

Abstract
Immortalization and malignant transformation are important steps in tumor development. The ability to induce these processes from normal human epithelial cells with genetic alterations frequently found in the corresponding human cancer would significantly enhance our understanding of tumor development. Alterations in several key intracellular regulatory pathways (the pRB, p53, and mitogenic signaling pathways and the telomere maintenance system) appear to be sufficient for the neoplastic transformation of normal human cells. Nevertheless, in vitro transformation models to date depend on viral oncogenes, most prominently the simian virus 40 early region, to induce immortalization and malignant transformation of normal human epithelial cells. Here, we demonstrate a transformation model creating oral–esophageal cancer cells by using a limited set of genetic alterations frequently observed in the corresponding human cancer. In a stepwise model, cyclin D1 overexpression and p53 inactivation led to immortalization of oral keratinocytes. Additional ectopic epithelial growth factor receptor overexpression followed by c-myc overexpression as well as consecutive reactivation of telomerase induced by epithelial growth factor receptor sufficed to transform oral epithelial cells, truly recapitulating the development of the corresponding human disease.
Keywords: cyclin D1, epithelial growth factor receptor, in vitro transformationl, telomerase, c-myc
Cultured oral–esophageal squamous epithelial cells provide a good model to study basic keratinocyte biology as well as processes of immortalization and malignant transformation, both of which are important steps in squamous carcinogenesis. Normal or primary oral keratinocytes display a restricted replicative life span in culture. Those cells initially proliferate but eventually enter a state of permanent growth arrest, called replicative senescence (1, 2). It has been suggested that senescence forms a barrier against tumorigenesis and that the acquisition of the ability to proliferate an unlimited number of times, termed immortalization, is therefore an essential step in the malignant transformation of cells. Immortalization is closely linked to the maintenance of telomeres either through activation of telomerase or alternative mechanisms to maintain telomeres (ALT). In addition to telomere maintenance, some of the most commonly known genetic alterations in cancer development, such as the inactivation of the p53 and pRB pathways, play a critical role in processes of immortalization (3, 4).
Overexpression of cyclin D1 is a common genetic alteration in human squamous cell carcinomas, especially of the oral–esophageal epithelium (5–7). It can be considered a cell-type-specific equivalent to pRB inactivation; consequently, inactivating mutations of pRB are not seen in oral squamous cell carcinomas. Additionally, p53 mutations are frequently observed in oral cancer. Furthermore, p53 is inactivated in a high proportion of oral dysplastic lesions, implicating a role for p53 inactivation in the induction of immortalization (8). By following these patterns of genetic events seen in in vivo tumor development, we defined the role of cyclin D1 overexpression and functional p53 inactivation in the process of immortalization of human oral squamous epithelial cells. Cyclin D1 alone and in combination with dominant negative p53 (dnp53) was ectopically expressed in normal human oral keratinocytes. Whereas cyclin D1 overexpression extended the replicative life span of oral keratinocytes, additional p53 inactivation resulted in the immortalization of these cells (9). Interestingly, immortalization was independent of telomerase activation. Instead, telomere length was maintained by an ALT mechanism (9), which was surprising because squamous cancer rarely show an ALT mechanism. Nevertheless, these cells did not display features of malignant transformation and therefore cannot be directly compared with squamous cancer cells.
In recent years, several groups have demonstrated that the serial introduction of the simian virus 40 (SV40) early region, H-rasV12 and human telomerase reverse transcriptase (hTERT) sufficed to transform several types of human cells, including epithelial cells (10–13). It was subsequently demonstrated that the large T antigen (LT) and small T antigen (st) of SV40 are required for the transforming activity of the SV40 early region (14, 15). Although the actions of SV40 LT are well characterized, the pathways altered by SV40 st remained undefined for a long time. Recently, it could be demonstrated that one critical target of SV40 st is the phosphatidyl-inositol 3-kinase (PI3K) pathway. Activation of PI3K signaling functionally mimics the expression of SV40 st and conferred anchorage-independent growth and tumorigenicity of human mammary epithelial cells (16). Other combinations of introduced genes have also been successfully used to transform human cells (17), but all of these in vitro transformation models required some viral oncogenes to induce the phenotype of malignant transformation or were done in fibroblasts (18). Nevertheless, these observations suggest that dysregulation of a limited set of pathways should be sufficient to transform normal human epithelial cells and that further dissection of the signaling pathways, perturbed by the respective genes introduced, will identify key carcinogenic steps of a specific tumor type.
Besides perturbation of the pRB and p53 pathways, overexpression of the epithelial growth factor receptor (EGFR) and c-myc oncogenes are common genetic alterations in oral–esophageal carcinomas. EGFR overexpression is observed in the majority of human oral–esophageal cancer (19–21) and frequently associated with an overexpression of EGFR ligands, such as EGF or TGF-α (22). The two major signal transduction pathways used by EGFR are the mitogen-activated protein kinase (MAPK) pathway and the PI3K pathway. Numerous studies have highlighted amplification and aberrant activation of PI3K and its downstream target AKT in many types of human cancers (23, 24). As a target of SV40 st the PI3K/AKT pathway has now also been implicated in human cell transformation. Together, these observations demonstrate the important role of PI3K signaling in malignant transformation.
The c-myc oncogene is also frequently altered in human oral–esophageal cancer (25, 26). c-myc regulates multiple biological processes, including cell proliferation, growth, and differentiation (27). Recent studies with conditional transgenic mice have shown that c-myc activation is especially important in the maintenance of solid tumors (28). Furthermore, several studies have shown that c-myc can cooperate with other genetic alterations, such as activated ras, in the transformation of rodent as well as human cells (10, 16). Interestingly, ras mutations do not play a role in oral–esophageal carcinogenesis (29).
Here, we demonstrate that serial introduction of cyclin D1, dnp53, EGFR, and c-myc, all important genetic alterations in oral–esophageal carcinogenesis, permits previously normal oral keratinocytes to grow in an anchorage-independent fashion as one key feature of malignant transformation. Furthermore, these cells induce tumors in nude mice. EGFR activates PI3K and phosphorylates AKT as well as induces a robust reactivation of telomerase in these cells. This in vitro transformation model demonstrates a stepwise generation of human cancer cells using the genetic alterations observed in the corresponding human tumor.
Methods
Retroviral Vectors, Infection, and Cell Culture. The retroviral expression vector pBPSTR-D1 was obtained from S. A. Reeves (Massachusetts General Hospital Cancer Center, Cambridge) and contained the puromycin resistance gene under the control of the 5′LTR promoter. The LXSN vector containing the p53 coding sequence with a V143A mutation was used to generate dnp53. Both vectors are described in ref. 9. For the EGFR-expressing retroviral vector (pFB-EGFR-neo) (30), the neomycin cassette was replaced with a hygromycin resistance gene. The retroviral vector pBabe-c-myc-blasto is described in ref. 16.
The amphotropic packaging cell line Phoenix A was grown in supplemented DMEM (Sigma) and transiently transfected with the respective retroviral vector to generate amphotropic retroviruses. Retroviral supernatant was harvested 48 h after transfection, filtered through a 0.45-μm filter, and freshly used for infection. Exponentially growing cells were infected in the presence of 4 μg/ml polybrene. After infection, successfully transduced polyclonal cell populations were obtained by selection with the appropriate antibiotic: 1 μg/ml puromycin, 400 μg/ml G418, 50 μg/ml hygromycin, and/or 2.5 μg/ml blasticidin, respectively. Infection frequencies were typically 20–30%.
Normal human oral keratinocytes were established from a biopsy of normal floor of the mouth (OKF6) or gingival mucosa (OKM1) of clinically and genetically normal tissue. OKF6 cells have been cryopreserved within their first two serial passages in culture and characterized extensively (9, 31, 32). OKM1 were established from a young male patient without any history of smoking. OKF6 and OKM1 as well as the lines generated thereof were grown in defined keratinocyte serum-free medium with defined growth supplement (Invitrogen) and final Ca2+ concentration of 0.4 mM. The medium was supplemented with antibiotics.
Determination of Replicative Life Span. Serial cultures of the different OKF6 and OKM1 lines were performed in 10-cm dishes by plating 105 cells, refeeding the cells every second day, and subculturing every 4–5 days. The doubling number of each passage was calculated with the formula PD = (nf/n0)/log2 where n0 is the initial number of cells and nf is the final number of cells. Immortalization was defined as cell growth of at least three times beyond the life span of the parental cells.
Western Blot Analysis. Lysates from exponentially growing cells were harvested in a buffer (50 mM Hepes, pH 7.4/0.1% Nonidet P-40/250 mM NaCl) with 1 mM protease and 10 mM phosphatase inhibitors. Of the total amount of protein, 10 μg was separated by 6–10% SDS/PAGE and transferred to immobilon membranes (Millipore). Blocking was performed in 5% milk/10 mM Tris·HCl, pH 7.4/150 mM NaCl/0.2% Tween 20 for 4 h, followed by incubation with primary antibodies (1:3,000). The secondary antibody was peroxidase-conjugated anti-mouse, anti-rabbit, or anti-goat Ig at 1:2,500 (Amersham Pharmacia). Detection was by enhanced chemiluminescence (ECL, Amersham Biosciences). Primary antibodies used were monoclonal cyclin D1 antibody (HD11), polyclonal cyclin D1 antibody (H295), monoclonal p53 antibodies (421 and 122), polyclonal EGFR antibody (1005), polyclonal AKT1/PKBα, and monoclonal phospho-AKT (Ser-473 and Thr-308) antibodies, polyclonal extracellular signal-regulated kinase (ERK)1/2 (C-14) and monoclonal phospho-ERK1/2 (N-4) antibodies as well as polyclonal c-myc (E-262) antibody. Quantification was done with aida 3.20 image software (Raytest, Straubenhault, Germany).
Telomeric Repeat Amplification Protocol (TRAP) Assays/Telomeric Length Assays. All generated oral keratinocytes were assayed for telomerase activity by using the PCR-based TRAP assay (11). Cellular extracts (100 ng) along with a heat-inactivated control were used for TRAP assays. Telomere length was measured by hybridizing a 32P-labeled telomeric (CCCTAA) probe to 10 μg of HinfI- and RsaI-digested genomic DNA as described in ref. 11.
Molecular HLA Analysis. HLA class I and II loci were amplified by PCR using locus- and sequence-specific primers in a tiered typing strategy. PCR-product DNA was hybridized with established sequence-specific oligonucleotide probes and immobilized probe arrays to identify specific HLA class I (HLA-A and HLA-B) and class II (HLA-DRB1) alleles (33) at the University of Freiburg Blood Bank.
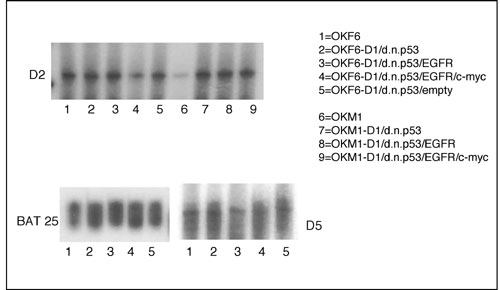
Microsatellite Analysis. Microsatellite analysis on all generated cells was performed by PCR amplification. Two dinucleotide repeats (D2S123 and D5S346) and two mononucleotide repeats (BAT25 and BAT26) were used as microsatellite polymorphic markers for the generated oral keratinocytes. For more information, see Fig. 7, which is published as supporting information on the PNAS web site.
Anchorage Independent Growth Assay. Cells (104 or 105) were seeded per 60-mm plate with a bottom layer of 0.6% Bacto agar in keratinocyte-serum-free medium (K-SFM) and a top layer of 0.3% Bacto agar containing K-SFM, with or without additional EGF (5 ng/ml). Fresh K-SFM (0.5 ml) was added after 1.5 weeks. Colonies were scored after 3 weeks. Only colonies >0.2 mm in diameter were counted. Such colonies typically contain 50–60 cells. At least two independent assays were performed in triplicate.
Tumorigenicity Assays. Six- to eight-week-old immunocompromised mice (severe combined immunodeficient mice, Charles River Laboratories) were used for s.c. injection with the generated OKF6 cells (2 × 106). To increase the probability of tumor formation, 1 × 106 BJ human foreskin fibroblasts or 10 ng/ml EGF was coinjected with OKF6-D1/dnp53/EGFR cells, respectively. Tumor size was monitored every 3 days. Mice were killed after 16 weeks of monitoring. SCC-15 or TE-12 cells (oral–esophageal squamous cancer cell lines) served as positive control. Total autopsies were done on all mice. Each tumor was dissected, measured, fixed in 4% paraformaldehyde, embedded in paraffin, and processed for hematoxylin/eosin staining. All histologic changes were assessed independently in a blinded fashion by three different observers.
Results
Growth Characteristics of Human Oral Keratinocytes Overexpressing Cyclin D1, dnp53, and EGFR. In an independent set of experiments, we confirmed that normal human oral keratinocytes (OKF6) are immortalized by overexpression of cyclin D1 and inactivation of wild-type p53. These immortalized oral epithelial cells (OKF6-D1/dnp53) show no increase in telomerase activity but recruit an ALT mechanism, as previously shown (9). To rule out a predisposing condition allowing OKF6 cells to be more readily immortalized, we confirmed the induction of immortalization by cyclin D1 overexpression and p53 inactivation with independent normal human oral keratinocytes (OKM1) established from a different patient. This patient was a young male without a history of smoking, making a predisposing genetic alteration unlikely. To ectopically overexpress EGFR in OKM1/OKF6-D1/dnp53 cells, we introduced pFB-EGFR-hyg by retrovirus-mediated gene transfer. To assess the growth characteristics of these OKM1/OKF6-D1/dnp53/EGFR cells, we observed serial passages in culture in the presence or absence of additional EGF. The population doubling (PD) rate in OKM1/OKF6-D1/dnp53/EGFR cells was significantly higher compared with the already immortalized OKM1/OKF6-D1/dnp53 cells. This PD rate could be further increased by the addition of exogenous EGF (Fig. 1; see also Fig. 5). The levels of endogenous and ectopically expressed EGFR in the different lines were determined using Western blot analysis (Fig. 2). All of these results along with those from the following experiments were confirmed in the respective OKM1 cells.
Fig. 1.

Replicative life span of parental and derived oral keratinocytes. Growth characteristics of OKF6, OKF6-D1, OKF6-dnp53, OKF6-D1/dnp53, OKF6-D1/dnp53/EGFR, and OKF6-D1/dnp53/EGFR plus EGF. The replicative life span was assessed by calculating the PDs of each cell line. Immortalization was assessed if cells grew at least three times beyond the life span of the parental cells.
Fig. 5.

Replicative life span of oral keratinocytes additionally overexpressing EGFR and c-myc. Growth characteristics of OKF6-D1/dnp53, OKF6-D1/dnp53/EGFR, OKF6-D1/dnp53/EGFR plus EGF, and OKF6-D1/dnp53/EGFR/c-myc cells (a), as well as OKM1, OKM1-D1/dnp53, OKM1-D1/dnp53/EGFR, OKM1-D1/dnp53/EGFR plus EGF, and OKM1-D1/dnp53/EGFR/c-myc cells (b) are displayed. The replicative life span was assessed by calculating the PD values.
Fig. 2.

Effects of EGFR overexpression in the generated oral keratinocytes. Equal amounts of protein for OKF6, OKF6-D1, OKF6-dnp53, OKF6-D1/dnp53, OKF6-D1/dnp53/empty, and OKF6-D1/dnp53/EGFR were separated on a 6% SDS/PAGE. The level of EGFR overexpression was compared by subsequent immunoblot analysis using a polyclonal EGFR antibody (1005). EGFR overexpression is indicated as fold increase compared to parental OKF6 cells.
In Vitro Transformation of Human Oral Keratinocytes (OKM1/OKF6-D1/dnp53/EGFR). Next, we assessed the ability of each of these cell lines to proliferate in an anchorage-independent fashion, a hallmark of in vitro transformation (34). Consistent with our previous observation (9), immortalized OKF6-D1/dnp53 cells were not able to grow in soft agar conditions even with the addition of EGF. Newly generated OKM1-D1/dnp53 cells were also not able to form colonies in soft agar. In contrast, OKM1/OKF6-D1/dnp53/EGFR cells formed colonies in soft agar. Interestingly, the number of colonies formed was to a similar rate as with oral or esophageal cancer cells (SCC-15 and TE-12), which served as positive control (Table 1). Next, we assessed in vivo tumorigenicity of the generated oral keratinocytes in immunocompromised mice. Early passage OKF6-D1/dnp53/EGFR cells were injected into severe combined immunodeficient mice with or without additional EGF or fibroblasts (Table 2), but no tumors arose after an observation period of 4 months. OKF6-D1/dnp53 and TE-12/SCC-15 served as negative and positive controls, respectively (Table 2).
Table 1. Colony formation rate in soft agar analysis.
| Colonies in soft agar
|
||
|---|---|---|
| Cell type | 104 cells | 105 cells |
| OKF6-D1/dnp53 (plus EGF) | 0 (2) | 2 (2) |
| OKM1-D1/dnp53 (plus EGF) | 0 (0) | 0 (2) |
| OKF6-D1/dnp53/empty | 1 | 1 |
| OKM1-D1/dnp53/empty | 1 | 2 |
| OKF6-D1/dnp53/EGFR | 50 | 163 |
| OKM1-D1/dnp53/EGFR | 55 | 135 |
| OKF6-D1/dnp53/EGFR/c-myc | 59 | 162 |
| OKM1-D1/dnp53/EGFR/c-myc | 53 | 149 |
| TE-12 | 61 | 153 |
Results from two individual clones derived from each set of experiments gave identical results. Experiments were done in triplicate. Colony formation was scored after 3 weeks.
Table 2. Formation of tumors in immunodeficient nude mice.
| Cell type | No. of tumors |
|---|---|
| OKF6-D1/dnp53 | 0 of 8 |
| OKF6-D1/dnp53/EGFR | 0 of 8 |
| OKF6-D1/dnp53/EGFR plus EGF | 0 of 4 |
| OKF6-D1/dnp53/EGFR plus fibroblasts | 0 of 4 |
| OKF6-D1/d.n.p53/EGFR/c-myc | 10 of 10 |
| SCC-15 | 8 of 8 |
Results from two individual clones derived from each set of experiments gave identical results. Tumor formation was scored after 4 months.
Telomere Biology. As described in ref. 9, OKF6-D1/dnp53 cells maintain their telomeres by an ALT mechanism in the absence of substantial telomerase activity. To assess telomerase activity in OKF6-D1/dnp53/EGFR cells, we assayed the generated oral keratinocytes in a TRAP assay. Telomerase activity was absent or very low in OKF6, OKF6-D1, OKF6-dnp53, and OKF6-D1/dnp53 cells, whereas it was robust in OKF6-D1/dnp53/EGFR cells (Fig. 3a). Accordingly, telomere length measurements revealed a progressive shortening of telomeres that correlated with the replicative life span extension of oral keratinocytes. The immortalized OKF6-D1/dnp53 (200 PD) revealed an elongation of telomere length with a very heterogeneous lengthening, including very long telomeres of up to 40 kilobase pairs, which is consistent with ALT. After the introduction of EGFR, this heterogenous telomere length was replaced by very homogenous telomeres at a length of 20 kilobase pairs in EGFR overexpressing oral squamous epithelial cells, correlating with reactivated telomerase (Fig. 3b).
Fig. 3.

Telomerase activity and telomere length in parental and derived oral keratinocytes. (a) Cellular extracts (100 ng) of OKF6-hTERT, OKF6-D1, OKF6-dnp53, OKF6-D1/dnp53, OKF6-D1/dnp53/EGFR, as well as OKM1-D1/dnp53, OKM1-D1/dnp53/EGFR, and OKM1-D1/dnp53/EGFR/c-myc cells were assayed for telomerase activity using the PCR-based TRAP assay. Heat-treated (heat) samples served as negative control, and OKF6-hTERT served as a positive control. IC is an internal PCR control. (b) Telomere length for preimmortal OKF6-D1/dnp53 (100 PD), immortal OKF6-D1/dnp53 (200 PD), OKF6-D1/dnp53/EGFR, OKF6-D1/dnp53/EGFR/empty, and OKF6-D1/dnp53/EGFR/c-myc cells was analyzed by hybridization of genomic DNA with a specific oligonucleotide probe. Two different clones of OKF6-D1/dnp53/hTERT cells served as positive control.
The PI3K/AKT Pathway Is Activated in EGFR-Overexpressing, Immortal Keratinocytes. Because EGFR overexpression had multiple cellular effects, we next asked which pathway is activated by EGFR overexpression in oral keratinocytes. The two major growth factor-stimulated signal transduction pathways used by EGFR are the MAPK–ERK and the PI3K-AKT pathways, respectively. Several lines of evidence suggested that PI3K and its downstream target AKT are activated in oral–esophageal carcinogenesis (35). To assess the effects of EGFR overexpression on those two signaling pathways, we measured the expression level and the level of phosphorylation of AKT and ERK in OKF6-D1/dnp53/EGFR cells. The level of AKT expression was unchanged compared with OKF6-D1/dnp53 cells as determined by Western blot analysis. Nevertheless, increased phosphorylation of AKT on Ser-473 was found in OKF6-D1/dnp53/EGFR cells (Fig. 4a). We observed similar results when we analyzed the phosphorylation status of AKT at Thr-308 (data not shown). In contrast, ERK1/2 levels and phosphorylation status remained unchanged in EGFR-overexpressing oral keratinocytes (Fig. 4a).
Fig. 4.

Western Blot analysis of AKT/phospho-AKT, ERK/phospho-ERK as well as c-myc in the generated oral keratinocytes. (a) Equal amounts of protein of OKF6-D1/dnp53, OKF6-D1/dnp53/empty, and OKF6-D1/dnp53/EGFR cells were separated with 10% SDS/PAGE, and a Western Blot was performed. The primary antibodies used were polyclonal AKT1/PKBa, monoclonal phospho-AKT (Ser-473), polyclonal ERK1/2 (C-14), and monoclonal phospho-ERK1/2 (E-4). (b) Equal amounts of protein for OKF6, OKF6-D1, OKF6-D1/dnp53, OKF6-D1/dnp53/EGFR, OKF6-D1/dnp53/EGFR/c-myc (two individual clones), and TE-12 were separated with 10% SDS/PAGE. The level of c-myc overexpression was compared by immunoblot analysis using a polyclonal c-myc antibody (N-262).
Additional c-myc Expression Allows in Vitro and in Vivo Transformation of Oral Keratinocytes. Because c-myc is an important oncogene in human and murine transformation systems, we sought to investigate whether c-myc can cooperate with cyclin D1 overexpression, p53 inactivation, and EGFR overexpression to induce a fully transformed phenotype in oral keratinocytes. c-myc was therefore retrovirally overexpressed in OKM1/OKF6-D1/dnp53/EGFR cells (Fig. 4b). As with each retroviral transduction, at least two different independent lines were generated. The generated OKF6-D1/dnp53/EGFR/c-myc cells displayed a culture growth to the same rate as OKF6-D1/dnp53/EGFR (Fig. 5a). The same holds true for OKM1-D1/dnp53/EGFR/c-myc cells, compared with their EGFR-overexpressing counterparts (Fig. 5b). These cells were then tested for their ability to grow in an anchorage-independent fashion. OKM1/OKF6-D1/dnp53/EGFR/c-myc cells displayed a colony formation rate in soft agar comparable with the OKM1/OKF6-D1/dnp53/EGFR cells or with oral–esophageal cancer cells (Table 1). Furthermore, additional c-myc overexpression permitted oral keratinocytes to promote tumor formation in vivo. OKF6-D1/dnp53/EGFR/c-myc cells injected into athymic nude mice induced small tumors in these mice corresponding with a poorly differentiated squamous-spindle cell histology (Table 2 and Fig. 6). Overexpression of c-myc in cells without EGFR overexpression did not allow colony formation in soft agar or tumor formation in mice (data not shown).
Fig. 6.

Nude mouse tumors induced by generated oral cancer cells. Tumors induced by the generated human oral cancer cells contain histologic features of poorly differentiated squamous cell cancer mixed with spindle cells. Depicted are representative photomicrographs of tumors from OKF6-D1/dnp53/EGFR/c-myc (two individual clones) shown with hematoxylin/eosin staining. (Magnification, ×400.)
Polymorphic Markers Confirm the Origin of the Generated Human Oral Cancer Cells. To rule out any contamination in the process of malignant transformation or the development of tumors in nude mice, we tested different polymorphic markers on OKF6 and OKM1 oral keratinocytes and their respective derivates. Four independent microsatellite markers (D2S123, D5S346, BAT25, and BAT26) were analyzed in a PCR-based assay. OKF6, OKM1, and all of the generated derivates stably expressed all of the tested microsatellite markers. Furthermore, HLA genotyping was performed on the genomic DNA of all generated oral keratinocytes and an OKF6-D1/dnp53/EGFR/c-myc-induced tumor. All cells derived from primary OKF6 and OKM1, respectively, up to oral cancer cells OKF6/OKM1-D1/dnp53/EGFR/c-myc as well as the tumor DNA derived from those cells displayed the same set of HLA types as their respective parental cells (Table 3, which is published as supporting information on the PNAS web site). This result additionally confirms the human origin of the tumor cells comprising the nude mouse tumors.
Discussion
Squamous oral–esophageal cancer is a common cancer worldwide. Cyclin D1 overexpression, p53 inactivation, EGFR overexpression, and c-myc amplification with consecutive overexpression are frequent genetic alterations observed in this type of cancer (6, 8, 19, 25). Nevertheless, it is not known how these genetic alterations interact during the transition from normal human oral or esophageal epithelial cells to immortalized epithelial cells and finally to squamous cancer cells. The ability to recapitulate this process of malignant transformation would therefore substantially enhance our understanding of squamous carcinogenesis. In this study, we demonstrate that sequential introduction of a limited set of genetic alterations frequently observed in the corresponding human cancer sufficed primary epithelial cells to transform into squamous cancer cells.
Malignant transformation in primary cells was first described in rodents by introducing viral or human oncogenes (36, 37). The transformation of such cultured primary cells, e.g., involved the introduction of at least two collaborating oncogenes, such as ras and c-myc or E1A (37, 38). Indications that the neoplastic transformation of human cells is even more complex came from numerous attempts to transform cultured normal human cells by introducing different genetic alterations. Invariably, such attempts failed (39, 40). These failures suggested that human cells require an even greater number of genetic alterations than rodent cells for transformation to a neoplastic state.
Studies in recent years have determined that alterations in a limited set of pathways may indeed be sufficient to transform human cells to tumor cells (11). The pathways required seem to involve the pRB and p53 pathways, the maintenance of telomeres through activation of telomerase or through ALT mechanisms, as well as regulatory mechanisms disrupting the dependence of normal cells on mitogenic stimulation, most frequently through the ras signaling pathway (41). Many in vitro transformation systems to date have been successfully used to transform human cells with different combinations of introduced genes, most prominently the combination of the SV40 early region, H-rasV12, and hTERT (10, 11, 17, 18, 42, 43). Nevertheless, all of these in vitro transformation models required some viral oncogenes to induce the phenotype of malignant transformation or were done in fibroblasts (17, 43). Thus, the genetic elements used did not correspond to those altered in a respective human cancer. Starting with primary oral keratinocytes isolated from a floor of the mouth biopsy or a gingival biopsy and termed OKF6 or OKM1, respectively, we have succeeded in transforming such cells to tumorigenicity through the introduction of a limited set of genetic alterations.
In most in vitro transformation systems to date, the SV40 early region, with one of its oncoproteins SV40 LT, is used to inactivate the pRB and p53 tumor suppressors (44). The pRB pathway plays a central part in determining whether a cell will proceed through the G1 phase of the cell cycle (45). Besides by viral oncoproteins, disruption of this pathway can be mediated by a number of alternative genetic and epigenetic alterations. Overexpression of the cyclin D1 oncogene, especially in squamous cell cancer, will have the identical outcome and was consequently used. As in most human tumors, the majority of oral–esophageal cancer cells harbor a p53 gene mutation. Elimination of functioning p53 is necessary to inactivate the apoptotic machinery in many types of cancer cells (46). One way to inactivate p53 is by using dominant negative mutants, which prevents p53's action on its target genes. We used a dnp53 mutant, V143A, to inactivate p53 in oral keratinocytes. The combination of cyclin D1 overexpression and p53 inactivation sufficed to immortalize these cells, as demonstrated in ref. 9. Interestingly, this immortalization was independent of telomerase activation. Instead, an ALT pathway of telomere maintenance was recruited. Ongoing maintenance of telomeres and not the activation of telomerase is the prerequisite for the indefinite proliferation of cells, the phenotype of immortalization, and is clearly an intrinsic part of neoplastic transformation. The immortalized cells also displayed multiple chromosomal rearrangements, including end-to-end fusions and translocations (9). This finding was confirmed by quantitative FISH analysis, which demonstrated a higher percentage of signal free ends in these cells, followed by very long and heterogeneous telomeres (M. Pantic and O.G.O, unpublished data).
Nevertheless, oral keratinocytes harboring a cyclin D1 overexpression, a p53 inactivation, and an ALT mechanism to maintain telomeres failed to form colonies in soft agar or produce tumors in nude mice. In most systems, cells overexpressing SV40 early region and hTERT are immortal but also fail to form tumors unless the ras pathway was additionally disrupted. The ras gene is mutated in many human cancers, but does not play a role in oral–esophageal cancer. Therefore, other pathways regulating the dependence of normal cells on mitogenic stimulation, needed to be altered in any appropriate model system of malignant transformation in human oral epithelial cells. EGFR overexpression is frequently observed in human oral–esophageal squamous cell cancer, and interestingly, pathways used for EGFR signaling, namely the MAPK–ERK pathway and the PI3K–AKT pathway, substantially overlap with those perturbed by activated ras. We could demonstrate that additional overexpression of EGFR allows oral keratinocytes with cyclin D1 overexpression and p53 inactivation to form colonies in soft agar, one hallmark of malignant transformation. The pathway recruited by EGFR is the PI3K-AKT pathway via phosphorylation of AKT at Ser-473 and Thr-308. The MAPK–ERK pathway remains unperturbed. Furthermore, additional overexpression of EGFR robustly reactivated telomerase in these cells. Consequently, telomeres become very stable in EGFR-overexpressing keratinocytes, which is also observed in quantitative FISH analysis (M. Pantic and O.G.O., unpublished data). This finding is of even greater interest with respect to a potential role of telomerase in tumor formation in addition to its function of elongating telomeres (17, 47, 48). This observation could also indicate that tumor cells can use an ALT mechanism in the process of immortalization, but telomerase activity is required for malignant transformation and tumor progression. The number of tumors or tumor cell lines being ALT positive (10–15%) might therefore not represent the role of ALT in the process of malignant transformation. Thus, ALT might play an important role in the progression from normal oral cells to oral cancer cells, even though it is rarely observed in oral–esophageal cancer.
There are some data reporting EGF as a potential transactivating molecule of hTERT expression (49, 50). Nevertheless, all these data are generated in telomerase-positive cells, and a de novo induction of telomerase by EGFR remains surprising. Studies that dissect the pathways involved in this robust reactivation of telomerase will be interesting to see. In this context, it is also surprising that only ectopic overexpression of EGFR produced high enough EGFR levels to induce the cellular effects observed, including the in vitro transformation phenotype, although EGFR is already up-regulated in highly proliferating immortal oral keratinocytes. Most likely, there is a threshold level of EGFR required to induce these cellular effects. Further studies to determine this threshold level are necessary.
Transformation studies using the SV40 early region have revealed that both the LT and the st of SV40 are required for malignant transformation of human cells (41, 51, 52). Given that SV40 LT binds and inactivates pRB and p53, SV40 st interacts with the serine threonine phosphatase PP2A. Target molecules of PP2A and SV40 st were largely unknown until recently. Recent findings demonstrate that SV40 st can perturb the PI3K pathway as a downstream target during the transformation of human mammary epithelial cells. In this setting, AKT phosphorylation is increased by EGF stimulation in SV40 st-expressing cells (16). Here, we demonstrate AKT phosphorylation in EGFR-overexpressing cells, and therefore unintentionally were able to mimic the effect of SV40 st overexpression. A very recent study demonstrated that PTEN inactivation upstream of PI3K also leads to the same result, mimicking SV40 st overexpression (43). Again, this in vitro transformation study was performed in fibroblasts. With the additional introduction of the c-myc oncogene we were able to fully transform our oral epithelial cells, then harboring a cyclin D1 overexpression, p53 inactivation, EGFR overexpression, telomerase reactivation and c-myc overexpression. These cells named OKF6-D1/dnp53/EGFR/c-myc or OKM1-D1/dnp53/EGFR/c-myc form colonies in soft agar and induce tumor growth in nude mice. Besides complementing ras in in vitro transformation systems (16, 37), it has been recently demonstrated that c-myc is also a downstream target of PP2A and therefore SV40 st (53). With the overexpression of EGFR acting through PI3K–AKT and the overexpression of c-myc as a new downstream target of PP2A, we circumvented the need for SV40 st and ras in our in vitro transformation system of oral epithelial cells, establishing a cellular cancer model truly recapitulating the development of the corresponding human tumor.
Supplementary Material
Acknowledgments
We thank S. A. Reeves for the cyclin D1 construct, C. N. Arnold and C. Schulz-Huotari for technical assistance, and members of the A.K.R. and O.G.O. laboratories for discussion. This work was supported by Deutsche Krebshilfe Grants 10-1656-Op 1 and 10-2209-Op 2.
Author contributions: W.C.H., A.K.R., and O.G.O. designed research; G.G., M.Q., H.H., S.H., Y.S., M.D., A.v.W., C.F., and O.G.O. performed research; W.C.H., H.H., and H.N. contributed new reagents/analytic tools; G.G., M.Q., W.C.H., H.H., S.H., H.N., A.K.R., H.E.B., and O.G.O. analyzed data; and O.G.O. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: LT, large T antigen; st, short T antigen; PI3K, phosphatidyl-inositol 3-kinase; SV40, simian virus 40; hTERT, human telomerase reverse transcriptase; EGFR, epithelial growth factor receptor; MAPK, mitogen-activated protein kinase; dnp53, dominant negative p53; ALT, alternative mechanism to maintain telomeres; OKM, human oral keratinocytes of the gingival mucosa; OKF, human oral keratinocytes of the normal floor of the mouth; ERK, extracellular signal-regulated kinase; TRAP, telomeric repeat amplification protocol; PD, population doubling.
References
- 1.Hayflick, L. (1965) Exp. Cell Res. 37, 614–636. [DOI] [PubMed] [Google Scholar]
- 2.Harada, H., Nakagawa, H., Oyama, K., Takaoka, M., Andl, C. D., Jacobmeier, B., von Werder, A., Enders, G. H., Opitz, O. G. & Rustgi, A. K. (2003) Mol. Cancer Res. 1, 729–738. [PubMed] [Google Scholar]
- 3.Reddel, R. R. (2000) Carcinogenesis 21, 477–484. [DOI] [PubMed] [Google Scholar]
- 4.Hahn, W. C. & Weinberg, R. A. (2002) Nat. Rev. Cancer 2, 331–341. [DOI] [PubMed] [Google Scholar]
- 5.Bartkova, J., Lukas, J., Muller, H., Strauss, M., Gusterson, B. & Bartek, J. (1995) Cancer Res. 55, 949–956. [PubMed] [Google Scholar]
- 6.Nakagawa, H., Zukerberg, L., Togawa, K., Meltzer, S. J., Nishihara, T. & Rustgi, A. K. (1995) Cancer 76, 541–549. [DOI] [PubMed] [Google Scholar]
- 7.Nakagawa, H., Wang, T. C., Zukerberg, L., Odze, R., Togawa, K., May, G. H., Wilson, J. & Rustgi, A. K. (1997) Oncogene 14, 1185–1190. [DOI] [PubMed] [Google Scholar]
- 8.Todd, R., Hinds, P. W., Munger, K., Rustgi, A. K., Opitz, O. G., Suliman, Y. & Wong, D. T. (2002) Crit. Rev. Oral. Biol. Med. 13, 51–61. [DOI] [PubMed] [Google Scholar]
- 9.Opitz, O. G., Suliman, Y., Hahn, W. C., Harada, H., Blum, H. E. & Rustgi, A. K. (2001) J. Clin. Invest. 108, 725–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elenbaas, B., Spirio, L., Koerner, F., Fleming, M. D., Zimonjic, D. B., Donaher, J. L., Popescu, N. C., Hahn, W. C. & Weinberg, R. A. (2001) Genes Dev. 15, 50–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hahn, W. C., Counter, C. M., Lundberg, A. S., Beijersbergen, R. L., Brooks, M. W. & Weinberg, R. A. (1999) Nature 400, 464–468. [DOI] [PubMed] [Google Scholar]
- 12.Lundberg, A. S., Randell, S. H., Stewart, S. A., Elenbaas, B., Hartwell, K. A., Brooks, M. W., Fleming, M. D., Olsen, J. C., Miller, S. W., Weinberg, R. A. & Hahn, W. C. (2002) Oncogene 21, 4577–4586. [DOI] [PubMed] [Google Scholar]
- 13.Zimonjic, D., Brooks, M. W., Popescu, N., Weinberg, R. A. & Hahn, W. C. (2001) Cancer Res. 61, 8838–8844. [PubMed] [Google Scholar]
- 14.Yu, J., Boyapati, A. & Rundell, K. (2001) Virology 290, 192–198. [DOI] [PubMed] [Google Scholar]
- 15.Hahn, W. C., Dessain, S. K., Brooks, M. W., King, J. E., Elenbaas, B., Sabatini, D. M., DeCaprio, J. A. & Weinberg, R. A. (2002) Mol. Cell. Biol. 22, 2111–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao, J. J., Gjoerup, O. V., Subramanian, R. R., Cheng, Y., Chen, W., Roberts, T. M. & Hahn, W. C. (2003) Cancer Cell 3, 483–495. [DOI] [PubMed] [Google Scholar]
- 17.Seger, Y. R., Garcia-Cao, M., Piccinin, S., Cunsolo, C. L., Doglioni, C., Blasco, M. A., Hannon, G. J. & Maestro, R. (2002) Cancer Cell 2, 401–413. [DOI] [PubMed] [Google Scholar]
- 18.Brookes, S., Rowe, J., Ruas, M., Llanos, S., Clark, P. A., Lomax, M., James, M. C., Vatcheva, R., Bates, S., Vousden, K. H., et al. (2002) EMBO J. 21, 2936–2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blume-Jensen, P. & Hunter, T. (2001) Nature 411, 355–365. [DOI] [PubMed] [Google Scholar]
- 20.Wells, A., Kassis, J., Solava, J., Turner, T. & Lauffenburger, D. A. (2002) Acta Oncol. 41, 124–130. [DOI] [PubMed] [Google Scholar]
- 21.Partridge, M., Gullick, W. J., Langdon, J. D. & Sherriff, M. (1988) Br. J. Oral Maxillofac. Surg. 26, 381–389. [DOI] [PubMed] [Google Scholar]
- 22.Grandis, J. R. & Tweardy, D. J. (1993) Cancer Res. 53, 3579–3584. [PubMed] [Google Scholar]
- 23.Li, J., Yen, C., Liaw, D., Podsypanina, K., Bose, S., Wang, S. I., Puc, J., Miliaresis, C., Rodgers, L., McCombie, R., et al. (1997) Science 275, 1943–1947. [DOI] [PubMed] [Google Scholar]
- 24.Steck, P. A., Lin, H., Langford, L. A., Jasser, S. A., Koul, D., Yung, W. K. & Pershouse, M. A. (1999) Genes Chromosomes Cancer 24, 135–143. [DOI] [PubMed] [Google Scholar]
- 25.Ishizuka, T., Tanabe, C., Sakamoto, H., Aoyagi, K., Maekawa, M., Matsukura, N., Tokunaga, A., Tajiri, T., Yoshida, T., Terada, M. & Sasaki, H. (2002) Biochem. Biophys. Res. Commun. 296, 152–155. [DOI] [PubMed] [Google Scholar]
- 26.Mandard, A. M., Hainaut, P. & Hollstein, M. (2000) Mutat. Res. 462, 335–342. [DOI] [PubMed] [Google Scholar]
- 27.Dang, C. V. (1999) Mol. Cell. Biol. 19, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pelengaris, S., Littlewood, T., Khan, M., Elia, G. & Evan, G. (1999) Mol. Cell 3, 565–577. [DOI] [PubMed] [Google Scholar]
- 29.Hollstein, M. C., Peri, L., Mandard, A. M., Welsh, J. A., Montesano, R., Metcalf, R. A., Bak, M. & Harris, C. C. (1991) Cancer Res. 51, 4102–4106. [PubMed] [Google Scholar]
- 30.Andl, C. D., Mizushima, T., Nakagawa, H., Oyama, K., Harada, H., Chruma, K., Herlyn, M. & Rustgi, A. K. (2003) J. Biol. Chem. 278, 1824–1830. [DOI] [PubMed] [Google Scholar]
- 31.Lindberg, K. & Rheinwald, J. G. (1990) Differentiation 45, 230–241. [DOI] [PubMed] [Google Scholar]
- 32.Rheinwald, J. G., Hahn, W. C., Ramsey, M. R., Wu, J. Y., Guo, Z., Tsao, H., De Luca, M., Catricala, C. & O'Toole, K. M. (2002) Mol. Cell. Biol. 22, 5157–5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Erlich, H. A. & Trachtenberg, E.A. (2001) in Molecular Epidemiology: A Practical Approach, eds. Carrington, M. & Hoelzel, A.R. (Oxford Univ. Press, Oxford), pp. 181–207.
- 34.Cifone, M. A. & Fidler, I. J. (1980) Proc. Natl. Acad. Sci. USA 77, 1039–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Okano, J., Gaslightwala, I., Birnbaum, M. J., Rustgi, A. K. & Nakagawa, H. (2000) J. Biol. Chem. 275, 30934–30942. [DOI] [PubMed] [Google Scholar]
- 36.Hanahan, D. & Weinberg, R. A. (2000) Cell 100, 57–70. [DOI] [PubMed] [Google Scholar]
- 37.Land, H., Parada, L. F. & Weinberg, R. A. (1983) Nature 304, 596–602. [DOI] [PubMed] [Google Scholar]
- 38.Ruley, H. E. (1983) Nature 304, 602–606. [DOI] [PubMed] [Google Scholar]
- 39.Stevenson, M. & Volsky, D. J. (1986) Mol. Cell. Biol. 6, 3410–3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sager, R., Tanaka, K., Lau, C. C., Ebina, Y. & Anisowicz, A. (1983) Proc. Natl. Acad. Sci. USA 80, 7601–7605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hahn, W. C. & Weinberg, R. A. (2002) N. Engl. J. Med. 347, 1593–1603. [DOI] [PubMed] [Google Scholar]
- 42.Drayton, S., Rowe, J., Jones, R., Vatcheva, R., Cuthbert-Heavens, D., Marshall, J., Fried, M. & Peters, G. (2003) Cancer Cell 4, 301–310. [DOI] [PubMed] [Google Scholar]
- 43.Boehm, J. S., Hession, M. T., Bulmer, S. E. & Hahn, W. C. (2005) Mol. Cell. Biol. 25, 6464–6474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sullivan, C. S. & Pipas, J. M. (2002) Microbiol. Mol. Biol. Rev. 66, 179–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaelin, W. G., Jr. (1999) BioEssays 21, 950–958. [DOI] [PubMed] [Google Scholar]
- 46.Shen, Y. & White, E. (2001) Adv. Cancer Res. 82, 55–84. [DOI] [PubMed] [Google Scholar]
- 47.Stewart, S. A., Hahn, W. C., O'Connor, B. F., Banner, E. N., Lundberg, A. S., Modha, P., Mizuno, H., Brooks, M. W., Fleming, M., Zimonjic, D. B., et al. (2002) Proc. Natl. Acad. Sci. USA 99, 12606–12611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chang, S. & DePinho, R. A. (2002) Proc. Natl. Acad. Sci. USA 99, 12520–12522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maida, Y., Kyo, S., Kanaya, T., Wang, Z., Yatabe, N., Tanaka, M., Nakamura, M., Ohmichi, M., Gotoh, N., Murakami, S. & Inoue, M. (2002) Oncogene 21, 4071–4079. [DOI] [PubMed] [Google Scholar]
- 50.Budiyanto, A., Bito, T., Kunisada, M., Ashida, M., Ichihashi, M. & Ueda, M. (2003) J. Invest. Dermatol. 121, 1088–1094. [DOI] [PubMed] [Google Scholar]
- 51.Ali, S. H. & DeCaprio, J. A. (2001) Semin. Cancer Biol. 11, 15–23. [DOI] [PubMed] [Google Scholar]
- 52.Rundell, K. & Parakati, R. (2001) Semin. Cancer Biol. 11, 5–13. [DOI] [PubMed] [Google Scholar]
- 53.Yeh, E., Cunningham, M., Arnold, H., Chasse, D., Monteith, T., Ivaldi, G., Hahn, W. C., Stukenberg, P. T., Shenolikar, S., Uchida, T., et al. (2004) Nat. Cell Biol. 6, 308–318. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}