uptake buffer for 2 min at room temperature and immediately spun through a layer of bromododecane (Sigma, St. Louis, MO) into 20% perchloric acid-8% sucrose, stopping the reaction, and separating the cells from unincorporated [3H]2-deoxyglucose. The perchloric acid-sucrose-T-cell layer was removed and analyzed by a liquid scintillation counter (Wallac, Turku, Finland). Measurement of glycolysis was performed as previously described (64).
Cell lysis, sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and Western blotting.
Cells were lysed in 1% NP-40 lysis buffer, and proteins were resolved by SDS-PAGE and transferred to nitrocellulose. Blots were probed with anti-phosphorylated Akt (anti-pAkt) and anti-phospho-glycogen synthase kinase 3α/β (GSK-3α/β) (Ser21/9) primary antibody (Cell Signaling Technology, Beverly, MA) and visualized with ECL+ (Amersham, Piscatway, NJ). Blots were stripped and reprobed with antibodies against the whole Akt to demonstrate equal loading of proteins. Densitometry was performed using ImageQuant 5.2 software (Amersham Biosciences Corp., Piscataway, NJ). Relative pAKT levels were determined by dividing the value determined for pAKT by the corresponding total AKT value and dividing this value by the value obtained in the resting cell lane.
Determination of PI3K activity.
Catalytic activity of PI3K was assessed by in vitro kinase assay as previously described (51). Briefly, samples were washed sequentially in lysis buffer, phosphate-buffered saline, LiCl, water, and finally kinase buffer. Samples were resuspended in 40 μl of kinase buffer before addition of 50 μl of a mixture of phosphatidylinositol with phosphatidylserine. Reactions were initiated by the addition of 20 μCi [γ-32P]ATP and 100 μM ATP and were terminated after 15 min at 30°C by the addition of 100 μl 1 M HCl. Lipids were extracted by partitioning in chloroform and were resolved by thin-layer chromatography using 1-propanol-acetic acid (65%:35%, vol/vol) as the solvent. Phosphorylated lipids were visualized by a PhosphorImager (Molecular Dynamics).
Creation and transduction of mCD28-PD-1 vectors.
The murine CD28 (mCD28) extracellular and transmembrane domain was amplified from an mCD28-encoding plasmid (generously provided by K. Lee, University of Miami, FL) by PCR and blunt ligated with a PCR product encoding the human PD-1 cytoplasmic tail (bases 645 to 935 of PD-1 [NM_005018]). These fragments were placed into the pCLPS lentiviral expression vector (51). Single tyrosine-to-phenylalanine mutations of mCD28/human PD-1 (mCD28/hPD-1) were produced as previously described (66). Double tyrosine-to-phenylalanine mutations of mCD28/hPD-1 were produced using a single mutation as a template. The fidelity of all constructs was confirmed by DNA sequencing. Lentiviral vectors were produced as described previously (51). CD4 T cells were stimulated for 24 h with CD3/hCD28 (CD3/h28)-coated immunobeads before infection. Three-hundred to 500 μl (∼5 × 107 to 5 × 108 IFU) of lentiviral vector was incubated with the activated CD4 T cells and centrifuged at 1,200 × g for 2 h. The following day the medium was exchanged and the cells were expanded until they returned to near resting cell volume as measured by a Coulter Counter Multisizer II (Hialeah, FL).
RNA extraction and reverse transcription-PCR (RT-PCR).
RNA was purified and reverse transcribed as described previously (56). Primers and probes to detect IL-2, Bcl-xL, and 28S were designed using Primer Express software and are available upon request (Applied Biosystems, Foster City, CA). Real-time PCR amplification and product detection were performed using the ABI Prism 7700 (Applied Biosystems) as recommended by the manufacturer. Results were normalized to 28S RNA levels, and relative expression was determined using the ΔΔct method as recommended by the manufacturer.
DNA microarray analysis.
Following stimulation, cells were harvested and total cellular RNA was isolated using an RNeasy kit (QIAGEN, Valencia, CA) including DNase treatment. Methods for mRNA amplification using in vitro transcription, cRNA labeling with Cy3 and Cy5, and hybridization have been described previously (30). cRNAs from stimulated and unstimulated cells were compared by competitive hybridization on hu25K ink jet oligonucleotide microarrays. Sequences represented on the microarrays were previously described (56). Hybridizations were performed in duplicate with fluor reversal. Slides were scanned using a confocal laser scanner from Agilent Technologies. Fluorescent intensities on images were quantified, corrected for background, and normalized.
RESULTS
CTLA-4 and PD-1 signaling regulate cellular metabolism.
To study whether CTLA-4- or PD-1-induced blockade used similar or distinct mechanisms to block primary human CD4 T-cell activation, a system to study the distinct effects of PD-1 or CTLA-4 signaling on T-cell activation was established. Previously we have used magnetic beads coated with anti-CD3, anti-CD28, and anti-CTLA-4 Ab (designated CD3/CD28/CTLA-4) as artificial antigen-presenting cells (aAPCs) to study the effects of CTLA-4 ligation (7). Inclusion of the anti-CD28 Ab is necessary because highly purified human cells do not expand or produce significant levels of IL-2 upon ligation of anti-CD3 and anti-MHC class I (designated CD3/MHC I)-coated beads (Fig. 1A and B). Anti-MHC I Ab is used as a binding, but nonsignaling, control Ab to ensure equivalent loading of Ab onto the aAPCs (56). Stimulation of primary CD4 T cells with anti-CD3-, anti-CD28-, and anti-MHC class I (designated CD3/CD28/MHC I)-coated beads leads to robust T-cell expansion and IL-2 production, as previously published (38). In contrast, similarly constructed CD3/CD28/PD-1 aAPCs inhibited T-cell expansion and IL-2 production as well as CD3/CD28/CTLA-4-coated beads. Thus, use of these aAPCs allows the study of primary human T cells under the influence of either CTLA-4 or PD-1 blockade.
FIG. 1.

CTLA-4 and PD-1 engagements inhibit T-cell activation and glucose metabolism. CD4 T cells were stimulated with immunobeads coated with the antibodies indicated, and (A) T-cell expansion, (B) IL-2 expression, (C) uptake of [3H]2-deoxyglucose, and (D) glycolysis were measured. IL-2 expression, uptake of [3H]2-deoxyglucose, and glycolysis were measured 18 h after activation. All data are representative of three independent experiments. CPM, counts per million.
The transition from a resting T cell to a rapidly dividing effector of the adaptive immune response requires an abrupt change in energy demand. A feature of CD28-mediated T-lymphocyte costimulation is an increase in glucose uptake and metabolism, which allows a lymphocyte to meet the increased metabolic demands associated with a sustained response. CTLA-4 ligation inhibits this increase in glucose metabolism (26), and we were interested in determining if PD-1 engagement also targeted the ability of T cells to import and utilize nutrients. The first point of regulation in glucose metabolism is the regulation of glucose uptake by cell surface receptors. Treatment of resting cells with CD3/MHC I-coated beads induced only a minor increase in glucose uptake. Stimulation of resting CD4 T cells with anti-CD3-, anti-CD28-, and anti-MHC I-coated beads (CD3/CD28/MHC I) rapidly induced glucose uptake, preparing the cells for the increased metabolic demands of cell division and cytokine secretion that accompany T-cell activation (Fig. 1C). The effect of CD28 costimulation was opposed by both CTLA-4 and PD-1 engagement such that levels of glucose uptake in cells stimulated by either CD3/CD28/CTLA-4- or CD3/CD28/PD-1-coated beads were similar to levels seen following CD3/MHC I-coated bead stimulation of cells. These data indicate that CTLA-4 and PD-1 ablate the effect of costimulation on glucose uptake in T lymphocytes.
We next investigated whether CTLA-4 and PD-1 engagement resulted in a decrease in the glycolytic rate. The dehydration reaction catalyzed by enolase is the penultimate step of glycolysis and can be measured to gauge the glycolytic rate. In accord with the glucose uptake data, the glycolytic rate was low in resting T cells. CD3/CD28/MHC I-coated bead stimulation of CD4 T cells increased the glycolytic rate fourfold, and as described above this increase was blocked by both CTLA-4 ligation and PD-1 ligation (Fig. 1D). Together these data demonstrate that both CTLA-4 and PD-1 block CD28-mediated increases in metabolism, as measured by glucose uptake and glycolytic rate, and suggest that restriction of cellular metabolism may be a widespread strategy in lymphocyte inhibition.
PD-1 signaling blocks CD28-mediated activation of PI3K and Akt.
Akt plays an important role in the regulation of cellular metabolism in human primary T lymphocytes. Previously, we demonstrated that CTLA-4 ligation potently inhibits Akt activation and that this lack of Akt phosphorylation coincided with a lack of increased glucose metabolism (26). Having already established that PD-1-mediated signaling exerted similar constraints on cellular metabolism, we wanted to determine if PD-1 achieved this effect by targeting Akt phosphorylation as well. Since PD-1 and CTLA-4 are poorly expressed on resting lymphocytes, we first cultured cells in the presence of phytohemagglutinin (PHA)-IL-2 to induce expression of these negative regulators of T-cell activation. To determine how CTLA-4 and PD-1 engagement alters the Akt induction activity in a side-by-side manner, we stimulated the T-cell blasts with either CD3/MHC I-, CD3/MHC I/CTLA-4-, or CD3/MHC I/PD-1-coated beads to measure the effect these receptors have on CD3 induced Akt phosphorylation or CD3/CD28/MHC I-, CD3/CD28/CTLA-4-, or CD3/CD28/PD-1-coated beads to measure the effect these receptors have on CD3/CD28-mediated Akt activation. After 30 min of stimulation, we examined whole-cell lysates for phosphorylated Akt. We observed that both CTLA-4 and PD-1 ligation interfered with both CD3- and CD3/CD28-induced activation of Akt, indicating that CD28 costimulation is not required to observe the CTLA-4 or PD-1 engagement (Fig. 2A). These data confirm the role of CTLA-4 engagement as an inhibitor of Akt activation (26, 50). Also, since PD-1 triggering also inhibited Akt phosphorylation, this suggests that targeting of Akt may be a conserved mechanism that negative regulators of T-cell activation use to block T-cell activation.
FIG. 2.

CTLA-4 ligation blocks Akt but not PI3K activation. (A) CD4 T cells were preactivated with 5 mg/ml PHA and 100 U/ml IL-2 for 3 days and stimulated with immunobeads coated with the antibodies indicated for 30 min. Cells were lysed and proteins were resolved by SDS-PAGE. Akt and GSK-3 phosphorylation was assessed by immunoblotting using phosphospecific Abs (upper panel). The blot was then stripped and reprobed with anti-Akt antibody (lower panel) to demonstrate equal loading. (B) The procedure described for panel A was used, except cells were harvested 30 min (30′), 4 h, and 24 h after activation. (C) PHA blasts were stimulated with immunobeads coated with antibodies as indicated for either 30 min or 60 min. Cells were lysed, and stimulatory beads were used to precipitate the appropriate receptors. Lipid kinase activity associated with receptors was assessed by in vitro kinase assays. Reaction products were resolved by thin-layer chromatography and were visualized using a PhosphorImager. Whole-cell lysate (5 μl) was used as a positive control for the assay. PtdIns, phosphatidylinositol.
The phosphorylation cascade that leads to Akt activation is a multiple-step process that is still being defined. While the Ser473 phosphospecific Akt Ab may be the best reagent to detect Akt phosphorylation, its ability to function as a surrogate for Akt activity is not clear (15). GSK-3 was the first substrate of AKT to be identified, and its phosphorylation by Akt at a noncatalytic residue is inhibitory to GSK-3 activity (21). Since Akt enzymatic activity is required to phosphorylate GSK-3, we probed the lysates described above for phosphorylated GSK-3 as a surrogate measure of Akt catalytic activity. These results mirrored the data obtained using the phosphospecific Akt Ab (Fig. 2A), suggesting that the phosphorylated Akt we detected has catalytic activity. Our previous studies demonstrated that maximal induction of Akt observed in primary human T cells after CD3/CD28 stimulation is observed 30 min after stimulation (26). To test whether CTLA-4 and/or PD-1 delayed rather than suppressed Akt activation, we extended the time course of the experiment to 24 h. In contrast to recent studies in a murine system (61), Akt activation in primary human T cells is transient, peaking at 30 min and undetectable after 4 h of stimulation (Fig. 2B). Akt phosphorylation in cells stimulated by CD3/CD28/CTLA-4 or CD3/CD28/PD-1 aAPCs did not have detectable phosphorylated Akt at any time point, indicating that CTLA-4 and PD-1 signaling blocked rather than delayed Akt activation.
We were interested in determining whether CTLA-4 and PD-1 targeted the same or distinct steps in the Akt activation process. Activation of Akt is dependent upon the prior activation of phosphatidylinositol 3-kinase, and these pathways are often referred to as the PI3K/Akt pathway (14). To determine whether CTLA-4 or PD-1 engagement altered CD28-mediated activation of PI3K, we assessed the ability of immunoprecipitated receptors to phosphorylate exogenous phosphatidylinositol. As described above, CD4 T cells were preactivated with PHA and IL-2 for 3 days to upregulate the expression of CTLA-4 and PD-1 on the cell surface. Stimulation of these cells with CD3/CD28/MHC I-coated beads resulted in a strong induction of PI3K activity detected in immunoprecipitates, in agreement with previous reports (67). Stimulation of cells with CD3/CD28/CTLA-4-coated beads resulted in slightly delayed but ultimately similar amounts of PI3K activity compared to that in cells stimulated with CD3/CD28/MHC I-coated beads, indicating that CTLA-4 ligation does not strongly oppose PI3K signaling (Fig. 2C). The delay could reflect either differences in the ability of CD3/CD28/MHC I beads to bind to cells with a higher affinity than CD3/CD28/CTLA-4 beads due to the higher surface expression of MHC class I than CTLA-4 or modest effects of CTLA-4 signal transduction on PI3K induction. In contrast, PD-1 engagement blocked the induction of PI3K activity. Since PI3K activity is required to activate Akt, PD-1's signaling that interferes with PI3K activation is likely to be the major reason one observes inhibition of Akt activation in cells under the influence of PD-1 blockage.
PD-1 suppression of PI3K/Akt is dependent upon factors binding to the ITSM motif in its cytoplasmic tail.
Little is known about how PD-1 generates a signal to block T-cell activation. PD-1 has two tyrosine residues in its cytoplasmic tail. The membrane-proximal tyrosine is located in an ITIM motif, and the C-terminal tyrosine is located in a newly described immunoreceptor tyrosine-based switch motif (ITSM) (60). To determine whether factors binding to either of these motifs were responsible for PD-1 inhibition of the PI3K/Akt pathway, we constructed chimeric molecules composed of the murine CD28 extracellular domain and either the wild-type human PD-1 cytoplasmic tail, the human PD-1 cytoplasmic tail with a mutation in the ITIM (Y223F), the human PD-1 cytoplasmic tail with a mutation in the ITSM (Y248F), or the human cytoplasmic tail that has both tyrosines mutated (Y223F, Y248F). Previously, we have demonstrated that the mCD28-hPD-1 chimeric molecule is able suppress T-cell expansion and cytokine production in a manner similar to that of the endogenous PD-1 molecule (16), and thus the mCD28-hPD-1 chimeric molecule is a faithful model to study PD-1 signal transduction. These constructs were introduced into primary human CD4 T cells via lentiviral transduction, and murine CD28 surface expression was determined 3 days posttransduction (Fig. 3A). These transduced cells were allowed to expand and rest until their cell volume approached levels near that of resting cells. Next, each of these populations of cells was restimulated with either CD3/MHC I, CD3/h28/MHC I, CD3/h28/mCD28, or CD3/h28/PD-1 aAPCs for 30 min, cell lysates were made, and the level of phosphorylated Akt was measured by Western blotting (Fig. 3B). No AKT phosphorylation was observed in unstimulated cells (lane 1); however, induction of AKT-P was observed in the anti-CD3-stimulated cells (lane 2) that was augmented by CD28 costimulation (lane 3). AKT phosphorylation is substantially reduced in cells restimulated with anti-CD3-, anti-CD28-, and anti-PD-1-coated beads (lane 5), indicating that engagement of the endogenous PD-1 receptor is able to suppress AKT phosphorylation in all cell types. This pattern of AKT phosphorylation is observed in all four cell populations, allowing us to compare the effects of the various PD-1 cytoplasmic tail mutants in a side-by-side manner.
FIG. 3.

PD-1's ITSM motif is required to mediate PD-1 suppression of the PI3K/Akt pathway. CD4 T cells were activated with CD3/CD28-coated beads and transduced with lentiviral vectors expressing mCD28-PD-1 wild type (WT), mCD28-hPD-1 Y223F, mCD28-PD-1 Y248F, and the double mutant mCD28-hPD-1 Y223F, Y248F. (A) Expression of each construct was examined by staining the cells with an Ab specific for mCD28 3 days after transduction. (B) Transduced cells were left unstimulated or were restimulated with the indicated aAPCs for 30 min. Cells were lysed, and proteins were resolved by SDS-PAGE. Akt activity was assessed by immunoblotting for serine-phosphorylated Akt (upper panel). The blot was then stripped and reprobed with anti-Akt antibody (lower panel) to demonstrate equal loading. Data are representative of three independent experiments.
Cells transduced with the mCD28-hPD-1 wild-type construct and subsequently restimulated with CD3/h28/mCD28-coated beads contained substantially reduced levels of AKT-P compared to the same cells restimulated with CD3/h28/MHC class I, indicating that this was an appropriate model to perform a structure-function analysis of the PD-1 cytoplasmic tail to determine which motif(s) was required to inhibit AKT-P. Cells transduced with mCD28-PD-1 223 F hPD-1 behave in a manner similar to those transduced with the mCD28-hPD-1 wild-type construct, indicating that the ITIM is not required to inhibit AKT-P. In contrast, in cells transduced with the mCD28 Y248F hPD-1 construct or in those cells transduced with the double mutant mCD28 Y223F, Y248F hPD-1 construct, AKT phosphorylation is not inhibited by CD3/h28/mCD28-coated bead stimulation. These results indicate that factors binding to the ITSM within the PD-1 cytoplasmic tail mediate PD-1 suppression of PI3K/Akt activation.
CTLA-4-mediated suppression of Akt phosphorylation is inhibited by the PP2A inhibitor okadaic acid.
In Fig. 2 we demonstrated that CTLA-4 engagement does not block CD28-mediated activation of PI3K and hence blocked Akt phosphorylation in a PI3K-independent manner. We postulated that CTLA-4 engagement activated a protein complex that was able to reverse PI3K-dependent phosphorylation of Akt. The type II serine/threonine phosphatase PP2A plays an important role in the regulation of Akt activity in fibroblasts (32) and insulin signaling (11). Moreover, this complex has been shown to be recruited to both the CD28 and CTLA-4 cytoplasmic tails (17), making it a likely candidate to mediate CTLA-4-induced inhibition of Akt phosphorylation. To determine whether PP2A played a role in mediating the suppression of Akt phosphorylation following either CTLA-4 or PD-1 engagement, we determined whether these receptors could block Akt phosphorylation in the presence of the PP2A inhibitor okadaic acid (OA). OA has a 50% inhibitory concentration of 1 nM in cell-free systems; however, much higher concentrations are required to observe effects of this inhibitor in experiments using intact cells due to the relatively inefficient transport of this molecule into the cell (6, 19, 43, 45). Concentrations of OA up to 1 μM can be used without any detectable intracellular inhibitory effect on PP1, PP2B, or PP2C (43), suggesting that treatment of cells with a concentration of less than 1 μM preferentially interferes with PP2A enzymatic activity. Pretreatment of CD3/CD28/MHC I-stimulated T cells with okadaic acid induced a slight increase in Akt phosphorylation (which was more defined in other replicates of this experiment), suggesting that PP2A recruitment to CD28 modestly opposes CD28-mediated activation of Akt (Fig. 4). Most importantly, CTLA-4-mediated inhibition of Akt phosphorylation was abrogated in the presence of okadaic acid. Moreover, these blots were stripped and reprobed for GSK-3 phosphorylation as described in Fig. 2. We observed a pattern of GSK-3 phosphorylation similar to that of Akt phosphorylation, demonstrating that OA treatment is able to render Akt functional in T cells undergoing CTLA-4-mediated suppression (data not shown). The effects of OA on Akt phosphorylation appear to be limited to CD28 and CTLA-4, as this compound had no effect on PD-1-mediated suppression of Akt phosphorylation. This result is consistent with our previous observations showing that PD-1 ligation blocks Akt at a more membrane-proximal step (PI3K). This observation that cells stimulated by CD3/CD28/CTLA-4 aAPCs produce high levels of PI3K activity in the absence of Akt phosphorylation is, we believe, the first time that PI3K activity and AKT phosphorylation have been dissociated in human T cells. Together these data demonstrate that CTLA-4 and PD-1 ligation affect Akt inhibition by biochemically distinct mechanisms.
FIG. 4.

CTLA-4-mediated suppression, but not PD-1-mediated suppression, of AKT is blocked by the presence of okadaic acid. PHA-IL-2-cultured CD4 T cells were stimulated with immunobeads coated with the indicated antibodies in either the presence or absence of okadaic acid. Akt activity was assessed by immunoblotting for serine-phosphorylated Akt [(p)Akt] (upper panel). The blot was then stripped and reprobed with anti-Akt antibody (lower panel) to demonstrate equal loading. The relative ratio of pAKT to total AKT is shown below each lane. Data are representative of three independent experiments. Unstim, unstimulated.
Expression of breakout gene Bcl-xL is dependent upon phosphatidylinositol 3-kinase.
CD28 costimulation increases expression of the survival factor Bcl-xL (8), and this CD28-mediated activation is relatively resistant to the effects of CTLA-4 ligation (7) but not to those of PD-1 ligation (16). Although Bcl-xL induction is known to have a PI3K-dependent component (37), it was important to determine if the CTLA-4-resistant component of Bcl-xL expression was dependent upon or independent of PI3K. Treatment of CD4 T lymphocytes for 24 h with CD3/CD28/CTLA-4-coated beads induced an increase in Bcl-xL expression that was inhibited by PI3K inhibitor LY294002 (65) in a dose-dependent manner (3 μM to 10 μM) to levels similar to those seen under PD-1-mediated inhibition (Fig. 5A). It is interesting that LY294002 inhibits CD3/CD28/CTLA-4-mediated upregulation of Bcl-xL to a greater degree than it inhibits CD3/CD28/MHC I upregulation of Bcl-xL. This difference may indicate that other signaling pathways that contribute to CD28-mediated upregulation of Bcl-xL are effectively being blocked by CTLA-4 engagement. These results demonstrate that the CTLA-4-resistant component of Bcl-xL expression is dependent upon PI3K activity and suggest that cells undergoing CTLA-4 and PD-1 blockage may have different susceptibilities to apoptosis.
FIG. 5.
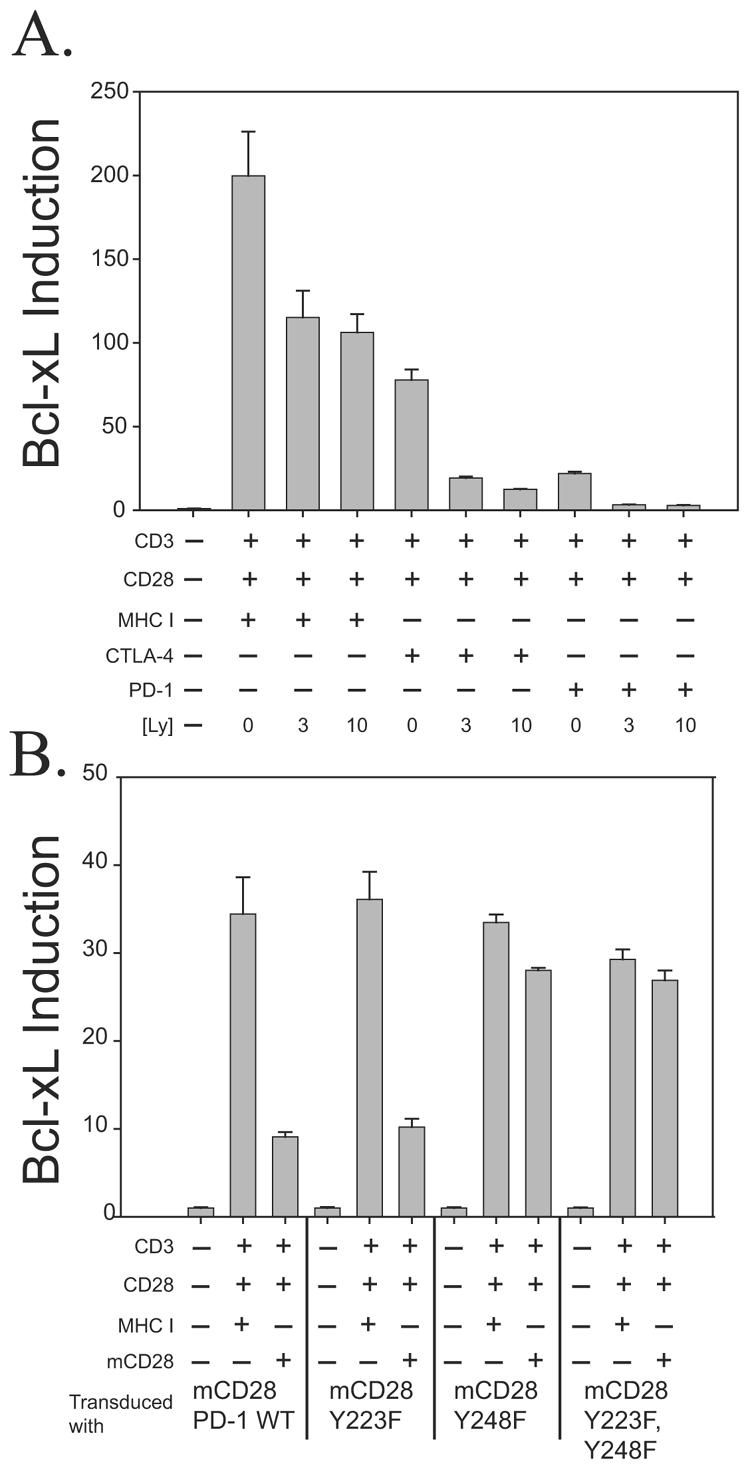
(A) PI3K is necessary to mediate CTLA-4-resistant Bcl-xL induction. CD4 T lymphocytes were stimulated with immunobeads coated with the antibodies indicated in either the absence or presence of LY294002 (3 μM or 10 μM) for 24 h. Bcl-xL expression was quantified by RT-PCR and expressed as a comparison to Bcl-xL expression in unstimulated cells. The error bars indicate the standard deviations of three replicates, and data are representative of three independent experiments. (B) An intact ITSM is required to mediate PD-1 suppression of Bcl-xL induction. The same set of transduced cells described in the legend to Fig. 3 was left either unstimulated or was restimulated with either CD3/CD28/MHC I- or CD3/CD28/mCD28-coated beads for 24 h. Quantitative RT-PCR to measure Bcl-xL expression was performed as described for panel A. WT, wild type.
It was also of interest to see if PD-1-mediated suppression of Bcl-xL was dependent upon an intact ITSM. As described in Fig. 3, we transduced primary CD4 T cells with lentiviral vectors expressing either mCD28-PD-1 wild-type or mCD28-PD-1 tyrosine mutant cytoplasmic tails. These transduced cells were cultured until their mean cell volume declined to a near resting level. At this point a portion of the cells were stimulated with CD3/CD28/MHC I or CD3/CD28/mCD28 aAPCs. After 24 h, cells were harvested and quantitative RT-PCR using Bcl-xL specific primers and probes was performed. As we observed in the case of Akt activation (Fig. 3B) and IL-2 induction (16), mutation of the ITSM (Y248F) completely abrogated the ability of PD-1 to block Bcl-xL induction (Fig. 5B). Taken together, these results suggest that factors recruited to the PD-1 ITSM block CD28-mediated induction of Bcl-xL and PI3K.
PD-1 engagement more effectively inhibited CD3/CD28-mediated changes in the T-cell transcriptional profile than CTLA-4 ligation.
Our data indicate that PD-1 and CTLA-4 target distinct steps in the PI3K/Akt signal transduction pathway. We used DNA microarrays to examine how this difference affects the transcriptional profile of T cells. This alternative approach to studying the effects of signal transduction pathways permits the systematic measurement of many thousands of discrete sequences. By comparing the amplitude and absolute number of transcripts altered as well as the type of genes being triggered, signal transduction pathways can be compared and information concerning the pathways can be deduced.
To determine whether PD-1 engagement and CTLA-4 engagement had similar effects on the T-cell transcriptional profile, freshly isolated CD4 T cells were mixed with CD3/CD28/CTLA-4- or CD3/CD28/PD-1-coated beads. After 24 h of stimulation, RNA was extracted from these cells and DNA microarray analysis was performed in which the transcriptional profiles of CD3/CD28/CTLA-4- and CD3/CD28/PD-1-coated-bead-stimulated cells were compared to those of unstimulated cells. Gene regulations induced by CD3/CD28/CTLA-4 and CD3/CD28/PD-1 were examined using a correlation plot (Fig. 6). In this method of analysis, the magnitude by which a particular transcript is affected by both CD3/CD28/CTLA-4 and CD3/CD28/PD-1 stimulation is plotted. Thus, if a transcript is regulated to the same extent in response to either CD3/CD28/CTLA-4 or CD3/CD28/PD-1 stimulation, it would appear on the diagonal line. While a strong correlation (r = 0.8) is observed between the transcriptional profiles of CD3/CD28/PD-1- and CD3/CD28/CTLA-4-stimulated cells, the slope of this line is less than 1, indicating that most transcripts were less regulated in CD3/CD28/PD-1-stimulated cells. In total, 1,403 transcripts were scored as being significantly regulated (P = 0.001; intensity, >−1), either up or down, by both CD3/CD28/CTLA-4 and CD3/CD28/PD-1 stimulations compared to unstimulated T cells. A total of 3,262 transcripts were regulated by CD3/CD28/CTLA-4 engagement and not CD3/CD28/PD-1 ligation, and 177 transcripts were regulated by CD3/CD28/PD-1 and not CD3/CD28/CTLA-4 (see Table S1 in the supplemental material). This large difference between transcripts regulated by CD3/CD28/CTLA-4 and not CD3/CD28/PD-1 and transcripts regulated in the opposite manner (3,262 versus 177 transcripts) suggests that PD-1 is a more potent suppressor of CD3/CD28-mediated changes in the T-cell transcriptional profile than CTLA-4.
FIG. 6.

PD-1 and CTLA-4 engagement each uniquely affect the T-cell transcriptional profile. Primary human CD4 T cells were cultured for 24 h with either CD3/CD28/CTLA-4- or CD3/CD28/PD-1-coated beads. Total RNA was isolated, amplified, and analyzed by DNA microarray hybridization. Shown are comparisons (correlations) of genes significantly regulated (P < 0.01; intensity, >−1) in CD3/CD28/CTLA-4-stimulated T cells (x axis) versus that in CD3/CD28/PD-1-stimulated T cells (y axis). The color scheme is as follows: red genes represent genes significantly regulated under both conditions; green genes represent genes significantly regulated in the x dimension; blue genes represent genes significantly regulated in the y dimension; brown genes represent genes showing opposite regulation in the two conditions; gray genes are transcripts not significantly regulated in either dimension. This experiment was repeated twice with equivalent results. The gene lists for each of these distinct regulations are found in Table S1 in the supplemental material.
Importantly, the vast majority of these differences are small (see below) and most likely represent quantitative rather than qualitative differences between the transcriptional profiles of CD3/CD28/CTLA-4- and CD3/CD28/PD-1-stimulated cells. Earlier time points (2 and 8 h after activation) were examined by DNA microarray analysis, but only RNA collected 24 h after activation displayed significant differences (data not shown), suggesting that the differences in gene expression profiles between CD3/CD28/CTLA-4- and CD3/CD28/PD-1-stimulated cells are due to genes that are regulated later rather than immediately following CD3/CD28 costimulation.
Another way to analyze these data is to focus on transcripts whose expression levels are altered greater than fivefold in relation to unstimulated T cells. Using this value, levels of 517 transcripts are altered by CD3/CD28/CTLA-4 ligation, whereas only 128 are regulated by CD3/CD28/PD-1 stimulation. To place these data in context, we compared them with data we previously generated comparing the gene expression profiles of CD3/MHC I-, CD3/CD28-, and CD3/CD28/CTLA-4-stimulated cells (56). Here, 447 transcripts were regulated at least fivefold by CD3/CD28/CTLA-4 stimulation over unstimulated cells after 24 h of stimulation, making the results of these identically performed studies comparable. CD3/MHC I stimulation resulted in 238 transcripts changing their expression levels at least fivefold, whereas after CD3/CD28 costimulation, 1,427 transcripts altered their expression levels by this fivefold threshold. Thus, CTLA-4 engagement reduced the number of transcripts that were regulated fivefold or more by CD3/CD28 costimulation by ∼67%, whereas after CD3/CD28/PD-1 engagement, ∼90% of transcripts fell below this threshold.
Lastly, it is important to point out that technical issues, including the absolute number of PD-1 and CTLA-4 molecules on the T-cell surface as well as the relative affinities the CTLA-4 and PD-1 Abs have for their respective ligands, could contribute to this finding. Unfortunately, the extent, if any, these factors play in influencing the T-cell transcriptional profile is currently unknown. Nonetheless, the observation that PD-1 blockade is more effective in attenuating changes in the T-cell transcriptional profile induced by CD3/CD28 costimulation than by CTLA-4 blockade supports our data that PD-1 ligation blocks more membrane-proximal signals, whereas CTLA-4 ligation interferes with more membrane-distal signals.
DISCUSSION
Negative regulators of T-cell activation restrict cellular metabolism.
The transition from a quiescent cell to a proliferating clonal effector places a challenging metabolic demand on lymphocytes (35). During this transition, T cells are vulnerable to inhibition by events that limit the supply of metabolites such as glucose and tryptophan (28). CD28-mediated activation of the PI3K/Akt pathway facilitates elevated glucose uptake and metabolism by T lymphocytes (26). The finding that both CTLA-4 and PD-1 limit glucose metabolism in the presence of suboptimal CD28 costimulation suggests that the disruption of cellular metabolism may be a widespread strategy to enforce T-lymphocyte inhibition. Moreover, these studies indicate that Akt is a key integrator of signals that activate and inhibit T-cell activation and further establish Akt's role in cellular metabolism.
In mouse systems, CTLA-4−/− and PD-1−/− T cells do not show augmented responses in the naive state but do so only after differentiating into effector cells (12, 13, 48), suggesting that T-cell differentiation may affect a cell's susceptibility to CTLA-4- and/or PD-1-mediated effects. In our studies, both resting cells (Fig. 6) and PHA blasts (Fig. 2 and 4) demonstrated differential responses to PD-1 and CTLA-4 signal transduction, suggesting that on a gross level one can observe differences in CTLA-4 and PD-1 signaling regardless of the differentiation state of the cell. As the pathways that mediate CTLA-4 and PD-1 effects become clearer, it will be interesting to more closely examine the effects of T-cell differentiation on CTLA-4 and PD-1 signal transduction.
CTLA-4 and PD-1 both limit T-cell activation, but they are not functionally redundant.
Although PD-1 and CTLA-4 have structurally different cytoplasmic domains, CTLA-4 and PD-1 each transmit inhibitory signals to T lymphocytes and regulate an overlapping set of signaling proteins (40, 47). Furthermore, our finding that both CTLA-4 and PD-1 ligation disrupt lymphocyte metabolism suggests that substantial overlap exists in their inhibitory effects. Nonetheless, we were able to observe differences between the gene expression profile of cells stimulated with CD3/CD28/CTLA-4-coated beads and those stimulated with CD3/CD28/PD-1-coated beads in which it appeared that PD-1 ligation was a much more effective inhibitor of T-cell activation than CTLA-4 engagement. Our results appear to be inconsistent with previous reports demonstrating that, in mice, CTLA-4 deficiency results in a more pronounced autoimmune phenotype than PD-1 deficiency (10, 27). One way to reconcile our biochemical data showing that PD-1 is a more effective inhibitor of T-cell activation with the observations of the CTLA-4−/− and PD-1−/− mice is that the ligands that engage these negative regulators of T-cell activation, namely CD80 and CD86 for CTLA-4 and PD-L1 and PD-L2 for PD-1, are differentially expressed. That is, the ligands for CTLA-4 rather than those for PD-1 are expressed in locations within the body in which induction of tolerance is more crucial. Another explanation is that the loss of CTLA-4-B7 interaction results in a non-T-cell defect. For example, it has been shown that CTLA-4-B7 interactions lead to the induction of indoleamine 2,3-dioxygenase in tolergenic dendritic cell (DC) populations (42, 44). Thus, loss of CTLA-4 would lead to a T-cell defect but would also result in the loss of tolergenic DCs, leading to a more severe form of immune dysregulation.
The implication of the DNA microarray data is that PD-1 engagement interferes with more pathways required for T-cell activation than does CTLA-4 engagement. Our previous studies indicated that CTLA-4 ligation preferentially targeted CD28- rather than CD3-generated changes to the T-cell transcriptional profile (56). This of course is not exclusive, as we present data in this work demonstrating that both CTLA-4 and PD-1 ligation inhibit CD3-induced Akt activation in preactivated T cells. However, the DNA microarray studies performed in this work, coupled with other studies which demonstrate that PD-1-mediated signal transduction dephosphorylates ZAP-70 (59), suggest that PD-1 ligation may interfere with more CD3-generated and costimulatory signals. Moreover, these differences in gene transcription correlate well with our observation that CTLA-4, but not PD-1, preserves the activity of PI3K, and that expression of at least one of the “breakout” genes, Bcl-xL, is dependent upon PI3K. We were, however, surprised not to see more pronounced differences (greater than fivefold) for many of the transcripts regulated by CD3/CD28/CTLA-4 but not CD3/CD28/PD-1 stimulation, given the key role PI3K is thought to play in T-cell activation. This observation suggests that in the absence of sustained Akt activation, PI3K activation alone has only a small effect on the majority of transcripts activated by TCR engagement or that CTLA-4 engagement interferes with other PI3K-dependent downstream signals. Most importantly, since PD-1 and CTLA-4 each inhibit the PI3K/Akt signaling pathway by distinct mechanisms, the effect of CTLA-4 and PD-1 in concert may be additive if not synergistic. This may provide a rationale for why there are multiple inhibitors of T-cell activation. If they inhibit T-cell activation at distinct, and in the case of PD-1 and CTLA-4, potentially synergetic points, then triggering by multiple negative regulators of T-cell activation may be much more effective at preventing T-cell activation than the actions of a single inhibitor. It should be stressed that our study did not define AKT and PI3K as the only targets of CTLA-4 and PD-1 engagement, and it is quite possible that other signaling pathways are also differentially regulated by CTLA-4 and PD-1. If so, then these additional pathways would strengthen the case that CTLA-4 and PD-1 control T-cell activation through synergistic mechanisms.
Functional involvement of PP2A in CTLA-4 signaling and of the ITSM in PD-1 signaling.
The role of PP2A in CTLA-4 signaling is unclear, since CD28 also binds PP2A (17), and a CTLA-4 mutant lacking lysine residues critical to PP2A binding appears more effective at inhibiting IL-2 transcription (3). Our demonstration that CTLA-4 does not inhibit Akt in the presence of the PP2A inhibitor okadaic acid establishes Akt as a cellular target of CTLA-4-activated PP2A. While these observations do not rule out roles for other signaling molecules such as SHP-2 (36) in mediating the effects of CTLA-4 engagement, they do provide evidence that PP2A plays a crucial role as an effector of CTLA-4 signaling. Strikingly, PD-1-mediated suppression of Akt activity is completely unaffected by the presence of okadaic acid, since PD-1 abrogates Akt phosphorylation by antagonizing its upstream activator, PI3K (see the model in Fig. 7). It should be noted that extended culture of primary T cells with OA was toxic, and after 4 h of culture, phosphorylation of Akt was observed in cells stimulated with CD3/CD28/PD-1. The delayed Akt phosphorylation did not result in GSK-3 phosphorylation, suggesting that Akt phosphorylated in this nonphysiologic manner did not have enzymatic activity. This toxicity prevented us from examining more downstream events, such as IL-2 production in CD3/CD28/CTLA-4-stimulated cells treated with OA.
FIG. 7.
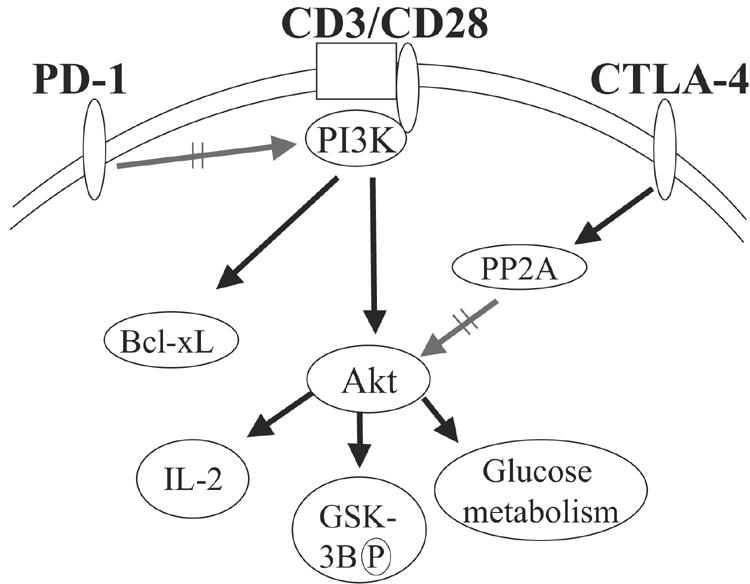
Model of CTLA-4- and PD-1-mediated T-lymphocyte inhibition. Signaling by CTLA-4 preserves PI3K activity, allowing expression of certain genes such as Bcl-xL, but inhibits Akt directly by activation of the phosphatase PP2A. In contrast, PD-1 antagonizes PI3K activity directly, perhaps by effecting a more global inhibition of T lymphocyte function. Since CTLA-4 and PD-1 target Akt by different mechanisms, their effect in concert may be additive or synergistic.
The mechanism of PD-1-mediated antagonism of the PI3K pathway remains unknown, but our study demonstrates that this inhibition is dependent on factors binding the ITSM. Our previous reports show that both SHP-1 and SHP-2 bind the PD-1 cytoplasmic tail in an ITSM-dependent manner, and thus these factors are likely candidates to mediate PD-1's suppression of PI3K in primary T cells (16). SHP-1 is generally thought to have a negative role in cell activation and has been shown to dephosphorylate p85 and limit PI3K activity (22), but more recent results have demonstrated that SHP-1 may dephosphorylate PTEN and hence potentiate PI3K activity (39). SHP-2 also appears to have a convoluted role in cell activation and PI3K regulation. Using a murine fibroblast model, SHP-2 was shown to be required for PI3K activation (70), whereas other reports indicate that SHP-2 interferes with PI3K activation (72). Interestingly, CD150, the molecule in which the ITSM was defined, recruits SHIP when the small adapter SH2D1A is present but recruits SHP-2 in the absence of SH2DIA (60). In contrast, the PD-1 cytoplasmic tail appears to recruit SHP-1 and SHP-2 but not SHIP regardless of whether SH2D1A is present (16), so it is not likely that SHIP, which is known to interfere with PI3K activation (29), is involved in PD-1-mediated suppression of PI3K activation. Thus, the factors binding to the PD-1 ITSM motif that are required to block PI3K activation remain to be defined.
Modification of negative signaling is potentially an important therapeutic strategy in the treatment of autoimmune diseases, cancer, and transplant rejection, but it has been achieved so far by only the relatively crude method of blocking receptor-ligand interactions (52). The repertoire of negative regulators expressed on T lymphocytes has increased in scope and complexity with the recent delineation of B- and T-lymphocyte attenuator (68) and a further as-yet-unidentified molecule bound by B7-H3 (62). A recent study demonstrated that CTLA-4 and PD-1 cooperate to maintain CD8 peripheral tolerance (53). Our data showing potential synergy between the signaling pathways generated by CTLA-4 and PD-1 may offer a molecular explanation for this observation. A detailed understanding of the negative signals these molecules transduce and of their interplay in T lymphocytes may facilitate the genesis of more powerful and specific strategies for therapeutic manipulation of the immune system.
Supplementary Material
Acknowledgments
We thank Carl June, Richard Carroll, Bob Vonderheide, Kim Nichols, and Bruce Levine for insightful discussions; Stephen Ward for assistance with kinase assays; Beatriz Carreno for the kind gift of PD-1 and CTLA-4 antibodies; Coral Haas for administrative support; Faraz Samadi for proofreading; Gwen Scott for graphing; the Rosetta Gene Expression Laboratory for performing the hybridizations; Hongyue Di for assistance with the gene expression analysis; and Bob Rutherford and Goro Osawa for isolation of T lymphocytes.
J.M.C. is supported by a grant from the Mildred Scheel Stiftung der Deutschen Krebshilfe. This work was supported by Public Health Service grant AI057838 from the National Institute of Allergy and Infectious Diseases.
Footnotes
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Agata, Y., A. Kawasaki, H. Nishimura, Y. Ishida, T. Tsubata, H. Yagita, and T. Honjo. 1996. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 8:765-772. [DOI] [PubMed] [Google Scholar]
- 2.Appleman, L. J., A. A. Van Puijenbroek, K. M. Shu, L. M. Nadler, and V. A. Boussiotis. 2002. CD28 costimulation mediates down-regulation of p27kip1 and cell cycle progression by activation of the PI3K/PKB signaling pathway in primary human T cells. J. Immunol. 168:2729-2736. [DOI] [PubMed] [Google Scholar]
- 3.Baroja, M. L., L. Vijayakrishnan, E. Bettelli, P. J. Darlington, T. A. Chau, V. Ling, M. Collins, B. M. Carreno, J. Madrenas, and V. K. Kuchroo. 2002. Inhibition of CTLA-4 function by the regulatory subunit of serine/threonine phosphatase 2A. J. Immunol. 168:5070-5078. [DOI] [PubMed] [Google Scholar]
- 4.Barthel, A., S. T. Okino, J. F. Liao, K. Nakatani, J. P. Li, J. P. Whitlock, and R. A. Roth. 1999. Regulation of GLUT1 gene transcription by the serine threonine kinase Akt1. J. Biol. Chem. 274:20281-20286. [DOI] [PubMed] [Google Scholar]
- 5.Bennett, F., D. Luxenberg, V. Ling, I. M. Wang, K. Marquette, D. Lowe, N. Khan, G. Veldman, K. A. Jacobs, V. E. Valge-Archer, M. Collins, and B. M. Carreno. 2003. Program death-1 engagement upon TCR activation has distinct effects on costimulation and cytokine-driven proliferation: attenuation of ICOS, IL-4, and IL-21, but not CD28, IL-7, and IL-15 responses. J. Immunol. 170:711-718. [DOI] [PubMed] [Google Scholar]
- 6.Bialojan, C., and A. Takai. 1988. Inhibitory effect of a marine-sponge toxin, okadaic acid, on protein phosphatases. Specificity and kinetics. Biochem. J. 256:283-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blair, P. J., J. L. Riley, B. L. Levine, K. P. Lee, N. Craighead, T. Francomano, S. J. Perfetto, G. S. Gray, B. M. Carreno, and C. H. June. 1998. CTLA-4 ligation delivers a unique signal to resting human CD4 T cells that inhibits interleukin-2 secretion but allows Bcl-X(L) induction. J. Immunol. 160:12-15. [PubMed] [Google Scholar]
- 8.Boise, L. H., A. J. Minn, P. J. Noel, C. H. June, M. A. Accavitti, T. Lindsten, and C. B. Thompson. 1995. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL. Immunity 3:87-98. [DOI] [PubMed] [Google Scholar]
- 9.Carreno, B. M., F. Bennett, T. A. Chau, V. Ling, D. Luxenberg, J. Jussif, M. L. Baroja, and J. Madrenas. 2000. CTLA-4 (CD152) can inhibit T cell activation by two different mechanisms depending on its level of cell surface expression. J. Immunol. 165:1352-1356. [DOI] [PubMed] [Google Scholar]
- 10.Carreno, B. M., and M. Collins. 2002. The B7 family of ligands and its receptors: new pathways for costimulation and inhibition of immune responses. Annu. Rev. Immunol. 20:29-53. [DOI] [PubMed] [Google Scholar]
- 11.Cazzolli, R., L. Carpenter, T. J. Biden, and C. Schmitz-Peiffer. 2001. A role for protein phosphatase 2A-like activity, but not atypical protein kinase Czeta, in the inhibition of protein kinase B/Akt and glycogen synthesis by palmitate. Diabetes 50:2210-2218. [DOI] [PubMed] [Google Scholar]
- 12.Chambers, C. A., M. S. Kuhns, and J. P. Allison. 1999. Cytotoxic T lymphocyte antigen-4 (CTLA-4) regulates primary and secondary peptide-specific CD4(+) T cell responses. Proc. Natl. Acad. Sci. USA 96:8603-8608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chambers, C. A., T. J. Sullivan, T. Truong, and J. P. Allison. 1998. Secondary but not primary T cell responses are enhanced in CTLA-4-deficient CD8+ T cells. Eur. J. Immunol. 28:3137-3143. [DOI] [PubMed] [Google Scholar]
- 14.Chan, T. O., S. E. Rittenhouse, and P. N. Tsichlis. 1999. AKT/PKB and other D3 phosphoinositide-regulated kinases: kinase activation by phosphoinositide-dependent phosphorylation. Annu. Rev. Biochem. 68:965-1014. [DOI] [PubMed] [Google Scholar]
- 15.Chan, T. O., and P. N. Tsichlis. 2001. PDK2: a complex tail in one Akt. Sci. STKE 2001:E1. [DOI] [PubMed] [Google Scholar]
- 16.Chemnitz, J. M., R. V. Parry, K. E. Nichols, C. H. June, and J. L. Riley. 2004. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J. Immunol. 173:945-954. [DOI] [PubMed] [Google Scholar]
- 17.Chuang, E., T. S. Fisher, R. W. Morgan, M. D. Robbins, J. M. Duerr, M. G. Vander Heiden, J. P. Gardner, J. E. Hambor, M. J. Neveu, and C. B. Thompson. 2000. The CD28 and CTLA-4 receptors associate with the serine/threonine phosphatase PP2A. Immunity 13:313-322. [DOI] [PubMed] [Google Scholar]
- 18.Cinek, T., A. Sadra, and J. B. Imboden. 2000. Cutting edge: tyrosine-independent transmission of inhibitory signals by CTLA-4. J. Immunol. 164:5-8. [DOI] [PubMed] [Google Scholar]
- 19.Cohen, P., S. Klumpp, and D. L. Schelling. 1989. An improved procedure for identifying and quantitating protein phosphatases in mammalian tissues. FEBS Lett. 250:596-600. [DOI] [PubMed] [Google Scholar]
- 20.Collins, A. V., D. W. Brodie, R. J. Gilbert, A. Iaboni, R. Manso-Sancho, B. Walse, D. I. Stuart, P. A. van der Merwe, and S. J. Davis. 2002. The interaction properties of costimulatory molecules revisited. Immunity 17:201-210. [DOI] [PubMed] [Google Scholar]
- 21.Cross, D. A., D. R. Alessi, P. Cohen, M. Andjelkovich, and B. A. Hemmings. 1995. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378:785-789. [DOI] [PubMed] [Google Scholar]
- 22.Cuevas, B., Y. Lu, S. Watt, R. Kumar, J. Zhang, K. A. Siminovitch, and G. B. Mills. 1999. SHP-1 regulates Lck-induced phosphatidylinositol 3-kinase phosphorylation and activity. J. Biol. Chem. 274:27583-27589. [DOI] [PubMed] [Google Scholar]
- 23.Daikh, D., D. Wofsy, and J. B. Imboden. 1997. The CD28-B7 costimulatory pathway and its role in autoimmune disease. J. Leukoc. Biol. 62:156-162. [DOI] [PubMed] [Google Scholar]
- 24.Datta, S. R., A. Brunet, and M. E. Greenberg. 1999. Cellular survival: a play in three Akts. Genes Dev. 13:2905-2927. [DOI] [PubMed] [Google Scholar]
- 25.Fallarino, F., P. E. Fields, and T. F. Gajewski. 1998. B7-1 engagement of cytotoxic T lymphocyte antigen 4 inhibits T cell activation in the absence of CD28. J. Exp. Med. 188:205-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frauwirth, K. A., J. L. Riley, M. H. Harris, R. V. Parry, J. C. Rathmell, D. R. Plas, R. L. Elstrom, C. H. June, and C. B. Thompson. 2002. The CD28 signaling pathway regulates glucose metabolism. Immunity 16:769-777. [DOI] [PubMed] [Google Scholar]
- 27.Greenwald, R. J., G. J. Freeman, and A. H. Sharpe. 2005. The B7 family revisited. Annu. Rev. Immunol. 23:515-548. [DOI] [PubMed] [Google Scholar]
- 28.Grohmann, U., C. Orabona, F. Fallarino, C. Vacca, F. Calcinaro, A. Falorni, P. Candeloro, M. L. Belladonna, R. Bianchi, M. C. Fioretti, and P. Puccetti. 2002. CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat. Immunol. 3:1097-1101. [DOI] [PubMed] [Google Scholar]
- 29.Gupta, N., A. M. Scharenberg, D. A. Fruman, L. C. Cantley, J. P. Kinet, and E. O. Long. 1999. The SH2 domain-containing inositol 5′-phosphatase (SHIP) recruits the p85 subunit of phosphoinositide 3-kinase during Fcγ RIIb1-mediated inhibition of B cell receptor signaling. J. Biol. Chem. 274:7489-7494. [DOI] [PubMed] [Google Scholar]
- 30.Hughes, T. R., M. Mao, A. R. Jones, J. Burchard, M. J. Marton, K. W. Shannon, S. M. Lefkowitz, M. Ziman, J. M. Schelter, M. R. Meyer, S. Kobayashi, C. Davis, H. Dai, Y. D. He, S. B. Stephaniants, G. Cavet, W. L. Walker, A. West, E. Coffey, D. D. Shoemaker, R. Stoughton, A. P. Blanchard, S. H. Friend, and P. S. Linsley. 2001. Expression profiling using microarrays fabricated by an ink-jet oligonucleotide synthesizer. Nat. Biotechnol. 19:342-347. [DOI] [PubMed] [Google Scholar]
- 31.Imboden, J. B., and G. A. Koretsky. 1995. Intracellular signalling. Switching off signals. Curr. Biol. 5:727-729. [DOI] [PubMed] [Google Scholar]
- 32.Ivaska, J., L. Nissinen, N. Immonen, J. E. Eriksson, V. M. Kahari, and J. Heino. 2002. Integrin α2β1 promotes activation of protein phosphatase 2A and dephosphorylation of Akt and glycogen synthase kinase 3β. Mol. Cell. Biol. 22:1352-1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones, R. G., M. Parsons, M. Bonnard, V. S. Chan, W. C. Yeh, J. R. Woodgett, and P. S. Ohashi. 2000. Protein kinase B regulates T lymphocyte survival, nuclear factor kappaB activation, and Bcl-X(L) levels in vivo. J. Exp. Med. 191:1721-1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kane, L. P., P. G. Andres, K. C. Howland, A. K. Abbas, and A. Weiss. 2001. Akt provides the CD28 costimulatory signal for up-regulation of IL-2 and IFN-gamma but not T(H)2 cytokines. Nat. Immunol. 2:37-44. [DOI] [PubMed] [Google Scholar]
- 35.Krauss, S., M. D. Brand, and F. Buttgereit. 2001. Signaling takes a breath-new quantitative perspectives on bioenergetics and signal transduction. Immunity 15:497-502. [DOI] [PubMed] [Google Scholar]
- 36.Lee, K. M., E. Chuang, M. Griffin, R. Khattri, D. K. Hong, W. G. Zhang, D. Straus, L. E. Samelson, C. B. Thompson, and J. A. Bluestone. 1998. Molecular basis of T cell inactivation by CTLA-4. Science 282:2263-2266. [DOI] [PubMed] [Google Scholar]
- 37.Leverrier, Y., J. Thomas, A. L. Mathieu, W. Low, B. Blanquier, and J. Marvel. 1999. Role of PI3-kinase in Bcl-X induction and apoptosis inhibition mediated by IL-3 or IGF-1 in Baf-3 cells. Cell Death Differ. 6:290-296. [DOI] [PubMed] [Google Scholar]
- 38.Levine, B. L., W. B. Bernstein, M. Connors, N. Craighead, T. Lindsten, C. B. Thompson, and C. H. June. 1997. Effects of CD28 costimulation on long-term proliferation of CD4(+) T cells in the absence of exogenous feeder cells. J. Immunol. 159:5921-5930. [PubMed] [Google Scholar]
- 39.Lu, Y., Q. Yu, J. H. Liu, J. Zhang, H. Wang, D. Koul, J. S. McMurray, X. Fang, W. K. A. Yung, K. A. Siminovitch, and G. B. Mills. 2003. Src family protein-tyrosine kinases alter the function of PTEN to regulate phosphatidylinositol 3-kinase/AKT cascades. J. Biol. Chem. 278:40057-40066. [DOI] [PubMed] [Google Scholar]
- 40.Marengere, L. E., P. Waterhouse, G. S. Duncan, H. W. Mittrucker, G. S. Feng, and T. W. Mak. 1996. Regulation of T cell receptor signaling by tyrosine phosphatase SYP association with CTLA-4. Science 272:1170-1173. [DOI] [PubMed] [Google Scholar]
- 41.Masteller, E. L., E. Chuang, A. C. Mullen, S. L. Reiner, and C. B. Thompson. 2000. Structural analysis of CTLA-4 function in vivo. J. Immunol. 164:5319-5327. [DOI] [PubMed] [Google Scholar]
- 42.Mellor, A. L., and D. H. Munn. 2004. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat. Rev. Immunol. 4:762-774. [DOI] [PubMed] [Google Scholar]
- 43.Millward, T. A., S. Zolnierowicz, and B. A. Hemmings. 1999. Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem. Sci. 24:186-191. [DOI] [PubMed] [Google Scholar]
- 44.Munn, D. H., M. D. Sharma, and A. L. Mellor. 2004. Ligation of B7-1/B7-2 by human CD4+ T cells triggers indoleamine 2,3-dioxygenase activity in dendritic cells. J. Immunol. 172:4100-4110. [DOI] [PubMed] [Google Scholar]
- 45.Namboodiripad, A. N., and M. L. Jennings. 1996. Permeability characteristics of erythrocyte membrane to okadaic acid and calyculin A. Am. J. Physiol. 270:C449-C456. [DOI] [PubMed] [Google Scholar]
- 46.Nishimura, H., N. Minato, T. Nakano, and T. Honjo. 1998. Immunological studies on PD-1-deficient mice: implication of PD-1 as a negative regulator for B cell responses. Int. Immunol. 10:1563-1572. [DOI] [PubMed] [Google Scholar]
- 47.Okazaki, T., A. Maeda, H. Nishimura, T. Kurosaki, and T. Honjo. 2001. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc. Natl. Acad. Sci. USA 98:13866-13871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oosterwegel, M. A., D. A. Mandelbrot, S. D. Boyd, R. B. Lorsbach, D. Y. Jarrett, A. K. Abbas, and A. H. Sharpe. 1999. The role of CTLA-4 in regulating Th2 differentiation. J. Immunol. 163:2634-2639. [PubMed] [Google Scholar]
- 49.Pages, F., M. Ragueneau, R. Rottapel, A. Truneh, J. Nunes, J. Imbert, and D. Olive. 1994. Binding of phosphatidylinositol-3-OH kinase to CD28 is required for T-cell signalling. Nature 369:327-329. [DOI] [PubMed] [Google Scholar]
- 50.Parry, R. V., K. Reif, G. Smith, D. M. Sansom, B. A. Hemmings, and S. G. Ward. 1997. Ligation of the T cell co-stimulatory receptor CD28 activates the serine-threonine protein kinase protein kinase B. Eur. J. Immunol. 27:2495-2501. [DOI] [PubMed] [Google Scholar]
- 51.Parry, R. V., C. A. Rumbley, L. H. Vandenberghe, C. H. June, and J. L. Riley. 2003. CD28 and inducible costimulatory protein Src homology 2 binding domains show distinct regulation of phosphatidylinositol 3-kinase, Bcl-x(L), and IL-2 expression in primary human CD4 T lymphocytes. J. Immunol. 171:166-174. [DOI] [PubMed] [Google Scholar]
- 52.Phan, G. Q., J. C. Yang, R. M. Sherry, P. Hwu, S. L. Topalian, D. J. Schwartzentruber, N. P. Restifo, L. R. Haworth, C. A. Seipp, L. J. Freezer, K. E. Morton, S. A. Mavroukakis, P. H. Duray, S. M. Steinberg, J. P. Allison, T. A. Davis, and S. A. Rosenberg. 2003. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc. Natl. Acad. Sci. USA 100:8372-8377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Probst, H. C., K. McCoy, T. Okazaki, T. Honjo, and B. M. van den. 2005. Resting dendritic cells induce peripheral CD8+ T cell tolerance through PD-1 and CTLA-4. Nat. Immunol. 6:280-286. [DOI] [PubMed] [Google Scholar]
- 54.Rathmell, J. C., R. L. Elstrom, R. M. Cinalli, and C. B. Thompson. 2003. Activated Akt promotes increased resting T cell size, CD28-independent T cell growth, and development of autoimmunity and lymphoma. Eur. J. Immunol. 33:2223-2232. [DOI] [PubMed] [Google Scholar]
- 55.Riley, J. L., and C. H. June. 2005. The CD28 family: a T-cell rheostat for therapeutic control of T-cell activation. Blood 105:13-21. [DOI] [PubMed] [Google Scholar]
- 56.Riley, J. L., M. Mao, S. Kobayashi, M. Biery, J. Burchard, G. Cavet, B. P. Gregson, C. H. June, and P. S. Linsley. 2002. Modulation of TCR-induced transcriptional profiles by ligation of CD28, ICOS, and CTLA-4 receptors. Proc. Natl. Acad. Sci. USA 99:11790-11795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rudd, C. E., and H. Schneider. 2003. Unifying concepts in CD28, ICOS and CTLA4 co-receptor signalling. Nat. Rev. Immunol. 3:544-556. [DOI] [PubMed] [Google Scholar]
- 58.Salama, A. D., T. Chitnis, J. Imitola, H. Akiba, F. Tushima, M. Azuma, H. Yagita, M. H. Sayegh, and S. J. Khoury. 2003. Critical role of the programmed death-1 (PD-1) pathway in regulation of experimental autoimmune encephalomyelitis. J. Exp. Med. 198:71-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sheppard, K. A., L. J. Fitz, J. M. Lee, C. Benander, J. A. George, J. Wooters, Y. Qiu, J. M. Jussif, L. L. Carter, C. R. Wood, and D. Chaudhary. 2004. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. 574:37-41. [DOI] [PubMed] [Google Scholar]
- 60.Shlapatska, L. M., S. V. Mikhalap, A. G. Berdova, O. M. Zelensky, T. J. Yun, K. E. Nichols, E. A. Clark, and S. P. Sidorenko. 2001. CD150 association with either the SH2-containing inositol phosphatase or the SH2-containing protein tyrosine phosphatase is regulated by the adaptor protein SH2D1A. J. Immunol. 166:5480-5487. [DOI] [PubMed] [Google Scholar]
- 61.Song, J., S. Salek-Ardakani, P. R. Rogers, M. Cheng, L. Van Parijs, and M. Croft. 2004. The costimulation-regulated duration of PKB activation controls T cell longevity. Nat. Immunol. 5:150-158. [DOI] [PubMed] [Google Scholar]
- 62.Suh, W. K., B. U. Gajewska, H. Okada, M. A. Gronski, E. M. Bertram, W. Dawicki, G. S. Duncan, J. Bukczynski, S. Plyte, A. Elia, A. Wakeham, A. Itie, S. Chung, J. Da Costa, S. Arya, T. Horan, P. Campbell, K. Gaida, P. S. Ohashi, T. H. Watts, S. K. Yoshinaga, M. R. Bray, M. Jordana, and T. W. Mak. 2003. The B7 family member B7-H3 preferentially down-regulates T helper type 1-mediated immune responses. Nat. Immunol. 4:899-906. [DOI] [PubMed] [Google Scholar]
- 63.Tivol, E. A., A. N. Schweitzer, and A. H. Sharpe. 1996. Costimulation and autoimmunity. Curr. Opin. Immunol. 8:822-830. [DOI] [PubMed] [Google Scholar]
- 64.Vander Heiden, M. G., D. R. Plas, J. C. Rathmell, C. J. Fox, M. H. Harris, and C. B. Thompson. 2001. Growth factors can influence cell growth and survival through effects on glucose metabolism. Mol. Cell. Biol. 21:5899-5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vlahos, C. J., W. F. Matter, K. Y. Hui, and R. F. Brown. 1994. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J. Biol. Chem. 269:5241-5248. [PubMed] [Google Scholar]
- 66.Wang, J., and M. F. Wilkinson. 2000. Site-directed mutagenesis of large (13-kb) plasmids in a single-PCR procedure. BioTechniques 29:976-978. [DOI] [PubMed] [Google Scholar]
- 67.Ward, S. G., J. Westwick, N. D. Hall, and D. M. Sansom. 1993. Ligation of CD28 receptor by B7 induces formation of D-3 phosphoinositides in T lymphocytes independently of T cell receptor/CD3 activation. Eur. J. Immunol. 23:2572-2577. [DOI] [PubMed] [Google Scholar]
- 68.Watanabe, N., M. Gavrieli, J. R. Sedy, J. Yang, F. Fallarino, S. K. Loftin, M. A. Hurchla, N. Zimmerman, J. Sim, X. Zang, T. L. Murphy, J. H. Russell, J. P. Allison, and K. M. Murphy. 2003. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat. Immunol. 4:670-679. [DOI] [PubMed] [Google Scholar]
- 69.Whetton, A. D., G. W. Bazill, and T. M. Dexter. 1984. Haemopoietic cell growth factor mediates cell survival via its action on glucose transport. EMBO J. 3:409-413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wu, C. J., D. M. O'Rourke, G. S. Feng, G. R. Johnson, Q. Wang, and M. I. Greene. 2001. The tyrosine phosphatase SHP-2 is required for mediating phosphatidylinositol 3-kinase/Akt activation by growth factors. Oncogene 20:6018-6025. [DOI] [PubMed] [Google Scholar]
- 71.Yi, L. A., S. Hajialiasgar, and E. Chuang. 2004. Tyrosine-mediated inhibitory signals contribute to CTLA-4 function in vivo. Int. Immunol. 16:539-547. [DOI] [PubMed] [Google Scholar]
- 72.Zhang, S. Q., W. G. Tsiaras, T. Araki, G. Wen, L. Minichiello, R. Klein, and B. G. Neel. 2002. Receptor-specific regulation of phosphatidylinositol 3′-kinase activation by the protein tyrosine phosphatase Shp2. Mol. Cell. Biol. 22:4062-4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.