

Short abstract
Relative mRNA levels from maize organs have been quantified by hybridization to maize arrays fabricated with 5,376 unique expressed sequence tag clones. mRNAs from embryos, three vegetative organs (leaf blades, leaf sheaths and roots), husk leaves and two types of floral organs (immature ears and silks) reveal that a majority of genes expressed in diverse organs with little difference in transcript levels.
Abstract
Background
A maize array was fabricated with 5,376 unique expressed sequence tag (EST) clones sequenced from 4-day-old roots, immature ears and adult organ cDNA libraries. To elucidate organ relationships, relative mRNA levels were quantified by hybridization with embryos, three maize vegetative organs (leaf blades, leaf sheaths and roots) from multiple developmental stages, husk leaves and two types of floral organs (immature ears and silks).
Results
Clustering analyses of the hybridization data suggest that maize utilizes both the PEPCK and NADP-ME C4 photosynthetic routes as genes in these pathways are co-regulated. Husk RNA has a gene-expression profile more similar to floral organs than to vegetative leaves. Only 7% of the genes were highly organ specific, showing over a fourfold difference in at least one of 12 comparisons and 37% showed a two- to fourfold difference. The majority of genes were expressed in diverse organs with little difference in transcript levels. Cross-hybridization among closely related genes within multigene families could obscure tissue specificity. As a first step in elucidating individual gene-expression patterns, we show that 45-nucleotide oligo probes produce signal intensities and signal ratios comparable to PCR probes on the same matrix.
Conclusions
Gene-expression profile studies with cDNA microarrays provide a new molecular tool for defining plant organs and their relationships and for discovering new biological processes in silico. cDNA microarrays are insufficient for differentiating recently duplicated genes. Gene-specific oligo probes printed along with cDNA probes can query individual gene-expression profiles and gene families simultaneously.
Background
Flowering plants are composed of diverse cell types organized into tissues and organs. To achieve the morphological and functional specialization of cells and tissues, suites of genes are expressed in spatial and temporal patterns determined by regulatory hierarchies responding to environmental cues. Plants reiteratively produce photosynthetic organs such as individual leaves that often differ in morphology and physiology depending on environmental cues and their positions on the plant. The fine-tuning of gene expression to permit such diversity remains largely uncharacterized. Flower development provides one example where organs of distinctive morphologies (sepals, petals, stamens, carpels) are produced in rapid succession; specification of each floral organ requires temporally and spatially refined expression of specific genes [1].
Although the functions and expression patterns for dozens of genes have been characterized in particular plant tissues and organs, the number of well studied examples is meager compared to the total number of genes [2].
Using information and material from genomic and high-throughput expressed sequence tag (EST) sequencing projects, several approaches have been devised to investigate global gene-expression profiles. In particular, spotted microarrays of ESTs have been used to initiate functional analyses of thousands of genes simultaneously. The first microarray contained only 45 Arabidopsis thaliana genes [3], but the demonstrated success of the method was quickly followed by studies of human [4] and yeast [5] gene expression. Microarray analysis has been used in plants for such diverse purposes as discovering genes responsible for strawberry flavor [6], comparing mutant to wild-type plants [7], and monitoring organism-level responses to environmental stimuli [8,9,10]. In most studies, treated and untreated tissues of the same age were compared. To date, there are just a few studies comparing distinct developmental stages. For example, Ruan et al. [11] surveyed expression in the three fundamental organ types - leaves, roots and flowers - of Arabidopsis, using microarrays containing 1,400 EST cDNA clones. Fernandes et al. [12] compared the hybridization of maize 14-day endosperm and immature ear (1-2 cm) RNA populations on separate endosperm and ear microarrays containing approximately 2,800 and 2,500 distinct genes, respectively. They found that nearly all probes on the ear array hybridized to cDNA prepared from endosperm or ear, whereas the endosperm array contained many apparently tissue-specific elements.
Using the 152,746 maize ESTs available, 31,858 tentative unique genes (TUGs) have been assembled as reported by the Zea mays database (April 14, 2002 [13]). Most ESTs are from the Maize Gene Discovery project [13], and a UniGene set is being developed from this resource. From the UniGene1 EST assembly 5,376 cDNA gene probes were used to fabricate a spotted cDNA microarray for a suite of hybridizations. In addition, 384 synthetic oligonucleotides 30, 40 or 45 nucleotides in length were printed to explore whether hybridization and washing conditions compatible with signal retention for both oligos and cDNA clones could be devised. Thirteen RNA samples from 7 distinct organs were hybridized on this array to ask how many genes were expressed in all or most organs, which genes had discrete patterns of expression, and whether gene-expression profiles could be used to understand the relationships among organs. Among > 5,000 genes selected for analysis, 56% showed less than a twofold difference and 37% showed a two- to fourfold difference in mRNA amounts compared to the reference sample - maize seedling RNA. These results imply that the differentiated state of maize tissues and organs is characterized by combinations of small numbers of differentially expressed (> 4-fold) tissue- or organ-specific genes among the 5,376 genes in this study. A complication of this interpretation is that some members of gene families will cross-hybridize, making it difficult to resolve whether there are large numbers of organ-specific genes within such families. As a first step in resolving expression patterns among recently duplicated genes, oligo probes for well characterized genes were printed on the same microarray slides as the cDNAs. The oligo probes generated more accurate information about gene expression than did cDNA probes for the genes examined in this study. We discuss how rationally designed oligo probes can distinguish expression patterns of individual genes in a gene family with similar sequences.
Results
Internal consistency of hybridization
To examine the consistency of experiments from the labeling reactions through to the scanning process, a pool of mRNA from 4-day-old roots (4DR) was used to synthesize two fluorescent cDNA targets using Cy3-dUTP or Cy5-dUTP. These labeled cDNAs were subsequently combined in equal proportion to perform control hybridization experiments. Signal intensities of the two fluorescence-measurement channels were linearly correlated for a majority of the 5,376 PCR probes, indicating that nearly all targets were labeled with each dye. The mean Cy5/Cy3 ratio of the hybridized group of 5,263 genes was 1.046 with a standard error of 0.0014 and standard deviation of ± 0.105. Nearly all probes (5,074 of 5,263, or 96%) were within the 0.75 to 1.25 range (-0.415 to 0.32 on the log2 scale), and only two probe ratios were slightly over 2 (1 on a log2 scale). Of 5,376 EST probes examined, 5,263 generated signal intensities exceeding 300 intensity units in each channel and > 1,500 in the summation of the two channels, indicating that the ESTs on the array correspond mainly to moderately expressed genes. The few ESTs with signal intensities below these values have been omitted from further analysis. Another data set selected with a lower standard (> 700 in the summation of two channels) generated very similar results (data not shown).
To investigate hybridization consistency further, aliquots of mRNAs from immature ears (IME) and 4DR were each labeled separately with Cy3-dUTP or Cy5-dUTP. A mixture of Cy3-labeled 4DR and Cy5-labeled IME was hybridized to one slide, and the same samples, but labeled oppositely, were hybridized to a second slide. Probes with a red color on one side appeared green on the other side and vice versa, as illustrated in Figure 1a,1b. The hybridization consistency was examined by evaluating signal ratios between the two channels produced from each 'dye-swapped' hybridization. Signal ratios were calculated by dividing the signal intensity of 4DR by that of the IME. Out of 5,016 probes, 4,536 hybridized successfully to produce signal intensities greater than the selection criteria in both hybridizations. One of 16 subarrays (360 probes) in one hybridization was excluded in this analysis, because there was a mechanical failure during array printing. Of high-quality probes, 4,334 (95.5%) showed a log2 ratio difference between 0.5 to -0.5. Only six (0.12%) showed a log2 ratio difference of more than 1 in the paired hybridizations (Figure 1c). These hybridization results appear to be similar (null hypothesis not rejected p < 0.01, t-test with an unequal variance p-value 0.819, two-tail t-distribution 41%). Therefore, hybridizations on two separate slides from the same printing generate highly reproducible results.
Figure 1.
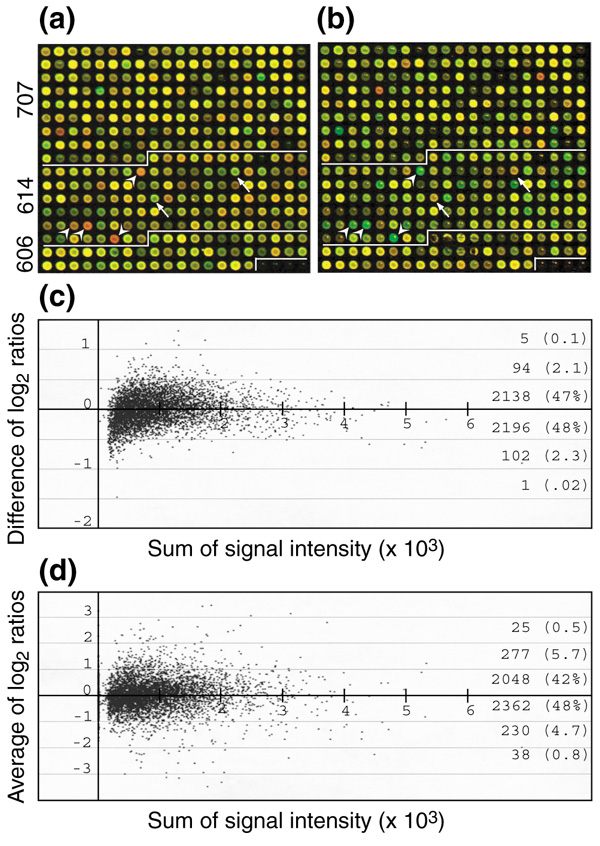
Dye-swap hybridization experiment. (a,b) Dye-swap hybridization protocol on microarrays fabricated with cDNAs from EST projects 606 (immature ear, IME), 614 (4-day seedling roots, 4DR), and 707+945 (mixture of adult tissues). Each image was obtained from co-hybridization of two dye-labeled targets on a single microarray. Two false-color images were superimposed to represent the relative amount of transcripts in the samples. (a) IME labeled with Cy3 (green) and 4DR with Cy5 (red); (b) samples labeled reciprocally. Arrows and arrowheads in (a) and (b) indicate a few obvious examples of organ-specific expression (see (d)). (c) Consistency of hybridization was examined by calculating signal-ratio differences from the dye-swapping experiment for each microarray element. Log2 signal ratios of 4DR over IME were calculated from each hybridization, followed by subtraction of the log2 ratios from slide (a) by those on slide (b). (d) Relative transcript abundances in the two samples were plotted against the sum of signal intensities from both channels. Log2 signal ratios of 4DR over IME were averaged from the dye-swap experiment. Difference of log2 ratio and average of log2 ratio are given by [log2(4DR/IME)a]/ [log2(4DR/IME)b] and [log2(4DR/IME)a + log2(4DR/IME)b]/2, respectively.
Expression-profile comparisons between 4-day-old roots and immature ears
The relative amounts of mRNA for each printed EST were inferred from hybridization results in the dye-swapping experiments of 4DR and IME (Figure 1d). The ratios were averaged from a set of dye-swapped hybridization experiments, and 4,878 EST probes were selected for further analyses. In the comparison, 3,222 (66%) of 4,880 probes registered within the range -1.4 to 1.4 (|log2| < 0.5), that is, within the range of signal variations intrinsic to the experimental procedures (Figure 1d, Table 1). Of the 4,880 probes, 1,188 (24%) showed a 1.4- to 2-fold difference (0.5 < |log2| < 1.0), and 407 (8.3%) showed a 2- to 4-fold (1 < |log2 ratio| < 2) difference between the two organs. Only 63, or 1.3%, of the EST probes showed more than a fourfold difference (|log2 ratio| > 2) (Figure 1d). Among the 63 most differentially expressed genes, 25 exhibited a more than fourfold higher level of expression and 38 a more than fourfold lower level in IME compared to 4DR. Among the 25 IME-abundant genes, there are five MADS-box gene family members. In plants with perfect flowers (male and female sexual organs, sepals and petals in the same flower), this gene family regulates inflorescence development, flower organ differentiation, flowering time and specification of floral cell type [1]. In the imperfect monoecious plant maize, some MADS-box genes are expressed specifically in the male tassel or in the female ear inflorescences whereas others are expressed in both [14,15]. Other ear-enriched transcripts were for three different heat-shock proteins, late elongated hypocotyl gene and the mudrA transposase. Genes exhibiting a more than 2.8-fold ratio difference are listed in the Additional data files.
Table 1.
Signal-ratio distribution according to the source libraries
| Log2 ratio* | 4-day roots | IM ears | Tissue mix | Total |
| <-2.5 | 4 (0.2)2 | 2 (0.3) | 4 (0.1) | 10 (0.2) |
| -2.5 ~ -2.0 | 2 (0.1) | 3 (0.4) | 10 (0.4) | 15 (0.3) |
| -2.0 ~ -1.5 | 4 (0.2) | 10 (1.3) | 22 (0.8) | 36 (0.7) |
| -1.5 ~ -1.0 | 40 (2.1) | 34 (4.4) | 67 (2.5) | 141 (2.6) |
| -1.0 ~ -0.5 | 166 (8.6) | 118 (15.4) | 233 (8.7) | 517 (9.6) |
| -0.5 ~ 0.0 | 497 (25.9) | 317 (41.3) | 717 (26.7) | 1,531 (28.5) |
| 0.0 ~ 0.5 | 609 (31.7) | 205 (26.7) | 877 (32.7) | 1,691 (31.5) |
| 0.5 ~ 1.0 | 327 (17.0) | 53 (6.9) | 291 (10.8) | 671 (12.5) |
| 1.0 ~ 1.5 | 94 (4.9) | 10 (1.3) | 82 (3.1) | 186 (3.5) |
| 1.5 ~ 2.0 | 25 (1.3) | 2 (0.3) | 17 (0.6) | 44 (0.8) |
| 2.0 ~ 2.5 | 13 (0.7) | 0 (0.0) | 7 (0.3) | 20 (0.4) |
| >2.5 | 15 (0.8) | 0 (0.0) | 3 (0.1) | 18 (0.3) |
| Weak hybridization | 124 (6.5) | 14 (1.8) | 353 (13.2) | 491 (9.1) |
| Total | 1,920 (100) | 768 (100) | 2,683 (100) | 5,371 (100) |
The log2 ratio is the average of log2 (4DR/IME) from the dye-swap experiment. Numbers within parentheses indicate percentiles.
Hybridization results were further analyzed by considering the cDNA library of origin. There were 1,920 probes on the slides from cDNA library 614 (4DR), 768 probes from 606 (IME), and 2,683 probes from 707 and 945 (two samples of the same mixed adult tissue cDNA library). In the hybridization control experiment (Figure 1), the behavior of 4DR dye-labeled cDNA sample on the 4DR (library 614) section of the slide was examined. Some probes of library 614 showed strong red on the slide in Figure 1a and strong green on the slide in Figure 1b in the 614 section (Figure 1, arrowheads), which would be expected if these clones were expressed in a tissue-specific pattern. Other EST elements, however, presented the opposite color pattern (Figure 1, arrows). Of 1,920 probes originating from a 4DR cDNA library (614), 1,106 (57.6%) showed log2 signal ratios between -0.5 and 0.5. Only 28 (15 + 13, 1.5%) showed over fourfold higher (log2 ratio > 2) mRNA amount in 4-day-old roots compared to immature ears (Table 1). Furthermore, 713 (37%) of 1,920 ESTs originating from a 4DR library were expressed at lower levels in 4DR than in IME. Similarly, probes originating from the IME (606) and mixed adult tissue (707 + 945) libraries showed only a small fraction of organ-specific expression elements (Table 1). These data, in part, reflect EST and UniGene1 consolidation methods (see Methods and materials). When an EST is chosen for a UniGene set, this does not mean that it was found only in one cDNA library source; contigs assembled from maize ESTs typically have contributions from several cDNA libraries.
Expression-profile comparisons among thirteen samples from seven organs
IME and 4DR are very distinctive in their developmental origin, location and functions in the maize life cycle. Despite this, the majority of the microarray elements appeared to be expressed in both organs. Interesting questions arose from this observation. How many genes are expressed abundantly in specific organs? How many genes are expressed in diverse organs? To answer these questions, seven different organ sources were analyzed. The signal ratios were averaged from two hybridization analyses for each organ in comparison to the reference organ - 4-day-old shoots with coleoptiles. The clustering by similarity of expression patterns is shown in Figure 2 for a subset of the data.
Figure 2.

Gene-expression cladograms showing the relative abundance of transcripts in 13 samples. The left cladogram was constructed with 4,531 elements, and the right one with 326 elements. The 326 elements exhibit >4-fold ratio difference in at least one of 12 comparisons. Trees at the left side of the cladogram present gene relationships, and the trees on the top of cladogram show organ relationships. Color codes and color ratios in each panel are: the brightest red is >4-fold higher, the brightest green is >4-fold lower, and black is same ratio in that comparison. Green colored names mark genes whose gene products are located in chloroplasts. Circled numbers match the numbers in Figure 5 which shows the place of these enzymes in the C4 photosynthetic pathways. Multiple genes in each gene family are marked with arrows, and other genes discussed in the text are marked with arrowheads. Numbers indicate Stanford identification numbers for individual EST clones. For the complete list of genes see Additional data files.
Analysis was restricted to 4,673 probes that generated intensities > 300 in both channels and > 1,500 summing both channels in 11 or all 12 pairs of hybridization experiments. Among these 4,673 genes, only 326 (7%) showed more than a fourfold (|log2 ratio | >2) and 1,731 (37%) had a 2-4-fold (1 < |log2 ratio| <2) signal ratio in at least one of the 12 organs compared to the reference. These are candidate tissue- or organ-specific genes. The majority (56%) of the genes showed signal ratios between -1.4- and 1.4-fold (|log2 ratio| < 0.5). In Figure 2 this observation is visually apparent from the high proportion of black elements depicting equal hybridization on the far-left cladogram composed of 4,531 ESTs. As shown in Table 2, over 90% of the genes showed a twofold range (|log2 ratio| < 1) in most comparisons, confirming and extending the initial comparisons between 4DR and IME. This consistency of hybridization results suggested that only a small number of genes were expressed differentially in specific organs and that the majority of printed ESTs had approximately similar expression levels in many organs. Among the 12 samples, adult leaf blades (L9-L10) showed the highest number of genes expressed at a lower level than the reference organ (Table 2, and see extensive green color within Figure 2), whereas the husks and silks showed the opposite trend (extensive red color in Figure 2).
Table 2.
Signal-ratio distribution for 12 samples
| Ratio | <(-2) | (-2) ~ (-1) | (-1) ~ 1 | 1 ~ 2 | >2 |
| 2-week roots | 36 (0.7)2 | 230 (4.6) | 4525 (90.0) | 251 (5.0) | 13 (0.3) |
| 8-day roots | 53 (1.0) | 314 (6.0) | 4483 (85.0) | 391 (7.5) | 10 (0.2) |
| 4-day roots | 49 (1.0) | 170 (3.4) | 4662 (94.0) | 53 (1.1) | 7 (0.1) |
| Husks | 6 (0.2) | 127 (3.2) | 3,574 (90.0) | 234 (5.9) | 22 (0.6) |
| Silks | 26 (0.5) | 325 (6.5) | 4,478 (90.0) | 120 (2.4) | 35 (0.7) |
| IM ears | 37 (0.7) | 184 (3.7) | 4,681 (94.0) | 49 (1.0) | 13 (0.3) |
| Embryos | 29 (0.6) | 147 (3.0) | 4,647 (94.0) | 80 (1.6) | 19 (0.4) |
| 2-week leaf sheaths | 4 (0.1) | 20 (0.4) | 4,885 (99.0) | 34 (0.7) | 0 (0.0) |
| 8-day leaf sheaths | 2 (0.0) | 32 (0.6) | 4,907 (99.0) | 35 (0.7) | 2 (0.0) |
| Adult leaves | 190 (4.0) | 1,041 (21.5) | 3,485 (72.0) | 99 (2.1) | 27 (0.6) |
| 2-week leaf blades | 62 (1.3) | 408 (8.2) | 4,326 (87.0) | 158 (3.2) | 17 (0.3) |
| 8-day leaf blades | 54 (1.1) | 427 (8.9) | 4,177 (87.0) | 138 (2.9) | 18 (0.4) |
| Sum | 548 (0.9) | 3,425 (5.8) | 52,830 (90.0) | 1,642 (2.8) | 183 (0.3) |
Log2 ratio is the average of log2 (experimental/reference) from the comparisons between an experimental tissue and reference tissue 4-day-old coleoptiles. Numbers within parentheses indicate percentiles.
Inferred hierarchy of organ similarity
Plant organs are classified on the basis of their ontogeny, a few key characteristics such as their ability to photosynthesize and the expression of genes required for this process, and the discrete impact of mutations on individual organs. Using the expression-profile data, organ relationships were analyzed using a large set of characters. Seven data sets were selected for clustering as described in Materials and methods, either followed by a median centering and normalization or without this process. As shown in Figure 3, both analytical treatments yielded nearly identical organ relationships. Overall branch lengths appeared longer, but tree topologies were more consistent among trees from the normalized method than from the unnormalized one.
Figure 3.
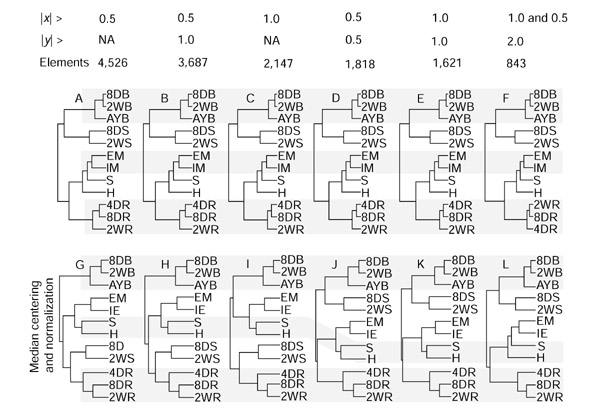
Organ relationships derived from gene-expression profiles. Each tree was constructed using Cluster [32] with various numbers of elements, selected on the basis of three criteria as shown at the top. |x| > indicates the absolute log2 ratio of each hybridization for a given element, and |y| > indicates absolute value of difference between maximum and minimum log2 ratios among 13 hybridizations. Elements indicate the numbers of probes that were selected by the given criteria, and then used to construct the trees in the column below. Trees A-F were constructed using the signal-ratio data without a secondary normalization process. Trees G-L were constructed with the same elements as the panel immediately above after a median centering and a normalization process. Results were indistinguishable between hybridization success rates of 80% and 50%; the diagrams shown are from the analysis of 80% of the data. A gray bar marks the same set of tissues. 8DB, 8-day leaf blade; 2WB, 2-week leaf blade; AYB, adult leaf blade; 8DS, 8-day leaf sheath; 2WS, 2-week leaf sheath; EM, embryo; IM, immature ear; S, silk; H, husk; 4DR, 4-day root; 8DR, 8-day root; 2WR, 2-week root; NA, not applicable.
Leaf blades at three developmental stages formed a tight group in all 14 tree diagrams (Figures 2, 3). Short terminal branches for these stages indicated a high similarity of their gene-expression profiles. Root profiles at three developmental stages also group together, although their internal relationships were less consistent among comparisons. Immature ears plus embryos or leaf sheaths from two developmental stages also grouped consistently. With more distant relationships, nodes were less well supported. For example, the leaf-sheath pair appeared as a sister group to the leaf blades in trees A-F but as sister to the leaf blades or roots in trees G-L (Figure 3). The extremely short, shared nodes imply a weak relationship of the leaf-sheath pair to either group. After collapsing short and inconsistent nodes, four distinct macrogroups remained with unresolved relationships among them (Figure 4): leaf blades, leaf sheaths, reproductive (ear, silk, embryo, husk) and roots. Within these four broad categories, each group has a unique set of expressed genes, and these patterns were relatively consistent during the developmental stages assayed.
Figure 4.
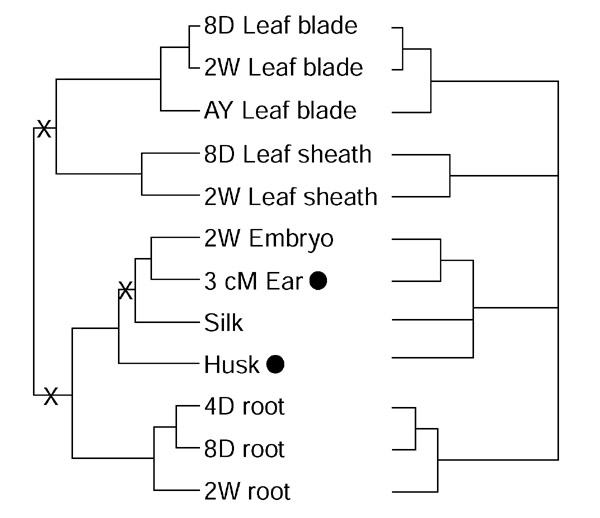
A consensus tissue tree based on the profiles shown in Figure 3. The left-hand tree is identical to B in Figure 3, and the right-hand tree is a majority consensus tree from the 14 analyses. X, internodes that are not consistent among the majority of the 14 trees.
The relationship of the husk, a photosynthetic 'leaf-like' organ [16] surrounding the ears of maize, to other organs merits special attention. It appeared as an immediate sister to the silk (Figure 3, bottom panel) or sister to the inflorescence organ group including the silk (Figure 3, upper panel). After collapsing ambiguous nodes, the husk remained within the reproductive group, and therefore distinctive from both leaf blade and sheath. Adjacent positions with the long terminal branches implied a loose relationship between husks and silks. Of the flower-related genes, 32 are expressed at high levels in husks (Figure 2, node c).
Organ-specific gene expression
To gain greater confidence in deciphering organ-specific gene-expression patterns, gene-product classification focused on 326 probes with a more than fourfold ratio difference in signal intensities in at least one organ compared to the reference (Figure 2). According to the criterion of E-value < e-10 in BLASTX searches [17], only 136 of the 326 genes predict similar proteins in public databases, 71 of 326 genes matched an Arabidopsis genome sequence, and 119 genes did not have significantly similar sequence in the public databases.
Flower-related genes included hydroxyproline-rich glycoprotein (hrgp [18]), β-amylase (PID9294660), pollen allergen (PID4006978), a bZIP transcription factor (PID6288682), four MADS-box genes [1,14], and several unknown genes (Figure 2). A relatively high expression of diverse transcription factors in embryo, immature ear and silk is consistent with microscopic observations that many stages of organ differentiation are occurring within the immature inflorescence and embryos. Because the husks were morphologically fully expanded leaf sheaths surrounding the ear, it was surprising that they expressed the same genes, such as MADS-box genes that are associated with early stages of flower development. Heat-shock proteins (16.9 kDa, 82 kDa, and 101 kDa) were abundant in silks, husks and IME, but not in embryos. The genes for the 82 kDa and 101 kDa proteins are expressed at raised temperatures [19,20]; however, in this study they appear to be part of a developmental program. Embryos and flowers are distinguished by large quantitative differences in expression of these three heat-shock genes. Root-specific genes included a nodulin homolog (PID3482914), putative lipid-transfer protein gene (PID10140658), physical impedance induced protein gene (PID2226329), and four additional unknown genes. The organ-specific expression pattern of these genes may spark interest in defining their physiological functions.
Genes expressed preferentially in the leaf blade
Twenty-six transcripts were comparatively abundant only in leaf blades (Figure 2, node a). The expression ratio of these genes was more than twofold higher in 8-day, 2-week and adult leaf blades, and more than fourfold lower in roots, immature ears and embryos, in comparison to the reference 4-day shoots. Twenty-three of these leaf-enriched transcripts shared high sequence similarity to previously published (identified or putative) coding sequences. Gene products from 17 of these 23 genes were previously characterized as located in or predicted to locate to plastids. Two well characterized genes in this leaf-blade group are for Rubisco small subunit (rbcS) and phosphoribulokinase, key enzymes for converting CO2 into carbohydrate via the Calvin cycle. Other genes represented components of light-harvesting complexes (photosystems I and II), chloroplastic aldolase [21], and the phosphoenolpyruvate translocator gene [22]. Most of these highly expressed leaf genes are encoded in the nuclear genome, and the proteins are imported into chloroplasts. Interestingly, three of the 26 'leaf-blade' genes are known to be encoded in the chloroplast genome in maize, as in other flowering plants [23]. None of them contained a poly(A)+ tail track in the EST sequences. Transcripts for these genes are so abundant in leaf blades that poly (A)+ selection apparently failed to remove them during mRNA purification for cDNA library construction and hybridization target labeling. These chloroplast-encoded genes consistently showed a co-regulated expression pattern, clustering with photosynthesis genes encoded in the nuclear genome. Thus, although they are contaminants, their expression patterns confirm the co-regulation of plastid and nuclear-encoded genes required to construct photosynthetically competent organelles.
A second group of genes was also expressed highly (> 4-fold) in leaf blades, especially adult leaf blades, and was about twofold lower in most other organs (Figure 2, node b). As listed in Figure 5, most of the annotated gene products are involved in the C4 pathway of CO2 fixation. Expression patterns of C4 genes are consistent with previous reports that they were highly expressed in leaves as well as at a low but detectable level in other organs ([24] and references therein, [25]). Abundant transcripts of carbonic anhydrase, phosphoenolpyruvate carboxylase (pepc), and NADP-malic enzyme genes in leaf blades are consistent with the NADP-ME type C4 pathway known to operate in maize [24,26]. In addition to the NADP-ME type C4 pathway genes, phosphoenolpyruvate carboxykinase and alanine aminotransferase genes were also highly expressed in leaf blades. These encode two of the three enzymes that distinguish the phosphoenolpyruvate carboxykinase (PEPCK) C4 pathway. The gene for the other key enzyme, aspartate aminotransferase, was not included in this array, however, EST sequences are well represented in diverse organs including the green tissues of 2-week-old shoot and mixed adult tissue libraries (ZmDB [13]). All genes involved in both the NADP-ME and PEPCK C4 pathways are regulated similarly in all organs surveyed in this study (Figure 2).
Figure 5.
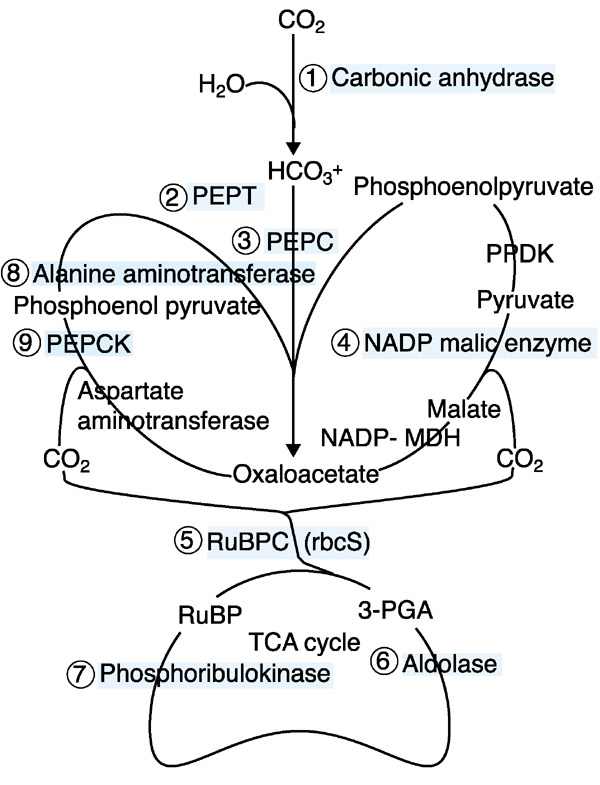
Proposed C4 pathways in maize. CO2 fixation genes on the cladogram of Figure 2 are mapped onto the biochemical pathway. NADP-MDH, NADP-malate dehydrogenase; PEPC, phosphoenolpyruate carboxylase; PEPCK, phosphoenolpyruvate carboxykinase; PEPT, PEP transferase; PPDK, pyruvate orthophosphate dikinase; RuBPC, Rubisco.
Verification of hybridization ratios and ratio interpretation
To estimate how well signal ratios reflect relative transcript amounts, an RNA blot hybridization was carried out with an hrgp probe. There are three hrgp TUGs which are > 94% identical along 860 nucleotides, and, more definitively, 6-, 9- and 15-nucleotide unique indels (insertions or deletions) distinguish them. As shown in Figure 6, blot hybridization with the hrgp probe produced results similar to the pattern of signal ratios in the 12 pairs of microarray hybridizations, which are listed by organ in Figure 7. Overall, individual signal ratios were lower on the microarray hybridization than with the blot hybridization measured by a Phosphor-Imager. For example, the ratio between IME and the reference was 2.5-fold on the array but was 5.6-fold by blot hybridization. Exceptionally, signal ratios for leaf blades were higher in the microarray analysis than by blot hybridization. To some extent these quantitative differences may reflect the different properties of microarray and RNA blot hybridizations. The microarray utilizes a complex mixture of labeled cDNA fragments (targets) to hybridize to one kind of tethered probe at each position, whereas an RNA blot applies one kind of labeled probe to a mixture of RNA targets separated by size using gel electrophoresis.
Figure 6.
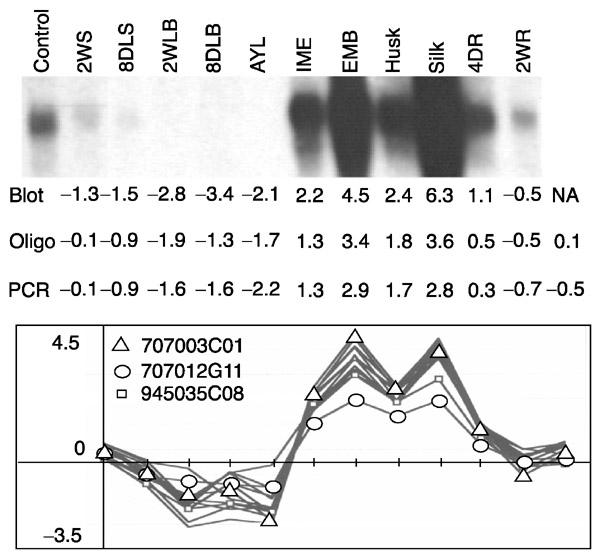
Signal-ratio comparisons between RNA blot and microarray hybridizations for oligos and the corresponding PCR products. Maize RNA samples were prepared from multiple tissues and probed with a hrgp cDNA clone. The blot ratios were calculated from the signal intensity of each lane divided by the signal intensity of the control lane. Signal ratios for individual probes were calculated from microarray hybridization. All calculations are presented as log2 ratios. Tissue abbreviations as in Figure 3.
Figure 7.
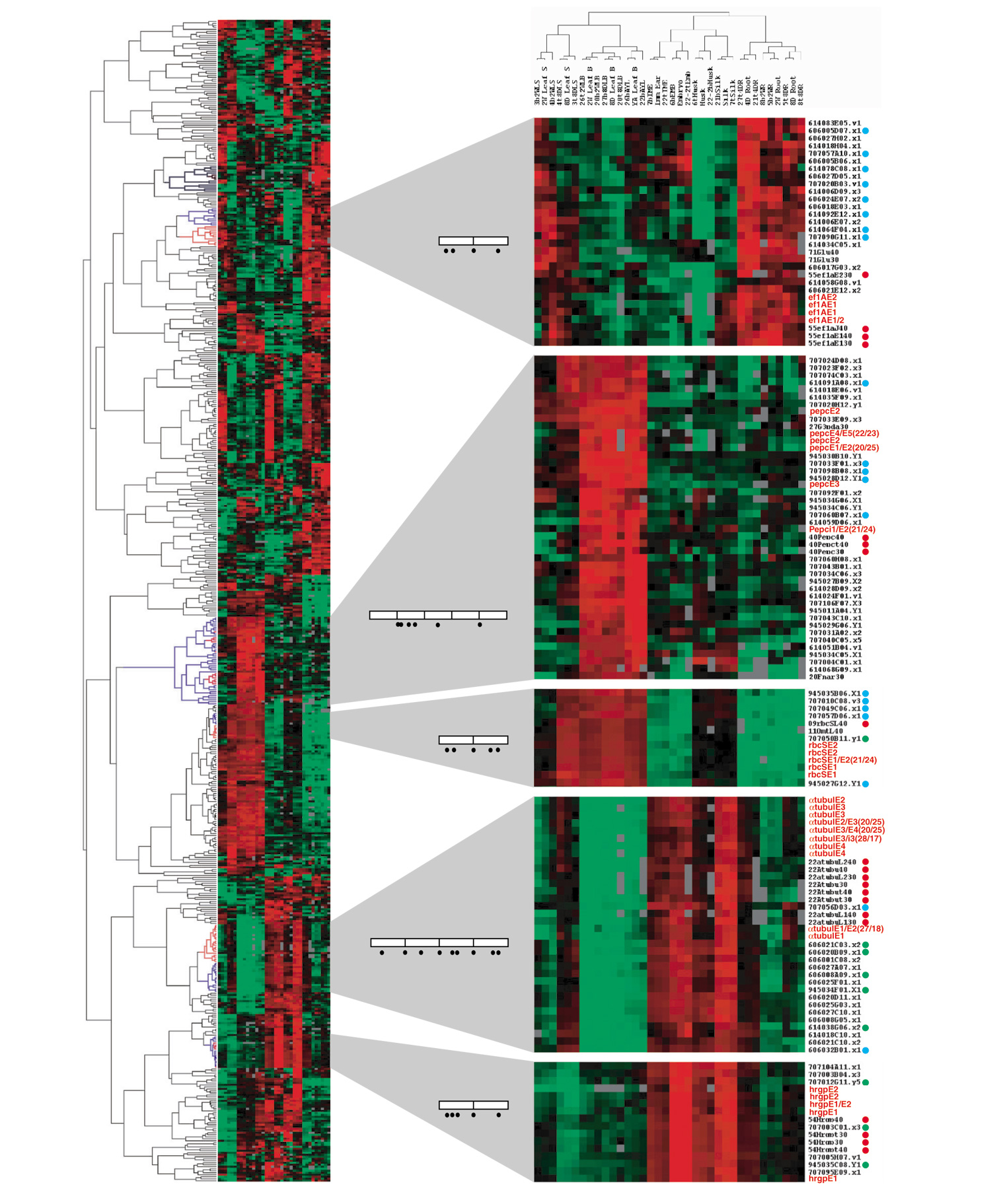
Gene-expression cladogram comparing hybridization patterns of oligos and PCR probes. 582 of 5,760 probes were selected for the analyses on the basis of hybridization success rate (>80%), absolute signal ratio (|log2 ratio| > 2 in a minimum of 1 of 24 pairs of hybridizations, and mean of duplicate hybridizations) and ratio difference (max|log2 ratio| - min|log2 ratio| > 2). Red terminal branches (in the cladogram on left) mark where oligo probes are distributed in each section. Blue branches mark the common terminal branches, which include all oligo probes and one or more related PCR probes. Oligos of 45 nucleotides are in red, and 30-nucleotide and 40-nucleotide oligos are marked with red dots beside each name; the corresponding cDNA probes are marked with green dots, and functionally related genes (according to annotations) with blue dots. Most other genes with EST numbers have not yet been assigned specific functions. Open boxes within gene diagrams represent exons and bars inside each box represent splicing junctions. Black dots depict the positions of oligo probes. The color code represents the relative mRNA amounts: red is high, green is low, and black is similar to the reference sample.
The blot hybridization also appears to report ratios over a wider range. The ratios from the microarrays for EMB and silks were about 7 for both organs, but they were 23 and 79 for EMB and silks in the blot hybridization, respectively. These observations may reflect two features of microarray analysis: the nonlinearity of fluorescence excitation and the saturation of signal intensities for abundant transcripts. The signal ratios from microarrays in this study probably underestimate the actual difference in amount for abundant transcripts. Therefore a small absolute ratio should be interpreted as a reliable indicator for the presence of the transcript type in both samples. In this experiment log2 ratios from -0.5 to 0.5 are interpreted as simply indicating transcript presence without ascribing a difference in absolute amount. Similarly, we conclude that a fourfold difference detected by microarray hybridization indicates more than a fourfold difference in RNA abundance and could indicate organ-specificity of expression.
Hybridization pattern comparisons between oligos and cDNA
There are five gene families on the 326-element cladogram shown in the middle of Figure 2: three αtubulin genes, three βtubulin genes, three carbonic anhydrase genes, four MADS box genes, and five putative cellulose synthase genes. Individual genes in each gene family displayed almost identical expression patterns in all 13 samples. A few individual α-tubulin [27], β-tubulin [28], MADS box [15,29], and cellulose synthase [30] genes have been reported to be differentially expressed in maize or other plants. For these gene families, individual gene expression profiles on microarrays are obscured by cross-hybridization among family members [12,31].
To test whether oligonucleotide probes can be utilized together with cDNA probes to resolve individual gene contributions, multiple oligos were printed on the same glass slide microarrays with the EST probes. We wished to determine whether multiple oligos designed to the same gene would exhibit a coherent hybridization pattern and whether the oligos from a particular gene would cluster with known examples of genes co-regulated in vivo, a powerful test of the microarray [32]. For this analysis, 582 probes were selected, a combination of oligo and EST probes. Oligo probes from five genes (α-tub, hrgp, rbcS, eEF1-α, pepc) met the selection criteria (see legend to Figure 7) of demonstrating a high ratio in at least one hybridization. Multiple oligo probes for each of these genes were printed, as illustrated in the gene models in Figure 7. After cluster analysis, the multiple oligos for each gene established well-separated groups in only one restricted branch of the cladogram of the 582-element data set (Figure 7, left panel). Within the rbcS block as an example, both 45-nucleotide probes from each of two exons appear as close neighbors; no other probes separate them. Such tight groups are characteristic of all 45-nucleotide oligos present in this cladogram. Similar results were produced from data that were neither median-centered nor normalized.
The multiple cDNAs for rbcS, hrgp, and α-tub genes cluster with the corresponding oligo probes. It is notable, however, that two of three hrgp cDNA probes showed fairly reduced ratios in several organs (Figure 6, triangles and squares). The signal ratios from these two PCR probes differ significantly from the other probe and from the oligo probes (p < 0.01 in a paired t-test). Differences between a cDNA and multiple oligo probes are particularly evident for pepc. Six of nine oligos are shown on the cladogram with functionally related genes (Figures 2, 7). Three others were excluded during the selection process because of weak hybridization. On the other hand, the cDNA probe was not selected, because it exhibited an insufficient absolute ratio difference. We suspect that cross-hybridization among pepc family members (or other genes) obscured the authentic gene expression from the cDNA probe. The ratio patterns of oligo probes in 12 pairs of duplicated hybridization experiments suggest that 45-mer oligos are a good alternative to gene-specific RNA blot hybridization to measure expression patterns of specific genes. Oligos of 30 and 40 nucleotides were also used successfully, although signal strength was weaker (red dots on the gene list in Figure 7). We conclude that the oligo hybridization patterns reflect transcript levels relatively accurately for the five genes presented in Figure 7. In fact, the representation is likely to be more accurate than that based on PCR products based on the RNA blot hybridization comparisons (Figure 6).
Discussion
Gene-expression profiles among thirteen samples from seven maize organs were analyzed using cDNA microarrays containing 5,376 unique genes. In addition, oligonucleotide elements included within the same microarrays yielded consistent hybridization patterns; oligo probes are a promising tool for resolving gene - or even allele-specific expression patterns. The majority of genes showed similar hybridization ratios among diverse maize organs, and only 326 (~ 7%) genes appeared highly organ-specific with > 4-fold ratio difference in comparison to the reference 4-day seedling sample. An organ hierarchy based on gene-expression profiles indicated a close relationship among silks, immature ears and embryos. These organs appeared distinct from vegetative organs such as leaf blades, leaf sheaths and roots. Surprisingly, husks were clustered in the floral organ group. In addition, analyses of coordinated expression patterns of photosynthetic genes strongly suggested the presence of two C4 pathways in maize leaf blades. As with other microarray experiments, the newly recognized patterns of gene expression are the springboard for additional genetic and molecular experiments.
Internal consistency of the microarray hybridization
Internal consistency of the array hybridization results was demonstrated by five pieces of evidence. First, a control hybridization with one type of mRNA for which aliquots were labeled with different dyes generated signal ratios within 0.75- to 1.25-fold for 96% of the genes. Second, a dye-swapping hybridization with two samples of mRNA hybridized on separate slides yielded very similar expression profiles (Figure 1). Third, multiple probes for each of several gene family members for five families deposited at random positions generated similar hybridization patterns (Figure 2), suggesting that local effects on hybridization were negligible. Fourth, functionally related genes clustered together, demonstrating a coherent pattern in 12 pairs of hybridization analysis (Figures 2, 7). Fifth, groups of oligos designed to match different positions within several genes generated similar signal ratios, and each oligo group clustered together (Figure 7). Collectively, these facts indicate that hybridization with these microarrays containing a mixture of cDNA and oligo probes was internally consistent.
Organ identity of husks
Each organ is expected to have a unique combination of expressed genes, allowing organ identification and assessment of similarity with other organs as shown by the cladograms in Figure 2. It is interesting that the highly expressed genes in husks parallel what is found in other floral organs. Anatomically, the husks around an ear are composed primarily of leaf sheath with a reduced ligule region subtending a highly reduced leaf blade in most maize inbred lines. Husks are usually classified as modified photosynthetic leaves, with the assumption that they are vegetative organs on a branch that terminates in an ear [16,33]. According the work of Langdale and colleagues [33], maize husks express mainly the C3 pathway of carbon fixation in contrast to leaf blades in which C4 fixation predominates. We found that husk gene-expression profiles are distinctive from both leaf blades and sheaths. For example, Rubisco subunit-binding protein (PID1345582) was expressed at a similar level in husks, but at > 2-fold lower levels in leaf blades compared to the reference 4-day-old shoot (Figure 2). On the other hand, all other photosynthetic genes expressed at high levels in leaf blades were at low levels in husks, relative to seedlings. Physiologically, photosynthetic rates in husks are consistently measured to be around 20-fold lower in leaf blades [33]. Both the expression pattern of photosynthetic genes and the low rate of carbon fixation in husks suggest that these are distinctive organs. In contrast, those genes that are highly expressed in silks and immature ears were expressed at comparable levels in husks (Figure 2 node c). They included hrgp [18], β-amylase (PID9294660), pollen allergen (PID4006978), a bZIP transcription factor (PID6288682), four MADS-box [1,14], three heat-shock proteins, and a dozen uncharacterized genes. Consistent with the hybridization results, one MADS box gene, ZAP1, has been reported to be expressed in the sterile organs of maize florets and in husks [15]. The Arabidopsis homolog AP1 is also expressed in non-reproductive organs such as sepal and petal primordia [34]. The close relationship of husks to maize floral organs shown by gene-expression profiles suggests that husks could be considered as photosynthetic floral organs arising from an inflorescence meristem.
Two types of C4 photosynthesis pathways in maize
C4 plants have been classified into three subgroups on the basis of the distinctive enzymes that decarboxylate C4 acids in the bundle sheath cells. Maize is a classic NADP-ME type C4 plant [35]. Interestingly, we found that the enzyme PEPCK is expressed in a pattern similar to NADP-ME and two additional universal C4 enzyme genes. PEPCK catalyzes the reversible decarboxylation of oxaloacetate (OAA) to PEP. This enzyme has several proposed functions, such as gluco-neogenesis in germinating seeds, carbon recovery during senescence, nitrogen assimilation during seed development and decarboxylation of OAA in PEPCK-type C4 photosynthesis [36,37]. The comparatively low expression level of the PEPCK gene in 4-day-old shoots weakens the hypothesis that its major function in maize is for gluconeogenesis in greening seedling parts. Similarly, high-level expression in seedling leaves and adult leaves cannot be for senescence-related carbon recovery. We concur with recent proposals that the major role of PEPCK in green tissues is decarboxylation of OAA during C4 photosynthesis in maize. Maize leaves have PEPCK activity equal to 45% of the activity levels of a 'pure' PEPCK-type C4 plant, Panicum maximum [34]. Furthermore, the enzyme activity was localized in bundle-sheath cells where CO2 is released from OAA for refixation in the Calvin cycle [38]. The cDNA was cloned from libraries enriched for maize bundle-sheath cells [26].
In addition to PEPCK, two additional genes for key enzymes of the PEPCK pathway were expressed coordinately: alanine aminotransferase and aspartate aminotransferase. Previously, the proteins were undetected by western analysis in purified maize bundle-sheath cells, using antibodies against Panicum maximum aspartate aminotransferase and Cucumis sativus alanine aminotransferase. Detection failure could reflect weak antibody cross-reactivity or enzyme degradation during bundle-sheath cell isolation [38]. By EST sequencing and microarray hybridizations all three PEPCK C4 pathway-specific genes are expressed similarly to the NADP-ME pathway genes. We therefore propose that the PEPCK-type C4 pathway is active in addition to the NADP-ME type C4 pathway for CO2 fixation in maize leaf blades (Figure 5).
Extensive hybridization overlap by diverse organs
The UniGene microarray contained 5,376 elements. On this microarray, most genes hybridized to RNA from diverse organ samples, and very few genes hybridized to RNA from just one sample. According to the array hybridization results, over 60% of the genes produced similar signal ratios (|log2| < 0.5) between the reference and each of 13 samples examined. Thus most transcript types appear to be present at near equivalent levels in many organs of the plant. The interpretation of organ differences reported here reflects results based on a subset (17%) of the current tentative unique genes defined by maize EST collections; many of the EST elements queried are likely to be moderately expressed genes. However, microarray hybridization result is consistent with DNA-RNA reassociation kinetic studies using multiple organs of tobacco plants [39]. About 40% of tobacco genes were expressed in all organs examined (leaf, petal, anther, ovary, root, stem) and 10-40% of the genes were tissue or organ-specific by the criterion of RNA complexity. The apparent low number of tissue- or organ-specific genes observed in maize is also consistent with other microarray studies indicating that only around 25% of Arabidopsis genes displayed significant (> 2-fold) difference in three organ comparisons: seed, root and leaf [40] or root, leaf and flower [11]. Similarly, only 24% of tested genes were distinguishable at three stages of strawberry ripening [6]. Studies of the same organ from different treatment regimes, such as dark-grown and light-grown seedlings of Arabidopsis [10] showed only a 16% difference. During a more complete study of the circadian cycle only 2% of genes examined showed differential expression with a circadian rhythm [41]. From the data available, it appears that plant organs differentially express only a small subset of unique genes and that physiological perturbations result in induction or repression of an even smaller number.
Why do so many genes appear to be expressed in diverse plant organs including those of maize? Some housekeeping genes are constitutively expressed in similar amounts in all organs to insure the maintenance of basic cellular processes. Differential expressions might be cell-type dependent; such differences might not be detected in this study because most tissues were mixtures of multiple cell types. In a gene-expression study during Poplar wood development, > 40% genes were differentially expressed in different development zones within the vascular meristem [42]. Some genes may be expressed at similar RNA levels but protein levels are controlled post-transcriptionally. On microarrays, the correspondence between individual gene expression and hybridization signal is not exact. Cross-hybridization among similar sequences is a major complication in microarrays fabricated with cDNAs. Substantial cross-hybridization has been reported among sequences showing 85% similarity over 30 nucleotides [31]. Cross-hybridization between related genes will be a profound problem in the analysis of gene-expression patterns in plants. About 70% of genes are duplicated in A. thaliana through both chromosome and local duplications [2,43]. Maize has undergone an allo-tetraploid chromosome duplication event within the past 11.4 million years, preceded by other genome-wide duplications [44,45]. In the available studies, however, there are many examples of maize duplicated genes expressed in different organs [46]. For example, two duplicated transcription factor genes (p1, p2) regulating pholaphene pigment synthesis are expressed fairly exclusively in two sets of organs. p1 and p2 arose following a local gene duplication and insertion of multiple retroelements between p1 and p2. Subsequently, p1 acquired a new regulatory sequence 5' of the gene, probably explaining its new expression pattern [47]. There are many retroelements and DNA transposons flanking maize genes, and they may contribute to the rapid divergence of transcriptional regulation [48]. Another example comes from duplicated chalcone synthase genes (C2, Whp). They share over 94% sequence similarity but are differentially expressed [46]. It is also evident that some duplicated genes are expressed redundantly at the same time in the same organ. For example, five copies of cellulose synthase genes [30] and five copies of eEF1-α genes [49] are coexpressed in diverse organs.
Redundant expression among duplicated genes
Sequence comparisons among TUGs assembled within individual EST sequencing projects 614 (4DR) and 606 (IME) provide anecdotal information about the expression modes of duplicated genes. TUGs sharing > 90% sequence similarity over 100 nucleotides were identified by BLAST [17]. Within library 606 (immature ear) 32% (963/3,032) of the TUGs are similar at this criterion, and 33% (1290/3,879) of the TUGs defined within library 614 (seedling root) appear to be duplicated genes. When TUGs assembled from libraries 606 and 614 are compared to each other, 20% appear to be duplicated. The sequence comparisons among TUGs probably underestimate the number of duplicated genes because sequence data are incomplete. Comparisons of full-length cDNA sequences of each gene would increase the fractions of gene families both within and between these two libraries. Because microarray hybridization conditions cannot resolve the precise expression patterns of gene > 90% similar, the true fraction of constitutively expressed genes cannot be calculated.
An important question is what fraction of closely related duplicated genes are expressed differentially during the maize life cycle. For the moderately expressed class of genes 'discovered' by EST sequencing of specific developmental stages, it is striking that so many gene families are expressed in all 13 samples examined here. Functional redundancy among individual genes within a gene family would produce no detectable phenotype until all functionally redundant genes are mutated (see examples in [1]). Yet, mutations in individual maize genes within a large gene family can produce a visible phenotype. This evidence indicates that functional specialization has occurred. By RNA blot hybridizations, it is often observed that the relative amount of transcripts varies among individual genes within a family, suggesting that promoter divergence produces quantitative differences [30,49]. In some cases, mutation that eliminates expression from one gene-family member may be compensated by higher expression of other members; even if there is no visible phenotype, a molecular phenotype is predicted. cDNA microarrays are not sensitive enough to detect minor changes of expression patterns or differential expression of recently duplicated genes with the current hybridization condition (65°C hybridization and 55°C washing). Oligo probes appear to be a good alternative for analysis of individual gene-expression profiles, either in conjunction with PCR products or by themselves. Oligo probes can be designed to represent individual genes by exploiting even small polymorphisms. Our results show that suitable hybridization and washing conditions can be used for the analysis of PCR and oligo probes on the same microarray slide.
Materials and methods
Sample organs
The maize strain in this study has the genetic background K55 (75%), W23 (20%), Robertson's Mutator (5%). Seedlings were grown under 100 μE/m2/sec constant illumination conditions of cool-white fluorescent light in a 27°C growth room. For the 4-day seedling with coleoptile reference sample, the shoot was harvested; other seedling samples were taken at 8 days or 14 days after planting. Field-grown plants were the source of most organ samples. The same genotype was planted in mid-June at the Stanford University Plant Growth Facility. Three immature ears (3-5 cm) were harvested after the tips of the husks had emerged from a leaf sheath; silks were excluded from immature ears. Husks were collected from the same ear. The two outermost husk layers were excluded, and all other inside layers were collected. Mature but unpollinated silks were harvested from two ears; the ears had been shoot-bagged to prevent pollen contamination on the silks. Adult leaf blades were taken from fully expanded leaves.
cDNA probe preparation
cDNA clones were from three, non-normalized cDNA libraries: a mixture of adult tissue (projects 707 and 945, W23 inbred line with active Mutator transposons), 4-day-old roots (project 614, W23 inbred line), and immature ears (project 606, Oh43 inbred line). The 5,376 cDNA clones chosen represent approximately 17% of the tentative unique genes in the April 2002 EST assembly [13]. They were designated as UniGene1 members after EST assembly of 73,000 available ESTs representing around 17,000 TUGs in September 2000. A clone for each TUG was selected on the basis of EST sequence length during UniGene1 consolidation. In many cases ESTs defining particular TUGs were recovered from multiple libraries. Clone identities were verified in UniGene1 by resequencing for approximately 50% of the clones to confirm well positions in the consolidation plates.
PCR amplifications of cDNA inserts were carried out at annealing temperatures of 50°C (614 and 606 libraries) or 60°C (707 library) in a 25 μl volume in a GeneAmp PCR system 9700 (Applied Biosystems, Foster City, CA) thermocycler for 35 cycles with a 2 min extension time. The reaction cocktail contained 1 ng EST plasmid DNA, 1.7 mM MgCl2, 1x reaction buffer (50 mM Tris pH 8.5, 20 mM KCl), and 0.1 units of Taq polymerase (GibcoBRL, Gaithersburg, MD). PCR-amplified products were purified with Gene Clean kits (BIO101, Carlsbad, CA), and eluted in 20 μl water. Samples of 3 μl were loaded onto a 1% agarose gel and electrophoresed to measure product size and yield. Of the 5,376 ESTs 197 produced multiple bands or smeared products and were excluded from the analyses. Samples of 10 μl were transferred to a 384-well plate and dried completely; the pellet was dissolved before printing in 5 μl of 150 mM phosphate pH 7.0 buffer to yield approximately 300 ng/μl DNA concentration. Probes were printed on 3D-link slides, followed by coupling and processing as recommended by the manufacturer (SurModics, Eden Prairie, MN).
Oligo probe preparation
A minimum of one oligo was synthesized within each exon, intron, and at exon/intron and exon1/exon2 junction regions from 17 selected maize genes. In most cases, two probes were synthesized in each exon and intron. Oligo design was based on double-stranded complete gene sequence, available from GenBank. Oligos were synthesized using phosphoramidite chemistry on an automated oligo-nucleotide synthesizer at the Stanford Genome Technology Center [50]. Oligos were synthesized from the 3' to 5' direction, and the 5' end of each oligo was modified by addition of a C6-amide group. A total 184 oligos of 45 nucleotides were synthesized from the selected 17 genes. In addition, 96 oligos of 40 nucleotides and 96 oligos of 30 nucleotides were synthesized from 45 different genes for comparison of their hybridization behavior to PCR probes; short oligos were also prepared for the 17 genes for which the 45-nucleotides oligos were designed. The calculated Tm of exon probes ranged from 92-109°C, while the Tm of intron and intron/exon junction probes ranged from 89-95°C. Synthesized oligos were dissolved in 150 mM phosphate buffer, pH 8.5 at 40 μm. Multiple 45-nucleotide oligo probes for 17 genes were printed on the same arrays as the cDNA probes. Mean signal intensities were calculated from each of 10 genes represented by these 45-mers that showed consistent hybridization in all organ comparisons. They were used to examine the consistency of hybridization as positive control elements, and they were included in the cluster analyses.
RNA purification, labeling, and hybridization
Total RNA was extracted from 13 samples, using the Trizol method (GibcoBRL). mRNA was further purified from total RNA with Oligotex mini-columns (Qiagen, Valencia, CA). mRNA quantity and quality were examined by UV absorption at 260 and 280 nm. RNA quality was also examined by agarose gel electrophoresis to monitor loss of ribosomal RNA after mRNA purification. About 2 μg of poly (A)+ RNA was used to synthesize fluorescently labeled cDNA targets. The reaction cocktail contained ~2 μg poly(A)+ RNA, 1x reaction buffer (50 mM Tris-HCl pH 8, 75 mM KCl, 3 mM MgCL2, 50 μM dNTP, 10 mM DTT), 1 μg oligo dT, 3 μg random hexamer, and 400 units Superscript II (GibcoBRL).
Hybridization was performed as described at [51]. Variations included hybridization at temperatures between 61-65°C and an initial washing at 55°C. mRNA from 4-day-old shoots with coleoptiles was labeled with Cy3-dUTP, which served as the common reference in all pairs of hybridization for the cluster analyses with Cy5-dUTP labeled samples from other stages. Microarray slides were scanned with an Axon400 scanner (Axon Instrument, Union City, CA). Signal was initially normalized during the image scanning process to adjust the average ratios between two channels. Grids were generated and adjusted automatically then refined manually to identify the microarray elements. Those probes whose signal intensity, subtracted by background, was lower than 300 in either channel or less than 1,500 in the sum of both channels were excluded from further analyses. Signal ratios for each probe element on each slide were calculated, using the mean intensity of pixels subtracted by median background for each channel. Array results are deposited at a public gene-expression database, Gene Expression Omnibus [52], and their accession numbers are GPL12 for the platform and GSM57-GSM80 for 24 samples.
Hierarchical clustering
Hierarchical clustering of the data was performed using the computer program Cluster [32]. The output was visualized using the program TreeView (available at [53]). Cluster analyses were carried out before and after a secondary normalization process to make the sum of the squares 1.0 in each row and column. Although the results were very similar, we prefer the results from the unnormalized data for three reasons. First, the reference sample is identical in all hybridizations. Second, unnormalized data produced organ relationships consistent with organ identities and the relationships inferred from normalized data. Third, the normalization could compound variation by combining an uncertainty from a computation method on top of the variations from hybridization.
RNA blot hybridization
A 15 μg sample of total RNA was loaded onto a glyoxal gel as described in [54]. Hybridization probes for RNA blots were prepared by the random primer labeling method to incorporate 32P. Blots were analyzed on a Phosphorlmager (Molecular Dynamics, Sunnyvale, CA).
Additional data files
A list of genes differentially expressed between 4-day-old roots and immature ears, and a list of the 326 microarray elements in Figure 2.
Supplementary Material
A list of genes differentially expressed between 4-day-old roots and immature ears
A list of the 326 microarray elements in Figure 2
Acknowledgments
Acknowledgements
We thank Brian Nakao, Gurpreet Randhawa and Khaled Sarsour for generating ESTs and UniGene1 verification sequencing, and ZmDB curators for database maintenance. We extend special thanks to Bret Schneider, Darren Morrow, Paula Casati, Mathew Fitzgerald and Dean Goodman for critical reading of the manuscript. The work is supported by National Science Foundation grant 98-72657 to V.W.
References
- Jack T. Plant development going MADS. Plant Mol Biol. 2001;46:515–520. doi: 10.1023/a:1010689126632. [DOI] [PubMed] [Google Scholar]
- The Arabidopsis Genome Initiative Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene-expression patterns with a complementary-DNA microarray. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- Schena M, Shalon D, Heller R, Chai A, Brown PO, Davis RW. Parallel human genome analysis: microarray-based expression monitoring of 1000 genes. Proc Natl Acad Sci USA. 1996;93:10614–10619. doi: 10.1073/pnas.93.20.10614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRisi JL, Iyer VR, Brown PO. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science. 1997;278:680–686. doi: 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- Aharoni A, Keizer LC, Bouwmeester HJ, Sun Z, Alvarez-Huerta M, Verhoeven HA, Blaas J, van Houwelingen AM, De Vos RC, van der Voet H, et al. Identification of the SAAT gene involved in strawberry flavor biogenesis by use of DNA microarrays. Plant Cell. 2000;12:647–662. doi: 10.1105/tpc.12.5.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helliwell CA, Chin-Atkins AN, Wilson IW, Chapple R, Dennis ES, Chaudhury A. The Arabidopsis amp1 gene encodes a putative glutamate carboxypeptidase. Plant Cell. 2001;13:2115–2125. doi: 10.1105/TPC.010146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reymond P, Weber H, Damond M, Farmer EE. Differential gene expression in response to mechanical wounding and insect feeding in Arabidopsis. Plant Cell. 2000;12:707–720. doi: 10.1105/tpc.12.5.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki M, Narusaka M, Abe H, Kasuga M, Yamaguchi-Shinozaki K, Carninci P, Hayashizaki Y, Shinozaki K. Monitoring the expression pattern of 1300 Arabidopsis genes under drought and cold stresses by using a full-length cDNA microarray. Plant Cell. 2001;13:61–72. doi: 10.1105/tpc.13.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desprez T, Amselem J, Caboche M, Hofte H. Differential gene expression in Arabidopsis monitored using cDNA arrays. Plant J. 1998;14:643–652. doi: 10.1046/j.1365-313X.1998.00160.x. [DOI] [PubMed] [Google Scholar]
- Ruan Y, Gilmore J, Conner T. Towards Arabidopsis genome analysis: monitoring expression profiles of 1400 genes using cDNA microarrays. Plant J. 1998;15:821–833. doi: 10.1046/j.1365-313X.1998.00254.x. [DOI] [PubMed] [Google Scholar]
- Fernandes J, Brendel V, Gai X, Lal S, Chandler VL, Elumalai R, Galbraith DW, Pierson E, Walbot V. Comparison of RNA expression profiles based on maize EST frequency analysis and microarray hybridization. Plant Physiol. 2002;128:896–910. doi: 10.1104/pp.010681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZmDB: a maize genome database: class browser http://www.zmdb.iastate.edu
- Heuer S, Hansen S, Bantin J, Brettschneider R, Kranz E, Lorz H, Dresselhaus T. The maize mads box gene zmmads3 affects node number and spikelet development and is co-expressed with zmmads1 during flower development, in egg cells, and early embryogenesis. Plant Physiol. 2001;127:33–45. doi: 10.1104/pp.127.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mena M, Mandel MA, Lerner DR, Yanofsky MF, Schmidt RJ. A characterization of the MADS-box gene family in maize. Plant J. 1995;8:845–854. doi: 10.1046/j.1365-313x.1995.8060845.x. [DOI] [PubMed] [Google Scholar]
- Cheng P-C, Pareddy D. Morphology and development of the tassel and ear. In: Freeling M, Walbot V, editor. In The Maize Handbook. New York: Springer-Verlag; 1993. pp. 37–47. [Google Scholar]
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood EE, Hood KR, Fritz SE. Hydroxyproline-rich glycoproteins in cell walls of pericarp from maize. Plant Sci. 1991;79:13–22. doi: 10.1104/pp.96.4.1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto-Sotelo J, Kannan KB, Martinez LM, Segal C. Characterization of a maize heat-shock protein 101 gene, HSP101, encoding a ClpB/Hsp100 protein homologue. Gene. 1999;230:187–195. doi: 10.1016/S0378-1119(99)00060-8. [DOI] [PubMed] [Google Scholar]
- Van Breusegem F, Dekeyser R, Garcia AB, Claes B, Gielen J, Van Montagu M, Caplan AB. Heat-inducible rice hsp82 and hsp70 are not always co-regulated. Planta. 1994;193:57–66. doi: 10.1007/BF00191607. [DOI] [PubMed] [Google Scholar]
- Michelis R, Gepstein S. Identification and characterization of a heat-induced isoform of aldolase in oat chloroplast. Plant Mol Biol. 2000;44:487–498. doi: 10.1023/a:1026528319769. [DOI] [PubMed] [Google Scholar]
- Fischer K, Arbinger B, Kammerer B, Busch C, Brink S, Wallmeier H, Sauer N, Eckerskorn C, Flugge UI. Cloning and in vivo expression of functional triose phosphate/phosphate translocators from C3- and C4-plants: evidence for the putative participation of specific amino acid residues in the recognition of phosphoenolpyruvate. Plant J. 1994;5:215–226. doi: 10.1046/j.1365-313X.1994.05020215.x. [DOI] [PubMed] [Google Scholar]
- Maier RM, Neckermann K, Igloi GL, Kossel H. Complete sequence of the maize chloroplast genome: gene content, hotspots of divergence and fine tuning of genetic information by transcript editing. J Mol Biol. 1995;251:614–628. doi: 10.1006/jmbi.1995.0460. [DOI] [PubMed] [Google Scholar]
- Ku MS, Kano-Murakami Y, Matsuoka M. Evolution and expression of C4 photosynthesis genes. Plant Physiol. 1996;111:949–957. doi: 10.1104/pp.111.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvez S, Hirsch AM, Wycoff KL, Hunt S, Layzell DB, Kondorosi A, Crespi M. Oxygen regulation of a nodule-located carbonic anhydrase in alfalfa. Plant Physiol. 2000;124:1059–1068. doi: 10.1104/pp.124.3.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furumoto T, Hata S, Izui K. cDNA cloning and characterization of maize phosphoenolpyruvate carboxykinase, a bundle sheath cell-specific enzyme. Plant Mol Biol. 1999;41:301–311. doi: 10.1023/a:1006317120460. [DOI] [PubMed] [Google Scholar]
- Uribe X, Torres MA, Capellades M, Puigdomenech P, Rigau J. Maize alpha-tubulin genes are expressed according to specific patterns of cell differentiation. Plant Mol Biol. 1998;37:1069–1078. doi: 10.1023/a:1006067710312. [DOI] [PubMed] [Google Scholar]
- Hussey PJ, Haas N, Hunsperger J, Larkin J, Snustad DP, Silflow CD. The beta-tubulin gene family in Zea mays: two differentially expressed beta-tubulin genes. Plant Mol Biol. 1990;15:957–972. doi: 10.1007/BF00039438. [DOI] [PubMed] [Google Scholar]
- Cordts S, Bantin J, Wittich PE, Kranz E, Lorz H, Dresselhaus T. ZmES genes encode peptides with structural homology to defensins and are specifically expressed in the female gametophyte of maize. Plant J. 2001;25:103–114. doi: 10.1046/j.0960-7412.2000.00944.x. [DOI] [PubMed] [Google Scholar]
- Holland N, Holland D, Helentjaris T, Dhugga KS, Xoconostle-Cazares B, Delmer DP. A comparative analysis of the plant cellulose synthase (CesA) gene family. Plant Physiol. 2000;123:1313–1324. doi: 10.1104/pp.123.4.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Bak S, Decker A, Paquette SM, Feyereisen R, Galbraith DW. Microarray-based analysis of gene expression in very large gene families: the cytochrome P450 gene superfamily of Arabidopsis thaliana. Gene. 2001;272:61–74. doi: 10.1016/S0378-1119(01)00516-9. [DOI] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langdale JA, Zelitch I, Miller E, Nelson T. Cell position and light influence C4 versus C3 patterns of photosynthetic gene expression in maize. EMBO J. 1998;7:3643–3651. doi: 10.1002/j.1460-2075.1988.tb03245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel MA, Gustafson-Brown C, Savidge B, Yanofsky MF. Molecular characterization of the Arabidopsis floral homeotic gene APETALA1. Nature. 1992;360:273–277. doi: 10.1038/360273a0. [DOI] [PubMed] [Google Scholar]
- Martin R, Niyogi K. Photosynthesis. In: Buchanan BB, Gruissem W, Jones RL, editor. In Biochemistry and Molecular Biology of Plants. Rockville MD: American Society of Plant Physiologists; 2000. pp. 619–628. [Google Scholar]
- Chen ZH, Walker RP, Acheson RM, Tecsi LI, Wingler A, Lea PJ, Leegood RC. Are isocitrate lyase and phosphoenolpyruvate carboxykinase involved in gluconeogenesis during senescence of barley leaves and cucumber cotyledons? Plant Cell Physiol. 2000;41:960–967. doi: 10.1093/pcp/pcd021. [DOI] [PubMed] [Google Scholar]
- Walker RP, Chen Z-H, Tecsi LI, Ramiani F, Lea PJ, Leegood RC. Phosphoenlpyruvate carboxykinase plays a role in interactions of carbon and nitrogen metabolism during grape seed development. Planta. 1999;210:9–18. doi: 10.1007/s004250050648. [DOI] [PubMed] [Google Scholar]
- Wingler A, Walker RP, Chen ZH, Leegood RC. Phosphoenolpyruvate carboxykinase is involved in the decarboxylation of aspartate in the bundle sheath of maize. Plant Physiol. 1999;120:539–546. doi: 10.1104/pp.120.2.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamalay JC, Goldberg RB. Organ-specific nuclear RNAs in tobacco. Proc Natl Acad Sci USA. 1984;81:2801–2805. doi: 10.1073/pnas.81.9.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girke T, Todd J, Ruuska S, White J, Benning C, Ohlrogge J. Microarray analysis of developing Arabidopsis seeds. Plant Physiol. 2000;124:1570–1581. doi: 10.1104/pp.124.4.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer R, Landgraf J, Accerbi M, Simon V, Larson M, Wisman E. Microarray analysis of diurnal and circadian-regulated genes in Arabidopsis. Plant Cell. 2001;13:113–123. doi: 10.1105/tpc.13.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertzberg M, Aspeborg H, Schrader J, Andersson A, Erlandsson R, Blomqvist K, Bhalerao R, Uhlen M, Teeri TT, Lundeberg J, et al. A transcriptional roadmap to wood formation. Proc Natl Acad Sci USA. 2001;98:14732–14737. doi: 10.1073/pnas.261293398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vision TJ, Brown DG, Tanksley SD. The origins of genomic duplications in Arabidopsis. Science. 2000;290:2114–2117. doi: 10.1126/science.290.5499.2114. [DOI] [PubMed] [Google Scholar]
- Gaut BS, Doebley JF. DNA sequence evidence for the segmental allotetraploid origin of maize. Proc Natl Acad Sci USA. 1997;94:6809–6814. doi: 10.1073/pnas.94.13.6809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson WA, Harrington SE, Woodman WL, Lee M, Sorrells ME, McCouch SR. Inferences on the genome structure of progenitor maize through comparative analysis of rice, maize and the domesticated panicoids. Genetics. 1999;153:453–473. doi: 10.1093/genetics/153.1.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franken P, Niesbach-Klosgen U, Weydemann U, Marechal-Drouard L, Saedler H, Wienand U. The duplicated chalcone synthase genes C2 and Whp (white pollen) of Zea mays are independently regulated: evidence for translational control of Whp expression by the anthocyanin intensifying gene in. EMBO J. 1991;10:2605–2612. doi: 10.1002/j.1460-2075.1991.tb07802.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Chopra S, Peterson T. A segmental gene duplication generated differentially expressed myb-homologous genes in maize. Plant Cell. 2000;12:2311–2322. doi: 10.1105/tpc.12.12.2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Arbuckle J, Wessler SR. Recent, extensive, and preferential insertion of members of the miniature inverted-repeat transposable element family Heartbreaker into genic regions of maize. Proc Natl Acad Sci USA. 2000;97:1160–1165. doi: 10.1073/pnas.97.3.1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carneiro NP, Hughes PA, Larkins BA. The eEFIA gene family is differentially expressed in maize endosperm. Plant Mol Biol. 1999;41:801–813. doi: 10.1023/a:1006391207980. [DOI] [PubMed] [Google Scholar]
- Lashkari DA, Hunicke-Smith SP, Norgren RM, Davis RW, Brennan T. An automated multiplex oligonucleotide synthesizer: development of high-throughput, low-cost DNA synthesis. Proc Natl Acad Sci USA. 1995;92:7912–7915. doi: 10.1073/pnas.92.17.7912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Brown Lab: protocols http://cmgm.stanford.edu/pbrown/protocols/
- Gene Expression Omnibus http://www.ncbi.nlm.nih.gov/geo/
- Eisen lab http://rana.stanford.edu/software/
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A list of genes differentially expressed between 4-day-old roots and immature ears
A list of the 326 microarray elements in Figure 2