

Short abstract
Overlapping but oppositely oriented transcripts have the potential to form sense-antisense perfect double-stranded (ds) RNA duplexes. A bioinformatics approach has identified over 217 candidate overlapping transcriptional units, bringing the total number of predicted and validated examples of overlapping but oppositely oriented transcripts to over 300.
Abstract
Background
Overlapping but oppositely oriented transcripts have the potential to form sense-antisense perfect double-stranded (ds) RNA duplexes. Over recent years, the number and variety of examples of mammalian gene-regulatory phenomena in which endogenous dsRNA duplexes have been proposed or demonstrated to participate has greatly increased. These include genomic imprinting, RNA interference, translational regulation, alternative splicing, X-inactivation and RNA editing. We computationally mined public mouse and human expressed sequence tag (EST) databases to search for additional examples of bidirectionally transcribed genomic regions.
Results
Our bioinformatics approach identified over 217 candidate overlapping transcriptional units, almost all of which are novel. From experimental validation of a subset of our predictions by orientation-specific RT-PCR, we estimate that our methodology has a specificity of 84% or greater. In many cases, regions of sense-antisense overlap within the 5'- or 3'-untranslated regions of a given transcript correlate with genomic patterns of mouse-human conservation.
Conclusions
Our results, in conjunction with the literature, bring the total number of predicted and validated examples of overlapping but oppositely oriented transcripts to over 300. Several of these cases support the hypothesis that a subset of the instances of substantial mouse-human conservation in the 5' and 3' UTRs of transcripts might be explained in part by functionality of an overlapping transcriptional unit.
Background
Characterized examples of endogenous antisense RNA in metazoans can be broadly divided into two categories (see [1,2] for extensive review). Antisense RNAs transcribed from loci distinct from their putative targets, such as lin-4 of Caenorhabditis elegans, are generally short and have the potential to form imperfect duplexes with complementary regions of their sense counterparts [3]. In contrast, antisense transcripts that originate from the same genomic region (but with opposing orientation) have, by virtue of their common but complementary origin, the potential to form long perfect duplexes.
Experimental evidence suggests a functional role for sense-antisense pairings at a surprising variety of levels in mammalian gene regulation, including genomic imprinting [4,5], RNA interference [6], translational regulation [7], alternative splicing [8], X-inactivation [9], and RNA editing [10]. Where the mode of regulation has been explored in detail each case has proved unique, so that it is difficult to make generalizations about mechanism or function. In transfection assays, full-length constructs of three splice variants of an endogenous coding transcript containing regions antisense to the FGF2 (fibroblast growth factor-2) mRNA can each suppress protein levels (but not mRNA levels) of FGF2 [7]. A non-coding transcript antisense to a homeobox gene, MSX1, has a conserved transcription initiation site and is expressed in inverse correlation to the MSX1 protein [11]. SCA8, an untranslated transcript implicated in spinocerebellar ataxia type 8, overlaps the 5' translation and transcription sites of KLHL1, a gene primarily expressed in the cerebellum [12]. XIST and TSIX are conserved, overlapping, but oppositely oriented non-coding transcripts, which serve crucial functions in X-inactivation [9]. Several imprinted loci generate multiple sense and antisense transcripts that are subject to reciprocal genomic imprinting, and a recent study demonstrated in vivo that premature termination of AIR, a non-coding imprinted antisense transcript, results in a failure to imprint several sense counterparts [5]. Lipman [13] suggested that this phenomenon might be much more widespread than previously believed, and hypothesized that the existence of functionally relevant overlapping antisense transcripts might explain a subset of the many cases in which strong evolutionary conservation is observed in 5' and 3' untranslated regions of mammalian genes.
Consistent with this, the number of known examples of pairs of RNA species with the potential to form long sense-antisense duplexes has increased steadily. We reviewed the literature and found 40 such cases (Table 1), and a recent analysis identified more than 80 additional pairs of annotated human mRNAs that originate from the same genomic locus and share regions of overlap [14]. We postulated that additional examples of this phenomenon might be obtainable by mining public human and mouse expressed sequence tag (EST) databases.
Table 1.
Overlapping transcriptional units in mammalian genomes previously described in the literature
| Sense | Antisense | References |
| IGF2R | AIR | [31] |
| ASE-1 | ERCC1 | [32] |
| COPG2 | COPG2AS | [33] |
| MADH5 | DAMS | [34] |
| DLX1 | DLX1AS | [35] |
| DLX6 | DLX6AS | [36] |
| GTROSA26 | GTROSA26AS | [37] |
| IGF2 | IGF2AS | [4] |
| KCNQ1 | KCNQ1OT1 | [38] |
| MCM3AP | MCM3APAS | [39] |
| MSX1 | MSX1AS | [11] |
| KLHL1 | SCA8 | [12] |
| GNAS | NESP-AS | [40] |
| FGF2 | NUDT6 | [41] |
| RFPL1 | RFPL1S | [42] |
| RFPL3 | RFPL3S | [42] |
| SLC22A1L | SLC22A1LS | [43] |
| ST7OT1 | ||
| ST7 | ST7OT2 | [44-46] |
| ST7OT3 | ||
| XIST | TSIX | [9] |
| MKRN3 | ZNF127AS | [47] |
| HOXA11 | HOXA11-AS | [48] |
| WT1 | WIT1 | [49] |
| EIF2S1 | Not named | [50] |
| GNRHR2 | RBM8A | [51] |
| MATN4 | RBPSUHL | [52] |
| PMCH | AROM | [53] |
| SFRS2 | ET | [54] |
| THRA | NR1D1 | [55] |
| SURF2 | SURF4 | [56] |
| TP53 | Not named | [57] |
| MYC | Not named | [58] |
| MYCN | NCYM | [59] |
| MBP | MBP | [60,61] |
| TNFRSF17 | Not named | [62] |
| GNRH1 | SH | [63] |
| HSPA1B | Not named | [64] |
| PTGER1 | PRKCL1 | [65] |
| MYB | Not named | [66] |
| COL1A1 | Not named | [67] |
| MRPL27 | Not named | [68] |
We searched reviews [1,2] and carried out keyword searches of PubMed [69], and NCBI LocusLink [70] for pairs of mammalian mRNA species known to share regions of overlap. This list does not include any of the 80 or so recently described examples of overlapping annotated mRNAs [14] ('cis-NATs') that were not observed elsewhere in the literature. A summary of these can be found in the 'cis-NATs' section of the authors' website [71].
Thousands of EST libraries, consisting in sum of millions of single-pass sequence reads, have been generated by investigators from around the world, using a variety of methods, and deposited into public sequence databases. UniGene [15] is an experimental algorithm developed at the NCBI, in which full-length mRNA and EST sequences are partitioned into a non-redundant set of gene-oriented clusters on the basis of nucleotide-level identity (using annotated mRNAs as initializing seeds), but these clusters are not further curated. The avoidance of spurious alignments by masking of transcribed repetitive elements, vector contaminants or low-complexity sequence is an important part of the UniGene build procedure. Each EST thus belongs to both an individual library from which it was sequenced (for example, a specific tissue from a specific individual) and a single UniGene cluster (along with other ESTs that are presumably derived from the same gene). We hypothesized that as a consequence of the automation of the UniGene build procedure, unannotated antisense transcripts might be found co-clustered with their sense counterparts. As nearly all annotated mRNAs in GenBank serve as UniGene cluster seeds (and therefore cannot be co-clustered with one another), such a strategy is strongly biased towards finding pairs of overlapping transcripts where one or both of the transcripts are unannotated.
Our bioinformatics and experimental strategy involved five steps. We first identified EST libraries that were directionally cloned and sequenced (that is, ESTs were cloned and sequenced in a defined orientation with respect to the mRNA transcript); then, focusing exclusively on ESTs from such libraries, we searched for UniGene clusters containing a statistically significant number of misoriented ESTs. We then mapped the mRNA and EST sequences from candidate UniGene clusters to their genomic coordinates and evaluated whether putative sense and antisense ESTs in a given UniGene cluster represented distinct RNA species, on the basis of differential exon-intron splicing structures, the locations of poly(A) signals and tails, and patterns of mouse-human sequence conservation. Finally, we experimentally validated a subset of the predictions by orientation-specific reverse transcription PCR (RT-PCR).
Results
A major obstacle to deducing the transcriptional orientation of individual ESTs is that not all cloning methods used to generate EST libraries are directional. For example, a substantial fraction of publicly available ESTs were generated by a random priming method that provides no information about directionality of transcription [16], whereas other methods that exploit the 3' polyadenylated tails of eukaryotic mRNAs do provide directional information. Rather than relying on fragmentary library annotation, we developed a simple measure of the quality of directional cloning of each EST library. A subset of UniGene clusters includes at least one full-length mRNA sequence for which the correct transcriptional orientation is known. Focusing on ESTs belonging to these clusters, we estimated in silico the fraction of ESTs from each library that were correctly oriented, relative to the mRNA-defined orientations (Figure 1). Our subsequent analysis focused exclusively on ESTs from 899 human and 176 mouse libraries for which we estimated that more than 95% of ESTs were in the correct orientation.
Figure 1.
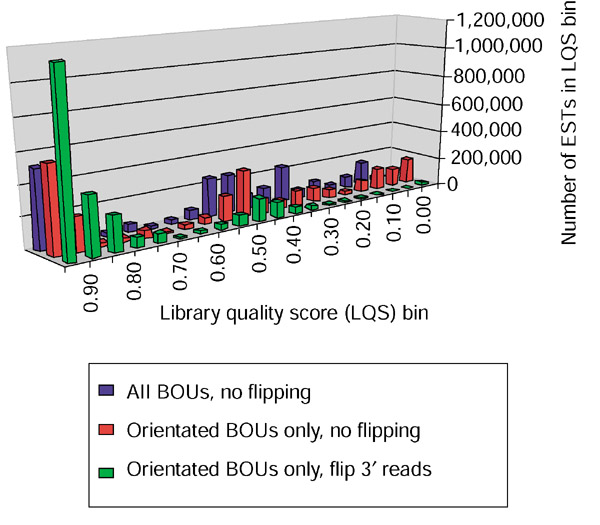
Assessment of the quality of directional cloning of individual EST libraries. Bins of library quality scores (LQS) at intervals of 0.05 are depicted along the x-axis. The heights of the bars reflect the number of human ESTs derived from libraries with an LQS that falls in a given bin. The LQS of each EST library was determined by calculating the fraction of ESTs from a given library that were deposited in the same orientation as the best-of-UniGene (BOU) representative of the UniGene cluster to which a given EST belonged. In our initial analysis, we assumed that all BOU representatives were correctly oriented (blue bars). As this is not the case, we repeated that analysis by calculating the LQS exclusively from ESTs belonging to UniGene clusters where the BOU representative possessed a defined ORF, indicating that it was correctly oriented (red bars). As a final improvement, we flipped in silico all ESTs annotated as 3' sequencing reads, as these are generally not reoriented before deposit in sequence databases. The result was a bimodal distribution of LQS scores (green bars) that appears to correspond broadly with directional (peak near LQS = 1.0) and non-directional (peak near LQS = 0.5) library generation protocols. A full list of both mouse and human EST libraries and their LQS scores is available at our website [22].
As expected, a small fraction of ESTs (around 1.5% on average) from these libraries were incorrectly oriented. Our null hypothesis was that these represented random artifacts that would be distributed across the full set of UniGene clusters in a manner proportional to the size of the individual UniGene clusters. In other words, we expected that 98.5% of the directionally cloned ESTs in each UniGene cluster would be correctly oriented. Binomial statistics were applied to identify UniGene clusters that contained a statistically significant overrepresentation of incorrectly oriented ESTs. For the substantial subset of UniGene clusters for which the dominant transcriptional orientation was unannotated, we required that the misoriented ESTs be significantly overrepresented regardless of the correct orientation of the cluster. In total, we were able to identify 549 mouse and human clusters that significantly deviated from the null hypothesis.
We next sought to address the possibility that a significant number of our candidates could represent systematic errors (for example, systematic bias for directional-cloning artifacts to occur in association with specific transcripts) or errors of the UniGene clustering algorithm. We postulated that if two distinct, overlapping but oppositely oriented RNA species were present in a single UniGene cluster, they should map to the same genomic region, but should possess significantly distinguishable exon-intron splicing structures.
We used publicly available tools (MEGABLAST [17] and SIM4 [18]) to map the exon-intron splicing structures of the relevant set of ESTs and mRNAs from candidate UniGene clusters onto draft assemblies of the human and mouse genomes [19,20]. Patterns of evolutionary conservation between these assemblies were determined by reference to a whole-genome set of approximately 1.15 million mouse-human sequence alignments [21]. Graphical representations were manually reviewed to evaluate whether the putative sense and antisense ESTs represented distinct RNA species, on the basis of differential exon-intron splicing structures, poly(A) signal and tail locations, and mouse-human conservation patterns. We identified 144 human and 73 mouse UniGene clusters that each appear to contain two distinct but oppositely oriented RNA species, co-clustered in UniGene as a consequence of a bidirectionally transcribed region of overlap. Figures 2,3,4,5,6,7 present several interesting representative examples of distinguishable exon-intron splicing structures of sense and antisense ESTs over the relevant genomic regions. For a data file containing a full tabulated list of the 217 candidates, see Additional data files and our website [22]. Graphical representations for all candidates, similar to those in Figures 2,3,4,5,6,7, are also available at [22].
Figure 2.
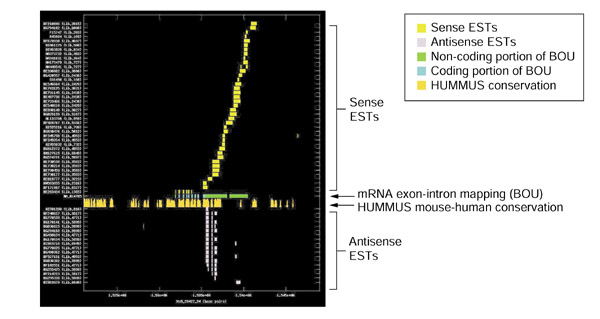
Splicing and mouse-human conservation patterns for sense and antisense ESTs from UniGene cluster Hs.47313. The graph depicts the exon-intron splicing structures of transcript sequences belonging to UniGene cluster Hs.47313. SIM4 [18] was used to map the exons of a single mRNA sequence (GenBank accession number NM_014785) and directionally cloned ESTs belonging to UniGene cluster Hs.47313 to genomic contig Hs9_28427_24 of the NCBI draft of the assembled human genome. The x-axis reflects base-pair positions along the genomic contig. Each position along the y-axis is assigned to a single EST or mRNA sequence. GenBank accession numbers are listed along with the UniLib ID of the library from which the EST was derived. Rectangular boxes indicate the locations of complete or partial exons. Individual exons of the BOU representative of this cluster (mRNA sequence NM_014785) are represented in blue and green, with annotated coding regions of the transcript shaded blue and untranslated regions shaded green. In this case, the mRNA is oriented from left to right with respect to the genomic contig. Immediately below the mRNA mapping, we have indicated the regions of the genome indicated to be highly conserved in HUMMUS [21], a set of around 1.15 million 'islands' of strong mouse-human conservation (in gold). The heights of individual bars in this row are proportional to the percent nucleotide identity over a 50-bp window centered on each base-pair. In the upper portion of the graph (all horizontal bars above the BOU mRNA sequence and HUMMUS rows), the exon mappings of sense ESTs are represented in yellow. In the lower portion of the graph (all horizontal bars below the BOU mRNA sequence and HUMMUS rows), exon mappings of antisense ESTs are represented in pink. Similar graphical representations for all 217 candidates (generated with GNUPLOT [27]) are available from our website [22]. The sense transcript (represented by the mRNA and sense ESTs) encodes KIAA0258, a protein of unknown function. Not unexpectedly, there is a strong correlation between the locations of sense transcript exons and the peaks in the strength of mouse-human conservation. It is also evident that the antisense ESTs are spliced in a consistent pattern that differs significantly from that of the mRNA and sense ESTs. This strengthens the claim that these represent a distinct RNA species inadvertently co-clustered into a single UniGene cluster by virtue of an antisense overlap. Observed regions of sense-antisense overlap are restricted to the 3' UTR of the sense transcript. Also striking is the observation that the islands of conservation in the 3' UTR of the BOU mRNA are largely coincident with the positions of exons of the putative antisense transcript, providing at least a potential explanation for the conserved elements observed in the 3' UTR of the sense mRNA. In this case, the antisense mRNA species does have strong homology to a known protein, suggesting that it is also a coding mRNA.
Figure 3.
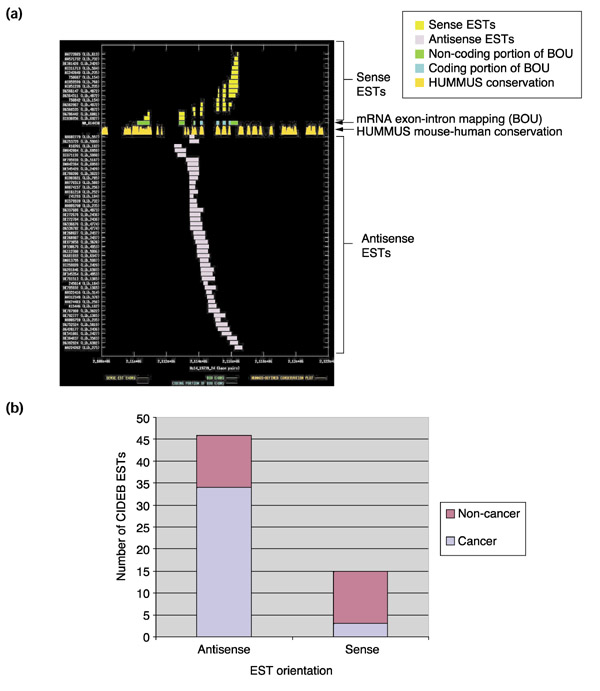
Splicing, mouse-human conservation patterns, and tissue origin of sense and antisense ESTs from UniGene cluster Hs.288835. (a) The graph depicts the exon-intron splicing structures of transcript sequences belonging to UniGene cluster Hs.288835. Organization of the figure as for Figure 2. The BOU mRNA (GenBank accession NM_014430) is oriented from left to right with respect to genomic contig Hs14_19739_24 of the NCBI human genome assembly. The mRNA encodes CIDEB (cell-death inducing DFFA-like effector B). With no exceptions, the sense-oriented ESTs have splicing patterns that are consistent with that of the mRNA. The antisense ESTs, however, consistently overlap with intronic sequence of the sense transcript, suggesting that they are derived from a distinct RNA species (presumably unspliced, at least in the region that we are observing). (b) A plot of EST numbers in the CIDEB cluster against orientation. The y-axis indicates the number of sense or antisense ESTs observed in the CIDEB cluster, and the relative proportions arising from neoplastic versus non-neoplastic tissues are indicated. A significantly greater fraction of the antisense ESTs (34/46 = ~0.74) than the sense ESTs (3/15 = ~0.2) were derived from neoplastic tissues (p = ~0.0002 by chi-squared statistic).
Figure 4.
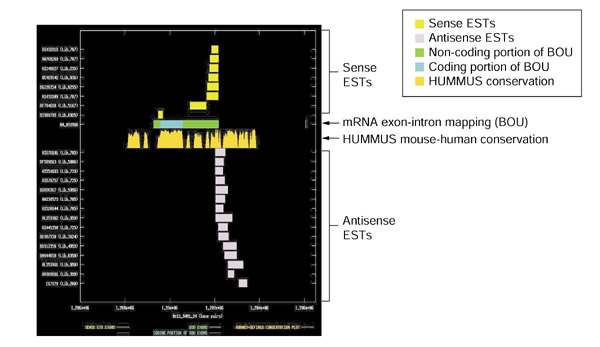
Splicing and mouse-human conservation patterns for sense and antisense ESTs from UniGene cluster Hs.113916. The graph depicts the exon-intron splicing structures of transcript sequences belonging to UniGene cluster Hs.113916. Organization of the figure as for Figure 2. The BOU mRNA (GenBank accession NM_032966) is oriented from left to right with respect to genomic contig Hs11_9491_24 of the NCBI human genome assembly. The mRNA encodes Burkitt lymphoma receptor 1, a GTP-binding protein. Although the transcript does not appear to be spliced, the sense ESTs terminate in a position consistent with that of the mRNA. Although the coding region of the sense transcript shows the highest degree of conservation between mouse and human, there are clearly islands of conservation within its 3' UTR. The antisense ESTs intersect with the most 3' portion of the sense transcript. They contain appropriately located polyadenylation signals, such that we are probably observing the 3' tail of an oppositely oriented transcript. The antisense ESTs have no protein homologies. It is worth noting that the most conserved stretch of the 3' UTR of the sense transcript is coincident with its region of overlap with the antisense RNA species.
Figure 5.
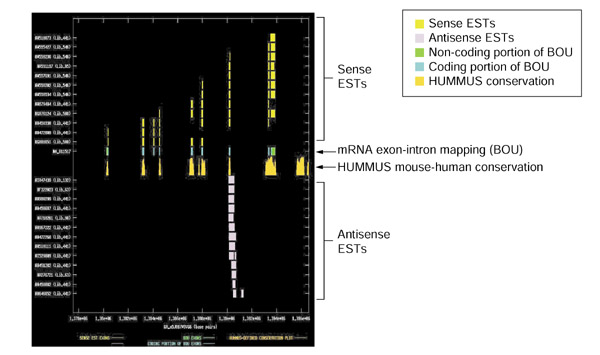
Splicing and mouse-human conservation patterns for sense and antisense ESTs from UniGene cluster Mm.148209. The graph depicts the exon-intron splicing structures of transcript sequences belonging to UniGene cluster Mm.148209. Organization of the figure as for Figure 2. The BOU mRNA (GenBank accession NM_011557) is oriented from left to right with respect to genomic contig GA_x5J8B7W5VG6 of the Celera mouse genome assembly. The mRNA encodes synaptonemal complex protein 3. The observed portion of the antisense species does not have protein-level homologies, and consistently overlaps a single internal coding exon of the sense transcript. Many of the antisense species are 3' reads containing an appropriately located poly(A) signal, suggesting that we are observing the 3' end of a larger transcript.
Figure 6.
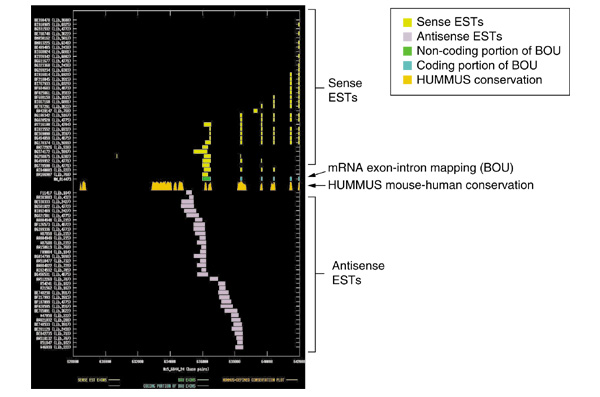
Splicing and mouse-human conservation patterns for sense and antisense ESTs from UniGene cluster Hs.125819. The graph depicts the exon-intron splicing structures of transcript sequences belonging to UniGene cluster Hs.125819. Organization of figure as for Figure 2. Note that the BOU mRNA (GenBank accession NM_014473) is in this case oriented from right to left with respect to genomic contig Hs5_6844_24 of the NCBI human genome assembly. The mRNA encodes a putative dimethyladenosine transferase. Notably, however, there appear to be two potential termini for the antisense ESTs (which are oriented from left to right), suggesting that we are observing either alternative termini of a single transcript or two distinct antisense RNA species. One terminus is coincident with an island of mouse-human conservation within the 3' UTR of the sense transcript. The second is coincident with the last internal coding exon of the sense transcript. In both cases, the sequence near each putative terminus contains an appropriately located polyadenylation signal. The antisense ESTs have no significant protein homologies, and do not appear to be spliced. However, the ESTs that we are observing may represent only the 3' terminus of a larger coding transcript. Notably, the islands of conservation immediately 'upstream' of the antisense ESTs also have no protein homologies, suggesting that this may not be the case.
Figure 7.
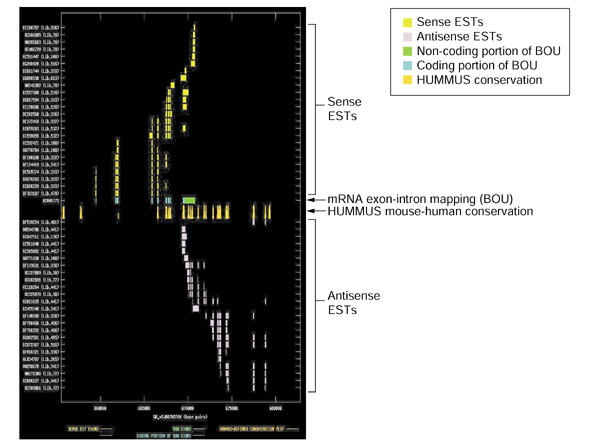
Splicing and mouse-human conservation patterns for sense and antisense ESTs from UniGene cluster Mm.10022. The graph depicts the exon-intron splicing structures of transcript sequences belonging to UniGene cluster Mm.10022. Organization of the figure as for Figure 2. The BOU mRNA (GenBank accession BC005773) is oriented from left to right with respect to genomic contig GA_x5J8B7W3T6H of the Celera mouse genome assembly. The mRNA is encoded by homer 3, a neuronal immediate early gene. The antisense species is homologous with a hypothetical human protein containing RNA helicase domains. This example is similar to Hs.47313 (Figure 2) in that the locations of strong mouse-human conservation in sub-regions within the 3' UTR of the sense transcript are coincident with the splicing structure of the antisense species.
To further validate our methodology, we sought to confirm a subset of our predictions experimentally. An orientation-specific RT-PCR assay was applied to test the directionality of transcription over the regions of predicted overlap (Figure 8). Primers were designed to amplify regions of predicted bidirectional transcription. The relative orientation of transcription was assessed by restricting which primers were present during the reverse transcriptase (RT) single-strand synthesis step of the reaction. Although total RNA from a series of different human tissues was used as template, only one tissue type was assayed per candidate, with the choice for each candidate geared towards the tissue types of libraries from which putative antisense ESTs were derived. We successfully detected the presence of antisense transcription over the queried region for 33 out of 39 candidates tested, and 0 out of 17 negative controls (Table 2). For 26 of these 33 candidates, both sense and antisense transcription was detected in the same tissue.
Figure 8.
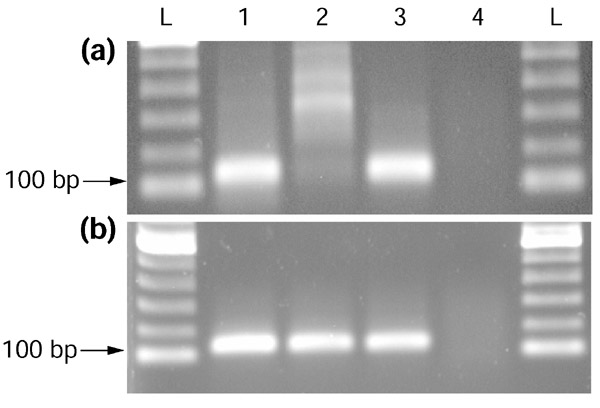
Assessment of transcriptional directionality by RT-PCR. Sample results from (a) a control and (b) a sense-antisense candidate. PCR primers were designed to amplify predicted regions of bidirectional transcription. Control primers were designed to amplify either non-overlapping regions of candidate transcripts or randomly selected regions of non-candidate transcripts. For each candidate or control, four RT-PCR reactions were carried out using total human RNA from a single tissue as template. Orientation of transcripts was assessed by restricting which primer was present during RT single-strand synthesis. 1, Both primers present during RT single-strand synthesis (positive control); 2, only antisense orientation-specific primer present during RT single-strand synthesis; 3, only sense-orientation-specific primer present during RT single-strand synthesis; 4, neither primer present during RT single-strand synthesis (negative control for genomic contamination). L, 100 bp DNA ladder (Gibco-BRL). In all four reactions, both primers were present during the subsequent PCR reactions. In these examples, the control primers in (a) targeted a 127 bp region of 'chromosome condensation-related SMC-associated protein 1' (NM_014865; Hs.5719) over which we did not observe bidirectional transcription, and the candidate primers in (b) targeted a 113 bp region of mannose-6-phosphate receptor (cation dependent) (NM_002355; Hs.75709) which our results suggested was shared by an overlapping RNA species. The template in both cases is total human placental RNA (Clontech). In the control (a) only sense transcription is detected over the queried region (the appropriately sized band in lane 3). In the candidate (b) both antisense and sense transcription are detected (appropriately sized bands in lanes 2 and 3, respectively).
Table 2.
Experimental evaluation of candidates by directional RT-PCR
| Sense (-)antisense (-) | Sense (+)antisense (+) | Sense (-)antisense (-) | Sense (+)antisense (+) | |
| Candidates | 0 | 6 | 7 | 26 |
| Negative controls | 1 | 17 | 0 | 0 |
Summary of results of directional RT-PCR reactions on 39 candidates and 18 controls (see Figure 8 and Materials and methods for description of the assay). PCR primers were designed to amplify predicted regions of bidirectional transcription. Control primers were designed to amplify either non-overlap regions of candidate transcripts or randomly selected regions of non-candidate transcripts. Six candidate primer sets and 17 negative control primer sets were positive for only sense transcription over the regions queried. Thirty-three candidate primer sets and no negative control primer sets were positive for antisense transcription over the regions queried. Of these 33 sets, 26 were also positive for sense transcription in the same tissue.
Discussion
Our bioinformatics approach identified 217 candidate instances of overlapping transcriptional units in the human and mouse genomes. We characterized the genomic arrangement of each pair of overlapping transcripts relative to one another (Table 3a, see also Additional data files). Our results are generally consistent with those of Lehner et al. [14], in that the majority of overlapping pairs can be described as having a tail-to-tail (3' to 3') arrangement. We were also interested in whether sense and antisense RNA species in these candidate clusters represented protein-coding or untranslated transcripts. For each candidate, we determined the best protein match to ESTs oriented in both the sense and antisense direction. Of the 217 candidate UniGene clusters, 116 contain ESTs with significant homology to a known protein on both strands, 95 on one strand, and six on neither strand. The identities of these best protein-level matches are provided as additional data with this paper and integrated with the graphical representation of each candidate at our website [22]. It is possible that a subset of the 101 candidates with no significant protein-level match on one or both strands include ESTs derived from non-coding transcripts. However, as ESTs represent fragments of the full transcripts, we cannot be certain about this until the full-length RNA species are cloned. The characteristics of each candidate pair with respect to observed coding potential and observed splicing (with the same caveat of being based on limited information) are listed in the additional data and are summarized in Table 3b.
Table 3.
Properties of overlapping pairs of transcripts
| (a) | Genomic arrangement | Number of pairs | |||
| Tail to tail (3' to 3') | 134 | ||||
| Head to head (5' to 5') | 12 | ||||
| Transcript starts in intron of the other transcript | 3 | ||||
| Transcript contained entirely within the other transcript | 39 | ||||
| Difficult to classify | 29 | ||||
| (b) | Candidate pairs | S/H | S/NH | NS/H | NS/NH |
| S/H | 92 | 24 | 18 | 65 | |
| S/NH | - | 0 | 3 | 2 | |
| NS/H | - | - | 1 | 8 | |
| NS/NH | - | - | - | 4 | |
(a) For each candidate, we characterized the genomic arrangement of the pair of transcripts relative to one another, based on the observed mappings of sense and antisense ESTs to genomic sequence. We utilized categories developed by Lehner et al. [14] to facilitate comparison. Categorizations of individual candidate pairs are provided in the additional data files. As ESTs represent fragments of the full transcripts, we cannot be conclusive about these categorizations until full-length RNA species are cloned. (b) The characteristics of each candidate pair with respect to observed coding potential (based on homologs in protein databases) and observed splicing were tabulated. A summary of the distribution of joint characteristics of each pair is presented here. S/H, splicing and protein homologies observed; NS/H, protein homologies observed, but splicing not observed; S/NH, splicing observed, but protein homologies not observed; NS/NH, neither splicing nor protein homologies observed. Categorizations of individual candidate pairs are provided in the additional data files. As ESTs represent fragments of the full transcripts, we cannot be conclusive about these categorizations until full-length RNA species are cloned.
The misinterpretation of genomic contaminants as putative antisense candidates is a clear concern. The observation of transcript splicing, protein homology and/or derivation from multiple independent libraries for any given set of putative antisense ESTs is evidence against genomic contamination. We have flagged (as requiring particular caution) 18 candidate cases where the set of antisense ESTs derive from one or a few libraries, and are not observed to be spliced or have protein homologies (see Additional data files).
Experimental validation of a subset of our predictions by orientation-specific RT-PCR supports our bioinformatics methodology. We estimate that our approach has a specificity of 84%, as we were able to detect antisense transcription over 33 of 39 regions queried. This may be a low-end estimate as we only queried one tissue per candidate, and cell type and/or temporal specificity of antisense transcript expression might explain our inability to confirm antisense transcription for six of the candidates experimentally. These same factors (differential temporal or cell-type distribution) may explain why the sense transcript (all of which are annotated mRNAs in the 39 cases that we attempted to experimentally verify) was not detected for seven candidates that were positive for antisense transcription.
The observation of highly conserved regions in the 5' and 3' untranslated regions (UTRs) of a large fraction of mammalian genes [23] has been an intriguing mystery. Lipman [13] hypothesized that the existence of functionally relevant overlapping antisense transcripts might explain a subset of these cases. Indeed, with a number of candidates we do observe interesting correlations between mouse-human nucleotide-level conservation patterns in UTRs and their region(s) of overlap with the putative antisense species. This includes both cases where the antisense species has homology to a known protein (Figures 2, 7) and cases where it does not (Figure 4; UniGene cluster Mm.41304; UniGene cluster Mm.183060).
There are seven cases in which a mouse candidate and human candidate are clear orthologs (Table 4). In six of these cases, the location, coding potential/identity and pattern of the overlap with the antisense RNA species is highly consistent between the mouse and human candidates, supporting the argument for functional relevance of these overlaps. Why is such little intersection observed between the human and mouse candidate sets? One possibility is that many of the examples of overlaps, whether functionally relevant or not, are not general to mammals but are lineage-specific in nature. A second possibility is that our method has a high frequency of false-negatives. Although EST databases are growing rapidly, it is clear that they are still under-sampling the full mammalian transcriptome. Undersampling of the transcriptome by either or both the mouse and human EST databases might be expected to contribute significantly to the false negative rate, and consequently to the relatively limited intersection between the mouse and human candidate sets. We are carrying out experiments to test mouse-human conservation of predicted overlaps more directly by focused and sensitive experimental assays such as quantitative RT-PCR and oligonucleotide arrays.
Table 4.
Orthologous mouse and human candidates
| Human candidate | Mouse candidate | Sense gene | Antisense gene or homolog |
| Hs.250697 | Mm.826 | TC10 | PIGF |
| Hs.211601 | Mm.4358 | MAP3K12 | No protein match |
| Hs.2210 | Mm.10167 | FLJ22865 | TRIP3 |
| Hs.296776 | Mm.20848 | RFXANK | LOC126382 |
| Hs.170263 | Mm.25231 | TP53BP1 | No protein match |
| Hs.330310 | Mm.176845 | KIAA0632 | G10 |
| Hs.343244 | Mm.10698 | AP1G2 | No protein match |
Seven pairs of mouse and human candidate UniGene clusters that contain clear orthologs are listed. In the first six, the location, coding potential/identity, and pattern of overlap of the sense and antisense RNA species are consistent between the mouse and human candidates. Thus, in the first example, a sub-region of the terminal exon of Ras-like protein overlaps the full terminal exon of a class F phosphatidylinositol glycan (Hs.250697 and Mm.826). In the second example, the 3' UTR of MAPKKK12 is overlapped by a transcript with no protein homologies in both mouse and human; moreover, this non-coding region of overlap is highly conserved at nucleotide level between mouse and human (Hs.211601 and Mm.4358).
As the proposed mechanisms by which the formation of long duplex dsRNA can potentially affect gene regulation are so varied [4,5,6,7,8,9,10,11,12], it is difficult to draw inferences regarding function without further experiments. One of the few areas where functionally relevant information on sequences is available relates to the neoplastic versus non-neoplastic nature of the tissue of origin of EST libraries. An interesting example is candidate UniGene cluster Hs.288835 (Figure 3), which contains CIDEB (cell-death inducing DFFA-like effector B). Noting that the sense transcript encoded a potential tumor suppressor, we checked the annotated tissue origin of these ESTs, and found that a significantly greater fraction of the antisense ESTs (34/46 = ~0.74) than the sense ESTs (3/15 = ~0.2) were derived from neoplastic tissues (p = ~0.0002 by chi-squared statistic). As the sense transcript codes for a pro-apoptotic gene, the result immediately suggests the interesting hypothesis that upregulation of the antisense RNA species in cancer tissues has functional relevance with respect to suppression of the potentially tumor-suppressing sense gene.
It is worth noting that stages of our methodology may also be useful for determining the correct transcriptional orientation of UniGene clusters that contain no annotated mRNA sequences. Many probes on orientation-sensitive oligonucleotide arrays for unknown genes are often based on such ESTs, and knowledge of the correct transcriptional orientation of each cluster may help circumvent problems such as those recently encountered in the design of an Affymetrix mouse chip [24].
We observed surprisingly little redundancy (10 out of 217 of our candidates) between our results and the literature (Table 5). As the sampling strategies applied seem more orthogonal than similar, it is difficult to assess how many more examples of overlapping transcriptional units in the human and mouse genomes remain to be discovered. Shoemaker and colleagues [25] carried out an experiment in which they queried the transcription of over 400,000 exon predictions using two strand-specific 60-mer oligonucleotide probe sets per exon. The negative controls, a set of probes that were the reverse complement of probes for 78,486 confirmed exons, indicated a 5% false-positive rate. We speculate that a subset of these false positives may have actually represented bidirectionally transcribed regions of the human genome.
Table 5.
Subset of 217 candidates for overlapping transcription previously described in the literature
| Candidate | Sense gene | Antisense gene or homolog |
| Hs.325978 | IL18BP | NUMA1 |
| Hs.330310 | KIAA0632 | G10 |
| Hs.283473 | PRO2900 | HDLBP |
| Hs.22116 | CDC14B | HAPB4 |
| Hs.279937 | KIAA1001 | FLJ10055 |
| Hs.301947 | SERHL | CGI-96 |
| Hs.172851 | ARG2 | VTI2 |
| Hs.276916 | NR1D1 | THRA |
| Hs.283061 | PRO1438 | LRMP |
| Hs.2182 | PMCH | AROM |
The set of 217 candidate mouse and human UniGene clusters was checked against examples from the literature (Table 1) and recently described cis-NATs [14,33]. This table lists ten candidates that have been previously described.
Conclusions
Our results, in conjunction with the literature, bring the total number of predicted and validated examples of overlapping but oppositely oriented transcripts to over 300. Given the variety of gene-regulatory phenomena that long-duplex dsRNA has been suggested or shown to influence [4,5,6,7,8,9,10,11,12], experimental approaches are required to query whether and how each of these overlaps is functionally relevant.
Materials and methods
Identification of high-quality directionally cloned ESTlibraries
Human UniGene (Build 146) and mouse UniGene (Build 100) datasets were downloaded from NCBI on 16 January, 2002. A useful feature of the UniGene resource is the identification of a single sequence in each cluster as its longest high-quality member. We refer to this set of representatives as the best-of-UniGene (BOU) sequences. To assess the quality of directional cloning in EST libraries, we applied the MEGABLAST tool [17] to align ESTs to the BOU of the UniGene cluster to which they belonged. For each EST library, we then calculated the fraction of member ESTs that were deposited in the same orientation as the BOU sequence of the UniGene cluster to which they belonged. This fraction, a metric of the quality of directional cloning of each EST library, is referred to as the library quality score (LQS).
Our original analysis was revised in two ways to improve its accuracy. Our goal of calculating library quality by estimating the 'correctness' of EST orientation is complicated by the fact that not every UniGene cluster contains an mRNA with a defined open reading frame (ORF), and not every BOU sequence is deposited in the correct orientation (in other words, the correct orientation of many UniGene clusters is not known definitively). We therefore revised our analysis to calculate LQS scores exclusively from UniGene clusters whose BOU representative was an mRNA with an annotated ORF window (indicating that the BOU is deposited in the correct orientation). We subsequently refer to these as 'oriented BOUs'. Another caveat arises in that 3' sequencing reads of directionally cloned ESTs are generally not reoriented before deposit of sequences in GenBank. We resolved this issue by 'flipping' in silico sequences annotated as 3' reads. The results of this analysis on the human EST dataset are shown in Figure 1. In our final analysis (green bars in Figure 1), the distribution of LQS scores across the full set of UniGene EST libraries is roughly bimodal, with a peak near LQS = 0.5 (random orientation of ESTs) and a peak near LQS = 1.0 (correct orientation of nearly all ESTs). These peaks correspond broadly to libraries generated by non-directional and directional cloning methods, respectively.
Of the 6,525 human and 566 mouse EST libraries considered, 899 and 176, respectively, had an LQS of greater than 0.95, indicating that these libraries were generated by an efficient method of directional cloning. The remainder of our analysis focused exclusively on the 1,151,724 human ESTs and 550,567 mouse ESTs derived from the libraries with LQS scores of greater than 0.95. A full list of the mouse and human EST libraries and their LQS scores is available at our website [22].
Statistically significant overrepresentation of misoriented ESTs in a subset of UniGene clusters
Our null hypothesis was that the relatively small fraction of misoriented ESTs from high-quality directionally cloned EST libraries (approximately 1.5%) represented random artifacts, leading to the expectation that they would be distributed across the full set of UniGene clusters in a manner proportional to the sizes of individual UniGene clusters. We applied binomial distribution probability analysis to identify clusters that significantly deviated from this expectation with a p-value of less than 0.00001 (roughly equivalent to the number of hypotheses being tested). This analysis was sufficient for UniGene clusters with an oriented BOU (see above). To avoid excluding from consideration UniGene clusters without oriented BOU sequences, we again applied the binomial distribution probability test, with an additional requirement; the result had to be significant (p < 0.00001) regardless of whether the BOU was correctly or incorrectly oriented. For example, the observation of a UniGene cluster with 100 ESTs deposited in the same orientation as the BOU and 100 ESTs oriented opposite to the BOU would deviate significantly from the null hypothesis expectation of 98.5% regardless of whether the BOU was correctly oriented or not. This approach identified 297 human and 252 mouse UniGene clusters that contained a statistically significant over-representation of incorrectly oriented ESTs.
Mapping of exon-intron organization of ESTs and mRNAs from candidate UniGene clusters to the mouse and human genomes
We downloaded the NCBI human genome draft assembly (Build 24) [19] and the Celera mouse genome draft assembly [20] in August 2001. Although the Celera mouse genome is not generally accessible, draft assemblies of the mouse genome based on the public sequencing effort have recently been released, and we anticipate that use of these assemblies would yield essentially equivalent results [26]. The MEGABLAST tool [17] was used to identify the approximate genomic coordinates for each UniGene cluster (for example, the contig on which a given gene appeared to be located). The SIM4 tool [18] was then applied to map the exon-intron splicing coordinates of individual BOU and EST sequences more precisely. We have exploited mouse-human synteny and the availability of draft assemblies of the mouse and human genomes to generate a set of around 1.15 million mouse-human sequence alignments. These have been used to create overlay versions of each genome, in which the most conserved sequences (around 10% of each genome) is overlaid with homologous sequence of the other species. More detailed descriptions of the methodology followed and general statistics on this resource (HUMMUS) is available over the web [21]. The graphical representations integrating information on transcript orientation, exon-intron structure, and mouse-human genomic conservation were generated using GNUPLOT [27]. Graphical representations for the curated set of 144 human and 73 mouse candidates is available from our website [22], and an Excel-format summary is available as additional data with this paper.
Assessment of transcriptional directionality via RT-PCR assay
Primers were designed with the PRIMER3 [28] algorithm and custom synthesized by Operon. For candidates, primers were selected to amplify a 100-200 base-pair (bp) sequence that was internal to a predicted region of transcriptional overlap. Control primers were designed to amplify 100-200 bp as well, either from non-overlapping regions of candidate transcripts or randomly selected regions of non-candidate transcripts. Templates included total human RNA from placenta, kidney, brain, thymus or uterus (Clontech). For each candidate or control, four RT-PCR reactions were carried out using total human RNA from a single tissue as template. We used the Qiagen One Step RT-PCR kit according to the manufacturer's protocol, except that reaction volume was reduced to 25 μl. Orientation of transcripts was assessed by restricting which primers were present during RT single-strand synthesis. The cycling parameters were as follows: (1) 50°C × 30 min, reverse transcription single-strand synthesis (with one, both or neither primer); (2) 95°C × 15 min, activate AmpliTaq polymerase, inactivate RT enzymes; (3) 4°C, add missing primers; (4) 94°C × 30 sec, commence PCR cycling; (5) 56°C × 30 sec; (6) 72°C × 30 sec; (7) go to step 4 (30 cycles in total); (8) 72°C × 10 min.
The inclusion of step (2) ensures that the RT will be inactivated before addition of missing primers. For each candidate or control primer pair, four RT-PCR reactions were carried out using total human RNA from a single tissue as template. In the first reaction, both primers were present during RT single-strand synthesis (positive control). In the second reaction, only the primer complementary to the antisense-orientation of the PCR product was present during RT single-strand synthesis (to assay for antisense transcription). In the third reaction, only the primer complementary to the sense-orientation of the PCR product was present during RT single-strand synthesis (to assay for sense transcription). In the fourth reaction, neither primer was present during RT single-strand synthesis (control for genomic contamination). Primers were designed to amplify regions of predicted bidirectional transcription for 39 of the human candidates and 18 negative controls. One of the 18 negative controls was discarded because no lane gave rise to a sharp band of the proper size. In all other cases, a sharp band of expected size was observed in one or more of the reactions.
Determination of protein homologies of sense and antisense oriented ESTs from candidate clusters
We applied the BLASTX tool [29,30] to blast each of the 45,588 relevant mRNA and EST sequences that belonged to both high-quality directionally cloned libraries and candidate UniGene clusters against the NCBI nr database (non-redundant database of protein sequences deposited in GenBank), with a threshold expectation value of 1e-10. Summary information for each candidate on the best protein alignment for ESTs oriented in each sense is available as additional data (see Additional data files).
Additional data files
Additional data including a full list of the 217 sense-antisense candidates, genomic data on overlapping transcripts, and data on protein-level homologies are available and at our website [22].
Supplementary Material
Additional data
Acknowledgments
Acknowledgements
We would like to thank Rob Mitra, Vasudeo Badarinarayana, and Yonatan Grad for helpful comments on the manuscript, and Fritz Roth for generously allowing us use of the LLAMA compute cluster.
References
- Kumar M, Carmichael GG. Antisense RNA: function and fate of duplex RNA in cells of higher eukaryotes. Microbiol Mol Biol Rev. 1998;62:1415–1434. doi: 10.1128/mmbr.62.4.1415-1434.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhee-Brossollet C, Vaquero C. Do natural antisense transcripts make sense in eukaryotes? Gene. 1998;211:1–9. doi: 10.1016/s0378-1119(98)00093-6. [DOI] [PubMed] [Google Scholar]
- Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- Moore T, Constancia M, Zubair M, Bailleul B, Feil R, Sasaki H, Reik W. Multiple imprinted sense and antisense transcripts, differential methylation and tandem repeats in a putative imprinting control region upstream of mouse Igf2. Proc Natl Acad Sci USA. 1997;94:12509–12514. doi: 10.1073/pnas.94.23.12509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleutels F, Zwart R, Barlow DP. The non-coding Air RNA is required for silencing autosomal imprinted genes. Nature. 2002;415:810–813. doi: 10.1038/415810a. [DOI] [PubMed] [Google Scholar]
- Billy E, Brondani V, Zhang H, Muller U, Filipowicz W. Specific interference with gene expression induced by long, double-stranded RNA in mouse embryonal teratocarcinoma cell lines. Proc Natl Acad Sci USA. 2001;98:14428–14433. doi: 10.1073/pnas.261562698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li AW, Murphy PR. Expression of alternatively spliced FGF-2 antisense RNA transcripts in the central nervous system: regulation of FGF-2 mRNA translation. Mol Cell Endocrinol. 2000;170:233–242. doi: 10.1016/s0303-7207(00)00440-8. [DOI] [PubMed] [Google Scholar]
- Munroe SH, Lazar MA. Inhibition of c-erbA mRNA splicing by a naturally occurring antisense RNA. J Biol Chem. 1991;266:22083–22086. [PubMed] [Google Scholar]
- Lee JT, Davidow LS, Warshawsky D. Tsix, a gene antisense to Xist at the X-inactivation centre. Nat Genet. 1999;21:400–404. doi: 10.1038/7734. [DOI] [PubMed] [Google Scholar]
- Kumar M, Carmichael GG. Nuclear antisense RNA induces extensive adenosine modifications and nuclear retention of target transcripts. Proc Natl Acad Sci USA. 1997;94:3542–3547. doi: 10.1073/pnas.94.8.3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blin-Wakkach C, Lezot F, Ghoul-Mazgar S, Hotton D, Monteiro S, Teillaud C, Pibouin L, Orestes-Cardoso S, Papagerakis P, Macdougall M, et al. Endogenous Msx1 antisense transcript: in vivo and in vitro evidences, structure, and potential involvement in skeleton development in mammals. Proc Natl Acad Sci USA. 2001;98:7336–7341. doi: 10.1073/pnas.131497098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemes JP, Benzow KA, Moseley ML, Ranum LP, Koob MD. The SCA8 transcript is an antisense RNA to a brain-specific transcript encoding a novel actin-binding protein (KLHL1). Hum Mol Genet. 2000;9:1543–1551. doi: 10.1093/hmg/9.10.1543. [DOI] [PubMed] [Google Scholar]
- Lipman DJ. Making (anti)sense of non-coding sequence conservation. Nucleic Acids Res. 1997;25:3580–3583. doi: 10.1093/nar/25.18.3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehner B, Williams G, Campbell RD, Sanderson CM. Antisense transcripts in the human genome. Trends Genet. 2002;18:63–65. doi: 10.1016/s0168-9525(02)02598-2. [DOI] [PubMed] [Google Scholar]
- NCBI UniGene http://www.ncbi.nlm.nih.gov/UniGene
- Camargo AA, Samaia HP, Dias-Neto E, Simao DF, Migotto IA, Briones MR, Costa FF, Nagai MA, Verjovski-Almeida S, Zago MA, et al. The contribution of 700,000 ORF sequence tags to the definition of the human transcriptome. Proc Natl Acad Sci USA. 2001;98:12103–12108. doi: 10.1073/pnas.201182798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Schwartz S, Wagner L, Miller WA. A greedy algorithm for aligning DNA sequences. J Comput Biol. 2000;7:203–214. doi: 10.1089/10665270050081478. [DOI] [PubMed] [Google Scholar]
- Florea L, Hartzell G, Zhang Z, Rubin GM, Miller W. A computer program for aligning a cDNA sequence with a genomic DNA sequence. Genome Res. 1998;8:967–974. doi: 10.1101/gr.8.9.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCBI Human Genome http://www.ncbi.nlm.nih.gov/genome/guide/human
- Celera http://www.celera.com
- HUMMUS http://arep.med.harvard.edu/hummus.html
- Sense and antisense (Church Lab) http://arep.med.harvard.edu/antisense.html
- Duret L, Dorkeld F, Gautier C. Strong conservation of non-coding sequences during vertebrate evolution: potential involvement in post-transcriptional regulation of gene expression. Nucleic Acids Res. 1993;21:2315–2322. doi: 10.1093/nar/21.10.2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Affymetrix Murine Genome U74 Array Set http://www.affymetrix.com/support/technical/product_updates/mgu74_product_bulletin.affx
- Shoemaker DD, Schadt EE, Armour CD, He YD, Garrett-Engele P, McDonagh PD, Loerch PM, Leonardson A, Lum PY, Cavet G, et al. Experimental annotation of the human genome using microarray technology. Nature. 2001;409:922–927. doi: 10.1038/35057141. [DOI] [PubMed] [Google Scholar]
- Ensembl Mouse Genome http://www.ensembl.org/Mus_musculus/
- Gnuplot Central http://www.gnuplot.info
- Primer 3 software distribution http://www-genome.wi.mit.edu/genome_software/other/primer3.html
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- NCBI BLAST http://www.ncbi.nlm.nih.gov/BLAST
- Wutz A, Smrzka OW, Schweifer N, Schellander K, Wagner EF, Barlow DP. Imprinted expression of the Igf2r gene depends on an intronic CpG island. Nature. 1997;389:745–749. doi: 10.1038/39631. [DOI] [PubMed] [Google Scholar]
- Whitehead CM, Winkfein RJ, Fritzler MJ, Rattner JB. ASE-1: a novel protein of the fibrillar centres of the nucleolus and nucleolus organizer region of mitotic chromosomes. Chromosoma. 1997;106:493–502. doi: 10.1007/s004120050271. [DOI] [PubMed] [Google Scholar]
- Lee YJ, Park CW, Hahn Y, Park J, Lee J, Yun JH, Hyun B, Chung JH. Mit1/Lb9 and Copg2, new members of mouse imprinted genes closely linked to Peg1/Mest(1). FEBS Lett. 2000;472:230–234. doi: 10.1016/s0014-5793(00)01461-7. [DOI] [PubMed] [Google Scholar]
- Zavadil J, Svoboda P, Liang H, Kottickal LV, Nagarajan L. An antisense transcript to SMAD5 expressed in fetal and tumor tissues. Biochem Biophys Res Commun. 1999;255:668–672. doi: 10.1006/bbrc.1999.0256. [DOI] [PubMed] [Google Scholar]
- McGuinness T, Porteus MH, Smiga S, Bulfone A, Kingsley C, Qiu M, Liu JK, Long JE, Xu D, Rubenstein JL. Sequence, organization, and transcription of the Dlx-1 and Dlx-2 locus. Genomics. 1996;35:473–485. doi: 10.1006/geno.1996.0387. [DOI] [PubMed] [Google Scholar]
- Liu JK, Ghattas I, Liu S, Chen S, Rubenstein JL. Dlx genes encode DNA-binding proteins that are expressed in an overlapping and sequential pattern during basal ganglia differentiation. Dev Dyn. 1997;210:498–512. doi: 10.1002/(SICI)1097-0177(199712)210:4<498::AID-AJA12>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Zambrowicz BP, Imamoto A, Fiering S, Herzenberg LA, Kerr WG, Soriano P. Disruption of overlapping transcripts in the ROSA beta geo 26 gene trap strain leads to widespread expression of beta-galactosidase in mouse embryos and hematopoietic cells. Proc Natl Acad Sci USA. 1997;94:3789–3794. doi: 10.1073/pnas.94.8.3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smilinich NJ, Day CD, Fitzpatrick GV, Caldwell GM, Lossie AC, Cooper PR, Smallwood AC, Joyce JA, Schofield PN, Reik W, et al. A maternally methylated CpG island in KvLQT1 is associated with an antisense paternal transcript and loss of imprinting in Beckwith-Wiedemann syndrome. Proc Natl Acad Sci USA. 1999;96:8064–8069. doi: 10.1073/pnas.96.14.8064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCBI LocusLink Record 114044 http://www.ncbi.nlm.nih.gov/LocusLink/LocRpt.cgi?l=114044
- Wroe SF, Kelsey G, Skinner JA, Bodle D, Ball ST, Beechey CV, Peters J, Williamson CM. An imprinted transcript, antisense to Nesp, adds complexity to the cluster of imprinted genes at the mouse Gnas locus. Proc Natl Acad Sci USA. 2000;97:3342–3346. doi: 10.1073/pnas.050015397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy PR, Knee RS. Identification and characterization of an antisense RNA transcript (gfg) from the human basic fibroblast growth factor gene. Mol Endocrinol. 1994;8:852–859. doi: 10.1210/mend.8.7.7984147. [DOI] [PubMed] [Google Scholar]
- Seroussi E, Kedra D, Pan HQ, Peyrard M, Schwartz C, Scambler P, Donnai D, Roe BA, Dumanski JP. Duplications on human chromosome 22 reveal a novel Ret Finger Protein-like gene family with sense and endogenous antisense transcripts. Genome Res. 1999;9:803–814. doi: 10.1101/gr.9.9.803. [DOI] [PubMed] [Google Scholar]
- Cooper PR, Smilinich NJ, Day CD, Nowak NJ, Reid LH, Pearsall RS, Reece M, Prawitt D, Landers J, Housman DE, et al. Divergently transcribed overlapping genes expressed in liver and kidney and located in the 11p15.5 imprinted domain. Genomics. 1998;49:38–51. doi: 10.1006/geno.1998.5221. [DOI] [PubMed] [Google Scholar]
- NCBI LocusLink Record 93653 http://www.ncbi.nlm.nih.gov/LocusLink/LocRpt.cgi?l=93653
- NCBI LocusLink Record 93654 http://www.ncbi.nlm.nih.gov/LocusLink/LocRpt.cgi?l=93654
- NCBI LocusLink Record 93655 http://www.ncbi.nlm.nih.gov/LocusLink/LocRpt.cgi?l=93655
- Jong MT, Gray TA, Ji Y, Glenn CC, Saitoh S, Driscoll DJ, Nicholls RD. A novel imprinted gene, encoding a RING zinc-finger protein, and overlapping antisense transcript in the Prader-Willi syndrome critical region. Hum Mol Genet. 1999;8:783–793. doi: 10.1093/hmg/8.5.783. [DOI] [PubMed] [Google Scholar]
- Potter SS, Branford WW. Evolutionary conservation and tissue-specific processing of Hoxa 11 antisense transcripts. Mamm Genome. 1998;9:799–806. doi: 10.1007/s003359900870. [DOI] [PubMed] [Google Scholar]
- Campbell CE, Huang A, Gurney AL, Kessler PM, Hewitt JA, Williams BR. Antisense transcripts and protein binding motifs within the Wilms tumour (WT1) locus. Oncogene. 1994;9:583–595. [PubMed] [Google Scholar]
- Silverman TA, Noguchi M, Safer B. Role of sequences within the first intron in the regulation of expression of eukaryotic initiation factor 2α. J Biol Chem. 1992;267:9738–9742. [PubMed] [Google Scholar]
- Faurholm B, Millar RP, Katz AA. The genes encoding the type II gonadotropin-releasing hormone receptor and the ribonucleoprotein RBM8A in humans overlap in two genomic loci. Genomics. 2001;78:15–18. doi: 10.1006/geno.2001.6650. [DOI] [PubMed] [Google Scholar]
- Wagener R, Kobbe B, Aszodi A, Aeschlimann D, Paulsson M. Characterization of the mouse matrilin-4 gene: a 5' antiparallel overlap with the gene encoding the transcription factor RBP-l. Genomics. 2001;76:89–98. doi: 10.1006/geno.2001.6589. [DOI] [PubMed] [Google Scholar]
- Borsu L, Presse F, Nahon JL. The AROM gene, spliced mRNAs encoding new DNA/RNA-binding proteins are transcribed from the opposite strand of the melanin-concentrating hormone gene in mammals. J Biol Chem. 2000;275:40576–40587. doi: 10.1074/jbc.M006524200. [DOI] [PubMed] [Google Scholar]
- Sureau A, Soret J, Guyon C, Gaillard C, Dumon S, Keller M, Crisanti P, Perbal B. Characterization of multiple alternative RNAs resulting from antisense transcription of the PR264/SC35 splicing factor gene. Nucleic Acids Res. 1997;25:4513–4522. doi: 10.1093/nar/25.22.4513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings ML, Milcarek C, Martincic K, Peterson ML, Munroe SH. Expression of the thyroid hormone receptor gene, erbAα, in B lymphocytes: alternative mRNA processing is independent of differentiation but correlates with antisense RNA levels. Nucleic Acids Res. 1997;25:4296–4300. doi: 10.1093/nar/25.21.4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo P, Yon J, Garson K, Fried M. Conservation of the organization of five tightly clustered genes over 600 million years of divergent evolution. Proc Natl Acad Sci USA. 1992;89:6358–6362. doi: 10.1073/pnas.89.14.6358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khochbin S, Lawrence JJ. An antisense RNA involved in p53 mRNA maturation in murine erythroleukemia cells induced to differentiate. EMBO J. 1989;8:4107–4114. doi: 10.1002/j.1460-2075.1989.tb08595.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celano P, Berchtold CM, Kizer DL, Weeraratna A, Nelkin BD, Baylin SB, Casero RA. Characterization of an endogenous RNA transcript with homology to the antisense strand of the human c-myc gene. J Biol Chem. 1992;267:15092–15096. [PubMed] [Google Scholar]
- Krystal GW, Armstrong BC, Battey JF. N-myc mRNA forms an RNA-RNA duplex with endogenous antisense transcripts. Mol Cell Biol. 1990;10:4180–4191. doi: 10.1128/mcb.10.8.4180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fremeau RT, Jr, Popko B. In situ analysis of myelin basic protein gene expression in myelin-deficient oligodendrocytes: antisense hnRNA and readthrough transcription. EMBO J. 1990;9:3533–3538. doi: 10.1002/j.1460-2075.1990.tb07562.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosic M, Roach A, de Rivaz JC, Dolivo M, Matthieu JM. Post-transcriptional events are responsible for low expression of myelin basic protein in myelin deficient mice: role of natural antisense RNA. EMBO J. 1990;9:401–406. doi: 10.1002/j.1460-2075.1990.tb08124.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laabi Y, Gras MP, Brouet JC, Berger R, Larsen CJ, Tsapis A. The BCMA gene, preferentially expressed during B lymphoid maturation, is bidirectionally transcribed. Nucleic Acids Res. 1994;22:1147–1154. doi: 10.1093/nar/22.7.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adelman JP, Bond CT, Douglass J, Herbert E. Two mammalian genes transcribed from opposite strands of the same DNA locus. Science. 1987;235:1514–1517. doi: 10.1126/science.3547652. [DOI] [PubMed] [Google Scholar]
- Murashov AK, Wolgemuth DJ. Sense and antisense transcripts of the developmentally regulated murine hsp70.2 gene are expressed in distinct and only partially overlapping areas in the adult brain. Brain Res Mol Brain Res. 1996;37:85–95. doi: 10.1016/0169-328x(95)00288-4. [DOI] [PubMed] [Google Scholar]
- Batshake B, Sundelin J. The mouse genes for the EP1 prostanoid receptor and the PKN protein kinase overlap. Biochem Biophys Res Commun. 1996;227:70–76. doi: 10.1006/bbrc.1996.1469. [DOI] [PubMed] [Google Scholar]
- Bender TP, Thompson CB, Kuehl WM. Differential expression of c-myb mRNA in murine B lymphomas by a block to transcription elongation. Science. 1987;237:1473–1476. doi: 10.1126/science.3498214. [DOI] [PubMed] [Google Scholar]
- Farrell CM, Lukens LN. Naturally occurring antisense transcripts are present in chick embryo chondrocytes simultaneously with the down-regulation of the alpha 1 (I) collagen gene. J Biol Chem. 1995;270:3400–3408. doi: 10.1074/jbc.270.7.3400. [DOI] [PubMed] [Google Scholar]
- Belhumeur P, Lussier M, Skup D. Expression of naturally occurring RNA molecules complementary to the murine L27' ribosomal protein mRNA. Gene. 1988;72:277–285. doi: 10.1016/0378-1119(88)90153-9. [DOI] [PubMed] [Google Scholar]
- PubMed http://www.ncbi.nlm.nih.gov/pubmed
- LocusLink http://www.ncbi.nlm.nih.gov/LocusLink
- Antisense transcripts in the human genome http://www.hgmp.mrc.ac.uk/Research/Antisense/ [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional data