

Abstract
An extrachromosomally replicating plasmid was used to investigate the specificity by which the origin recognition complex (ORC) interacts with DNA sequences in mammalian cells in vivo. We first showed that the plasmid pEPI-1 replicates semiconservatively in a once-per-cell-cycle manner and is stably transmitted over many cell generations in culture without selection. Chromatin immunoprecipitations and quantitative polymerase chain reaction analysis revealed that, in G1-phase cells, Orc1 and Orc2, as well as Mcm3, another component of the prereplication complex, are bound to multiple sites on the plasmid. These binding sites are functional because they show the S-phase-dependent dissociation of Orc1 and Mcm3 known to be characteristic for prereplication complexes in mammalian cells. In addition, we identified replicative nascent strands and showed that they correspond to many plasmid DNA regions. This work has implications for current models of replication origins in mammalian systems. It indicates that specific DNA sequences are not required for the chromatin binding of ORC in vivo. The conclusion is that epigenetic mechanisms determine the sites where mammalian DNA replication is initiated.
Keywords: initiation of DNA replication, origin of replication, mammalian episome
Introduction
The initiation of genome replication starts with the binding of specific initiator proteins to genomic sites, termed origins, and results in the localized unwinding of the DNA duplex and the establishment of replication forks. These interactions are best understood for prokaryotes and some eukaryotic viruses such as simian virus 40 (SV40) expressing one single initiator protein, T antigen, which binds with high specificity to the viral origin, a specific 60-bp-long DNA segment, and functions as a helicase separating the complementary DNA strands. This simple initiation mechanism has evolved to serve the purpose of ‘run-away' replication of the viral genome (for a review, see Simmons, 2000).
In contrast, the replication of cellular genomes is strictly coordinated within the cell cycle and effectively controlled such that the genome is replicated completely, but without over-replication, ‘once and only once' per cell cycle (Coverley and Laskey, 1994). This is achieved by several levels of control beginning with the stepwise assembly of prereplication complexes (pre-RC) in the early G1 phase of the cell cycle consisting of the DNA-bound multisubunit origin recognition complex (ORC with its components Orc1–Orc6), which serves as a platform for the association of additional proteins, Cdc6, Cdt1 and the six Mcm proteins (MCM2–MCM7). Transition into the S phase is initiated by the timely activation of specific protein kinases and is marked by the conversion of pre-RCs to initiation complexes (for reviews, see Bell and Dutta, 2002; Blow and Hodgson, 2002).
Re-replication during the S phase is prevented by a partial decomposition of the pre-RC including the loss of Mcm proteins, which move away from their original binding sites probably in close association with replication forks (Aparicio et al, 1997; Labib et al, 2000; Alexandrow et al, 2002; Schaarschmidt et al, 2002) where they may be involved in DNA unwinding (You et al, 1999) and eventually dissociate from chromatin (for a review, see Tye, 1999). An additional mechanism preventing the re-replication of DNA is the dissociation of the Orc1p subunit of ORC in mammalian cells (for a review, see DePamphilis, 2003).
In the budding yeast, Saccharomyces cerevisiae, pre-RCs assemble at defined origin elements known as autonomously replicating sequences (ARSs) because they direct the extrachromosomal replication of ARS-bearing DNA circles. ARS sites are composed of several short sequences including an AT-rich ARS consensus which, together with adjacent elements, form binding sites for ORC (Bell and Stillman, 1992; Lee and Bell, 1997) and are start sites for bidirectional DNA replication (for a review, see Bielinsky and Gerbi, 2001). In contrast, origins of the fission yeast, Saccharomyces pombe, are longer, 500–1000 bp in size, and do not contain a consensus sequence, but possess elements rich in AT base pairs which serve as binding sites for ORC (Chuang and Kelly, 1999). Moreover, the two dozen or so metazoan origins characterized so far share no obvious sequence elements, except perhaps short stretches of AT base pairs (for reviews, see DePamphilis, 1999; Gilbert, 2001). This lack of sequence homology contrasts with the finding that identical replication start sites are used in successive mammalian cell cycles. In fact, a transfer of authentic origins to ectopic sites in the same nucleus showed that replication does not start at random genomic sites, and, moreover, that a given origin is composed of discrete functional elements which, however, differ among individual origins (Altman and Fanning, 2001; Liu et al, 2003). It has therefore been proposed that replication start sites in mammalian genomes are not so much determined by the underlying DNA sequence, but by epigenetic factors such as the presence of specifically bound transcription factors or the conformation of chromatin (for a review, see Mechali, 2001).
For a detailed investigation of the origin problem in metazoa and particularly in mammalian cells, it would be useful to possess an assay system that is comparable to the ARS system in yeast. However, searches for ARS-like elements in cultured mammalian cells were usually not successful and did not generally lead to specific sequences that confer a replication advantage over extrachromosomal DNA circles without the sequence. In fact, it has been observed that all DNA segments >15 kb replicate with similar efficiencies in a once-per-cell-cycle manner in cultured mammalian (Caddle and Calos, 1992; Krysan et al, 1993) and Drosophila cells (Smith and Calos, 1995). However, it has also been reported that plasmids with an authentic origin sequence possess a replicative advantage over plasmids lacking this sequence (McWhinney and Leffak, 1990), and a loose potential consensus sequence has recently been described for DNA circles that replicate extrachromosomally in cultured mammalian cells (Price et al, 2003). In either case, many of these observations are difficult to interpret because they depend on transient replication assays with extrachromosomal elements that were either gradually lost upon subsequent cell divisions or integrated into the cellular genome.
To investigate the in vivo DNA-binding specificities of mammalian ORC, we use a plasmid, pEPI-1, with a unique combination of genetic elements including a strong eukaryotic promoter and a scaffold/matrix attachment region (S/MAR) (Figure 1A). This DNA circle of ca. 6.7 kb has been shown to replicate extrachromosomally over hundreds of cell generations in Chinese hamster ovary (CHO) cells (Piechaczek et al, 1999) and HeLa cells (see below). It associates with mitotic chromosomes (Baiker et al, 2000), most probably via an interaction of its S/MAR element with the scaffold attachment factor SAF-A in cellular chromosomes (Jenke et al, 2002), and is therefore correctly transmitted from parental to progeny cells.
Figure 1.
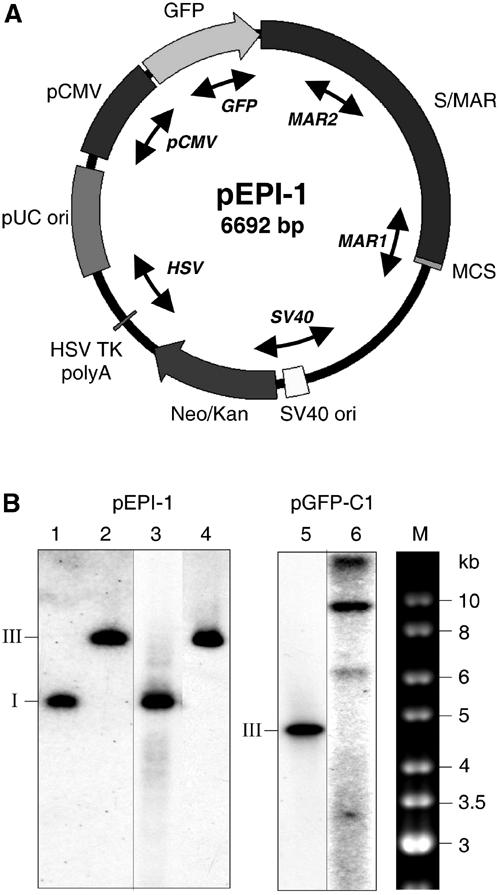
pEPI-1 replicates episomally in HeLa cells. (A) Map of pEPI-1. The human S/MAR was cloned into the multiple cloning site of the vector pGFP-C1 (Piechaczek et al, 1999). The divergent arrow heads indicate the regions analyzed by PCR. (B) Southern blots of DNA from the HeLa cell clones used for ChIP. DNA was separated on 0.8% agarose gels, blotted and hybridized with 32P-labeled pGFP-C1 probe. Lanes 1 and 2, 20 pg (about 3 × 106 molecules) of undigested (supercoiled form I) or EcoRI-digested (linearized form III) pEPI-1 DNA. Lanes 3 and 4, undigested and EcoRI-digested DNA from a Hirt supernatant of 106 pEPI-1 transfected HeLa cells. Lane 5, 20 pg (about 4 × 106 molecules) of EcoRI-digested (linearized form III) pGFP-C1 DNA. Lane 6, EcoRI-digested total genomic DNA of about 3 × 106 pGFP-C1-transfected HeLa cells. M, 1-kb ladder (MBI) as marker.
Here, we report that pEPI-1 replicates early in S phase in a once-per-cell-cycle manner like a chromosomal DNA segment, and carries ORC and other components of the pre-RC. Importantly, we demonstrate by chromatin immunoprecipitation (ChIP) and quantitative polymerase chain reaction (PCR) that, in the G1 phase, members of ORC and of the Mcm proteins are bound to many regions of the plasmid. These sequences are enriched in a fraction of nascent DNA, suggesting that the bound ORCs coincide with regions where replication is initiated. Furthermore, Orc1p and Mcm3p dissociate during the S phase and thus behave just like chromosomally bound Orc1p and Mcm proteins as previously described (Kreitz et al, 2001). These results show that ORC is able to bind productively to many DNA sequences in vivo, and imply that, in principle, many, if not random, sites in the mammalian genome could serve as origins of DNA replication.
Results
pEPI-1 replicates as an episome in mammalian cells
The construction of pEPI-1 (Figure 1A) has been described by Piechaczek et al. (1999), who showed that the presence of S/MAR (from the 5′ region of the human interferon-β gene; Bode et al, 1992) is essential for extrachromosomal replication and for its stable transmission at mitosis, because a plasmid, pGFP-C1, identical to pEPI-1, but lacking S/MAR, integrates into the cellular genome. More recently, Stehle et al. (2003) demonstrated that an active cytomegalovirus promoter (pCMV in Figure 1A), which directs the transcription through the GFP gene and well into the S/MAR region, is an essential element for autonomous replication whereas all SV40-derived sequences in pEPI-1 are dispensable (IM Stehle and HJ Lipps, unpublished data). It thus appears that two regions are important for extrachromosomal replication, an active CMV promoter with the adjacent downstream sequences and at least part of S/MAR, which seems to be required for mitotic transmission. In the absence of one or the other element, the plasmid is rapidly incorporated into the host genome or lost.
To demonstrate the episomal state of pEPI-1 in the cell lines under study, we prepared extrachromosomal DNA from pEPI-1-transfected CHO cells using a procedure that Hirt had originally developed to separate the large cellular DNA from small viral DNA (see Supplementary Data). pEPI-1 plasmids in the Hirt supernatants were amplified in bacteria and used to transfect CHO cells and human HeLa cells. The S/MAR-lacking plasmid pGFP-C1 served as a control in parallel transfections. Transfected cells were selected with the kanamycin derivative G418. Several of the resulting G418-resistant colonies were subcloned and then expanded in media without the drug. After approximately 30 cell generations in the absence of selection, Hirt supernatants were analyzed by Southern blotting for free extrachromosomal pEPI-1 DNA circles. These were detected in nine out of the 10 CHO cell clones investigated as well as in all five HeLa cell clones. Figure 1B shows the result of a HeLa clone that was further investigated (see below). The number of extrachromosomal pEPI-1 molecules was determined by comparing the Southern blot bands of Hirt supernatants with those of known quantities of pEPI-1 and by quantitative real-time PCR (see below). We measured six–nine copies per cell in HeLa as well as in CHO cells in agreement with previous results (Piechaczek et al, 1999; Baiker et al, 2000). The mitotic stability and integrity of these extrachromosomal elements were tested by fluctuation assays and bacterial retransformation experiments (see Table SI in Supplementary data, available at The EMBO Journal Online).
In contrast, the Hirt supernatants of the six CHO and four HeLa cell clones transfected with the control plasmid pGFP-C1 were free of plasmid DNA. However, plasmid sequences could be detected in Southern blots of EcoRI-digested cellular DNA, indicating that pGFP-C1 had integrated during selection in the presence of G418 (Figure 1B, lane 6).
Once-per-cell-cycle replication
Being convinced that pEPI-1 is episomal in the cells under study, we wished to determine whether the plasmid replicates concomitantly with cellular DNA during the S phase. For this purpose, CHO cells were transferred to growth medium with 5-bromodeoxyuridine (BrdU) to increase the buoyant density of replicated DNA for CsCl equilibrium gradient centrifugation in a Meselson–Stahl density shift experiment. To ensure that the incorporation of BrdU was successful, we measured the distribution of total cellular DNA in the CsCl gradients, and found that essentially all DNA had shifted from a light (unsubstituted) position to a heavy–light (hemisubstituted) position within 17 h, the approximate doubling time of CHO cells under the growth conditions (HL in Figure 2A). After another 17-h growth period in the presence of BrdU, the HL DNA had divided into an HL peak and an HH peak of fully substituted DNA (Figure 2A) exactly as expected for semiconservative DNA replication.
Figure 2.
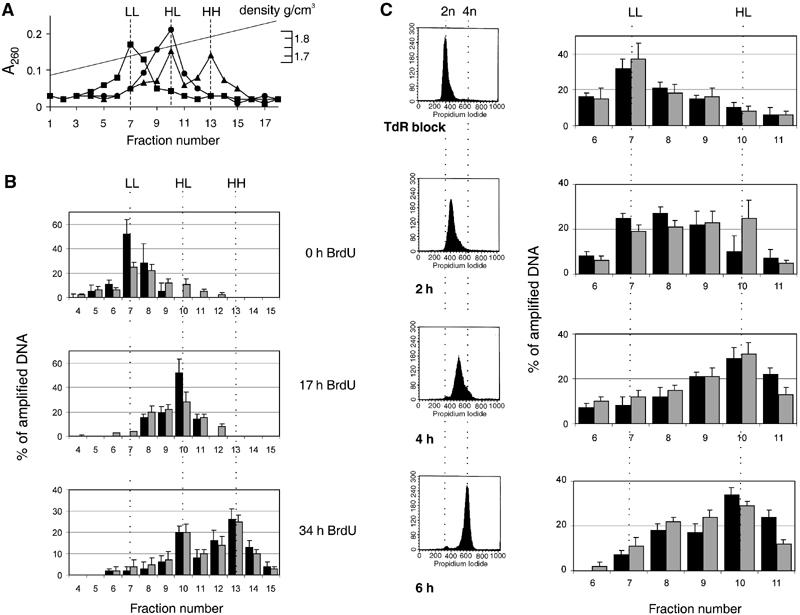
pEPI-1 replicates in a once-per-cell-cycle manner synchronously with cellular DNA and early in the S phase. Asynchronously growing CHO cells were labeled with 30 μg/ml BrdU for 0, 17 or 34 h. After shearing and digestion, total DNA was separated by CsCl gradient centrifugation. (A) The distribution of total DNA was determined from A260 measurements. Squares, 0 h; circles, 17 h; triangles, 34 h. Three peaks of A260-absorbing material were seen at densities of 1.7, 1.75 and 1.8 g/cm3 corresponding to light–light (LL), heavy–light (HL) and heavy–heavy (HH) DNA strands. (B) Samples were dialyzed against TE, precipitated and further analyzed by quantitative PCR using primer pairs SV40 for the plasmid pEPI-1 and DHFRex1 for genomic CHO DNA. The distribution of pEPI-1 (gray bars) or cellular DNA (black bars) is presented as a percentage of total amplified DNA. (C) CHO cells were synchronized by the excess-thymidine procedure at the G1-to-S-phase transition and then released into thymidine-free medium containing BrdU. Left, FACS analysis of isolated nuclei. Right, CsCl density gradient centrifugation followed by quantitative PCR to determine the distribution of cellular DNA (black bars) and plasmid DNA (gray bars).
The density shifts of pEPI-1 DNA were investigated by quantitative PCR assays using the Light Cycler instrument. The internal PCR control was a region from the well-studied cellular DHFR gene locus. The data in Figure 2B, expressed as relative units (percentage of total amplified DNA), clearly show that pEPI-1 (gray bars) replicated in synchrony with the cellular sequence (black bars), in a once-per-cell-cycle manner and without over-replication.
To determine the time of replication within the S phase, cells were first arrested in isoleucine-free medium for 48 h, and then released into medium with high thymidine concentration to achieve a block at the G1/S-phase border. Cells were released with normal medium which, however, contained BrdU as a density label. According to FACS analyses, the cells traversed the S phase with reasonable synchrony until the total DNA content was duplicated at approximately 6 h after release from the TdR block (left panels in Figure 2C). DNA was extracted before and at 2, 4 and 6 h after the release and analyzed by CsCl gradient centrifugation. Quantitative PCR was used to measure the amount of pEPI-1 DNA and of cellular DHFR sequences in individual fractions of the equilibrium gradients. At 2 h within the S phase, pEPI-1 DNA (gray bars in Figure 2C, right panels) and DHFR DNA (black bars) had partially shifted to the peak of semisubstituted HL DNA, and this shift was virtually complete at 4 h after release. The plasmid sequence shifted to the position of replicated (semisubstituted) DNA in parallel with the DHFR sequence, and since the DHFR locus had been classified as early replicating (Dijkwel and Hamlin, 1992), pEPI-1 can also be considered to be an early replicon. Early replicating regions are believed to be sections of the genome with relatively open chromatin structures as found in and around active genes (Gilbert, 2002; Lin et al, 2003).
We conclude that episomal pEPI-1 plasmids are replicated once and only once during the S phase of the cell cycle. Their replication should therefore be regulated much like the replication of the cellular genome including an assembly of the pre-RC before and its disassembly later in the S phase.
Orc2p and Mcm3p on episomal pEPI-1
To determine whether episomal pEPI-1 DNA in CHO cells carries components of the pre-RC, we used a ChIP procedure that had previously been successful in the identification of ORC-binding sites in human HeLa cells (Keller et al, 2002; Ladenburger et al, 2002). This procedure involves a short treatment of proliferating cells with formaldehyde, the preparation of crosslinked nucleoprotein and shearing to fragments of 0.2–1 kb in size. This is followed by immunoprecipitation of the resulting nucleoprotein fragments and an analysis by Western blotting or, after deproteinization, by quantitative PCR for specific DNA sequences.
In the experiment of Figure 3, the nucleoprotein fragments were immunoprecipitated with control antibodies or with antibodies directed against human Orc2p and Mcm3p. Antibodies against human Mcm3p react well with the corresponding CHO proteins (Alexandrow et al, 2002), as do the antibodies against human Orc2p. Indeed, the Orc2-specific as well as the Mcm3-specific antibodies precipitated essentially all the Orc2p and most of Mcm3p in the input sample (Figure 3A).
Figure 3.
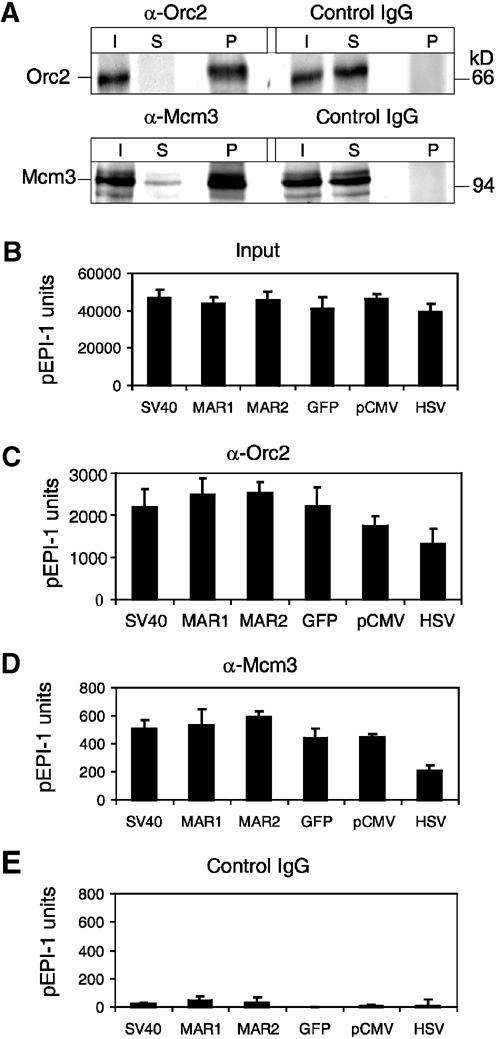
Orc2p and Mcm3p on pEPI-1. (A) Immunoprecipitation with antibodies against human Orc2p (α-Orc2) (upper) and human Mcm3p (α-Mcm3) (lower). IgG, nonspecific control antibodies. Inputs (I, crosslinked CHO chromatin before immunoprecipitation), supernatants (S, remaining chromatin after immunoprecipitations) and immunoprecipitates (P) were analyzed by Western blotting with Orc2p-specific (ORC2) and Mcm3p-specific (Mcm3) antibodies. (B) Input DNA (10 000 calculated genomic copies) was extracted from crosslinked CHO chromatin and analyzed by quantitative PCR using the primer sets indicated (Figure 1A, Table I) In parallel, quantitative PCR was performed with DNA extracted from immunoprecipitated chromatin using antibodies against Orc2p (C), Mcm3p (D) and IgG control antibodies (E). The data output obtained with each primer pair was calculated relative to the input control. The DNA measured in the control immunoprecipitation with IgG antibodies was subtracted. These numbers are expressed in ‘units' relative to known amounts of serially diluted pEPI-1 control DNA.
Next, we determined the presence of pEPI-1 sequences in the input (sheared crosslinked chromatin before immunoprecipitation) using the six primer pairs indicated in Figure 1A. The parallel PCR assays gave similar results, 4–5 × 104 copies of pEPI-1 in the nucleoprotein prepared under standard conditions (Figure 3B). An estimate based on the number of cells used for the preparation of the input sample gave a value of 6–9 episomal pEPI-1 copies per cell. This estimate takes into account that some of the amplifiable sequences are destroyed during sonication. We have determined this fraction in control experiments by mixing known amounts of pEPI-1 with crosslinked nucleoprotein from pEPI-1-free cells. Routinely, 75% of added plasmid sequences survived and could be amplified.
We then analyzed for the presence of pEPI-1 sequences in the Orc2 immunoprecipitates. Originally, we had expected to find an enrichment of specific pEPI-1 fragments such as those around the CMV promoter, but to our initial surprise, we detected that all segments of the replicon were present in the immunoprecipitate in approximately equal amounts (2000–2500 copies), with the exception of the MAR sequences (which were slightly more abundant) and the HSV region (which was less abundant than the other regions) (Figure 3C). Likewise, Mcm3p became crosslinked to multiple sites in the replicon just like Orc2p (Figure 3D). We note though that the absolute numbers of DNA fragments in the sample immunoprecipitated with Mcm3-specific antibodies were always lower than those in the Orc2 immunoprecipitates. This has been noted in previous experiments investigating cellular chromatin amplification (Schaarschmidt et al, 2002). One reason for this could be that the asynchronously proliferating cell population included a substantial fraction of late S-phase and post-S-phase cells that have no chromatin-bound Mcms. Another reason may be that Mcm-bearing DNA is partially unwound or otherwise structurally altered and is therefore less amenable to PCR.
The experiment in Figure 3 was controlled in different ways. First, control antibodies failed to enrich for pEPI-1 sequences in immunoprecpitates (Figure 3E). Second, HinfI digestion separates the regions of pEPI-1 selected for PCR amplification and cuts the MAR1 target side. Consequently, we were unable to amplify MAR1 of HinfI-restricted chromatin, whereas all the other pEPI-1 regions could be amplified just as in an untreated sample (not shown). This experiment underlines the reliability of the PCR procedure. Third, Orc2-specific antibodies were used to precipitate crosslinked chromatin from the pGFP-C1-transfected control cells. In this case, we were unable to detect plasmid DNA sequences in the immunoprecipitates, showing that Orc2p was not bound to the integrated plasmid sequences (not shown).
The interesting conclusion from Figure 3 is that Orc2p, Mcm3p and, by implication, other components of pre-RC are able to associate with many sequence sites in the extrachromosomal replicon. It should be noted that the interactions determined with these assays are not measured for a single replicon, but are a population average. It is therefore likely that each individual replicon carries just one or a few pre-RCs.
Abundance of nascent DNA strands
An obvious question is where the start points of replication are located on pEPI-1. We addressed this point using an assay to determine the abundance of DNA strands of 0.8–1.4 kb lengths extracted from denatured genomic DNA (Giacca et al, 1997). Strands of this size class exclusively occur in the vicinity of replication start sites because leading strands at more advanced replication forks are much longer, and lagging strands possess Okazaki fragments of 0.1–0.2 kb lengths. Accordingly, we sedimented denatured DNA and, for comparisons, sheared genomic DNA through neutral sucrose gradients (not shown), collected DNA fragments of 0.8–1.4 kb length, and treated these fractions with polynucleotide kinase and λ-exonuclease to eliminate artefactually broken DNA but not short nascent strands (Gerbi and Bielinsky, 1997).
To check the quality of our nascent strands preparation, we analyzed the best characterized origin region in the CHO genome, the DHFR locus with the ori-β (Kobayashi et al, 1998) (Figure 4A). We detected by quantitative PCR a 13-fold enrichment of ori-β sequences and a three-fold enrichment of closely adjacent sequences (ori-β 2.5 kb) in the fraction of nascent DNA compared to the sheared genomic DNA. In contrast, origin–distal sequences were not enriched in the 0.8–1.4 kb fraction (Figure 4A). Therefore, the preparation of denatured CHO DNA was suitable to search for nascent pEPI-1 DNA. The results showed that the abundance of nascent plasmid DNA was 1.4–1.9-fold for all pEPI-1 regions in relation to the HSV region which was set as 1 (Figure 4B). The abundance of nascent plasmid DNA was statistically significant and reproducible although rather modest in comparison to the ori-β sequences. An explanation is that replication initiation was not restricted to a single plasmid DNA site, but occurred at the many sites with the bound ORC.
Figure 4.
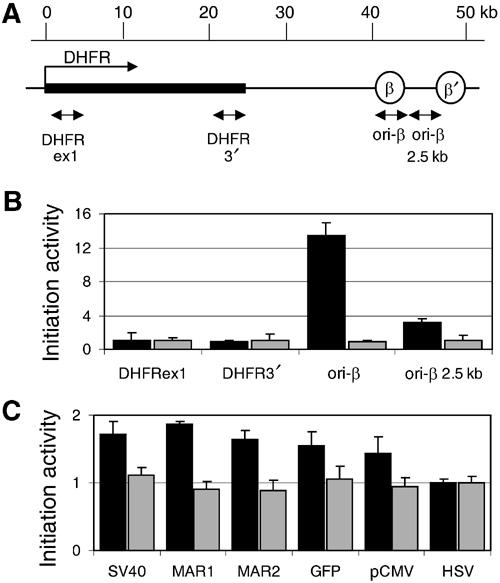
Measuring the relative abundance of nascent DNA strands on pEPI-1. (A) Genomic organization of the DHFR locus analyzed: ori-β and ori-β′ are the preferred sites of replication initiation (Kobayashi et al, 1998). Arrows indicate the positions of the primers used for quantitative PCR analysis. (B) Nascent strand abundance for four sequences from the DHFR region. Quantitative PCR analysis was performed either with nascent DNA (black bars) or sheared total genomic DNA (gray bars) from asynchronous CHO cells. The average of three independent assays is shown. (C) Nascent strand abundance assay with pEPI-1 sequences. Quantitative PCR analysis was again performed either with nascent DNA (black bars) or sheared total genomic DNA (gray bars) from asynchronous CHO cells carrying several copies of episomal pEPI-1. The data are averages from three independent experiments. To account for the different copy numbers, the values of the amplified DHFRex1 and HSV target sequences were set as 1.
Variations during the S phase
If pre-RCs on the extrachromosomal plasmids were functional, as the nascent strand abundance assays seem to suggest, we would expect that Orc2p remains stationary while other proteins such as Orc1p and the Mcm proteins dissociate from chromatin during the S phase. We have tested this prediction with CHO cells, which were arrested by the TdR procedure at the G1-to-S-phase transition and were then synchronously released into the S phase (Figure 5A, left panels).
Figure 5.
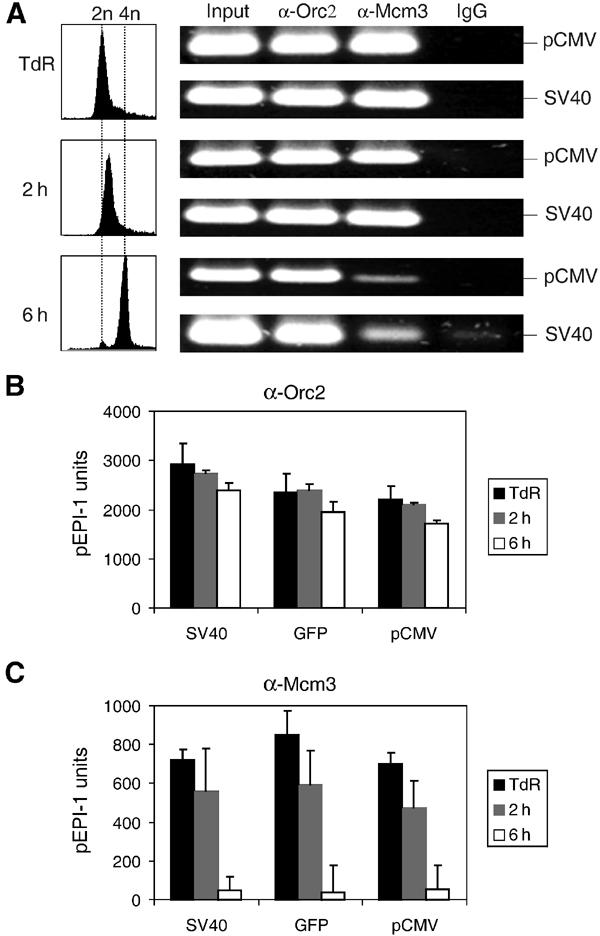
Mcm3p is released from pEPI-1 during the S phase. (A) ChIP assays with 400 μg nucleoprotein from CHO cells blocked at the G1-to-S-phase border by a thymidine block (TdR) and from cells released into the S phase for 2 or 6 h. Left, FACS analysis of the isolated nuclei. Right, conventional PCR was performed with 1/50th of the recovered DNA after immunoprecipitation with Orc2p (α-Orc2), MCM3p (α-Mcm3) and nonspecific control antibodies (IgG) using primer pairs SV40 and pCMV. Input, 5 ng crosslinked nucleoprotein before immunoprecipitation. (B,C) Quantitative real-time PCR was performed using three different primer pairs (SV40, GFP and pCMV) with DNA templates extracted from chromatin precipitated with antibodies against Orc2p or Mcm3p.
ChIP assays were performed with Orc2p- and Mcm3p-specific antibodies. The presence of plasmid sequences in the immunoprecipitates was first assayed by conventional PCR using two primer pairs corresponding to opposite regions in pEPI-1 (pCMV and SV40; see Figure 1A). As shown in Figure 5A, Orc2 antibodies precipitated plasmid sequences before and at two time points within the S phase, whereas Mcm3 antibodies precipitated equal amounts of plasmid sequences before and at 2 h, but less at 6 h after the beginning of the S phase. These results were supported by real-time PCR showing again that pEPI-1 sequences in the Orc2 precipitates decreased only slightly during the S phase (Figure 5B), while pEPI-1 sequences in the Mcm3 precipitates were reduced in the 2-h samples and almost absent at the end of the S phase (Figure 5C). Thus, Mcm proteins dissociated from the plasmid replicon in parallel to its semiconservative replication during the S phase (see Figure 2C).
Another pre-RC protein known to dissociate during the S phase is Orc1p (Kreitz et al, 2001). This, however, cannot be assayed in the CHO cell system because the available antibodies are directed against the human Orc1p and do not react with hamster Orc1p (not shown). Therefore, to investigate the behavior of Orc1p, we used a HeLa cell clone harboring extrachromosomal pEPI-1 (Figure 1B). These HeLa cells were synchronized by a 2 × TdR procedure (Ritzi et al, 1998) and processed for ChIP after treatment with formaldehyde before and at 3 h after entry into the S phase.
In an exploratory experiment, we performed conventional PCR and found amplifiable sequences in the Orc1, Orc2 and Mcm3 precipitates before the S phase, and a strong reduction of amplifiable DNA in the Orc1 and Mcm3 precipitates at 3 h after the beginning of the S phase, but no significant changes in the Orc2 precipitates (Figure 6A). The immunoprecipitated chromatin was then investigated by quantitative PCR using the six-primer pairs (Figure 1A). We found that the immunoprecipitated pre-S-phase chromatin contained sequences corresponding to all plasmid regions regardless of which of the three antibodies was used (black bars in Figure 6B–D). Thus, components of pre-RCs in HeLa cells assemble on multiple sites on the plasmid replicon, just as they do in CHO cells. The data also indicate that Orc1p as well as Mcm3p dissociated while Orc2p remained on chromatin during the S phase (gray bars in Figure 6B–D).
Figure 6.
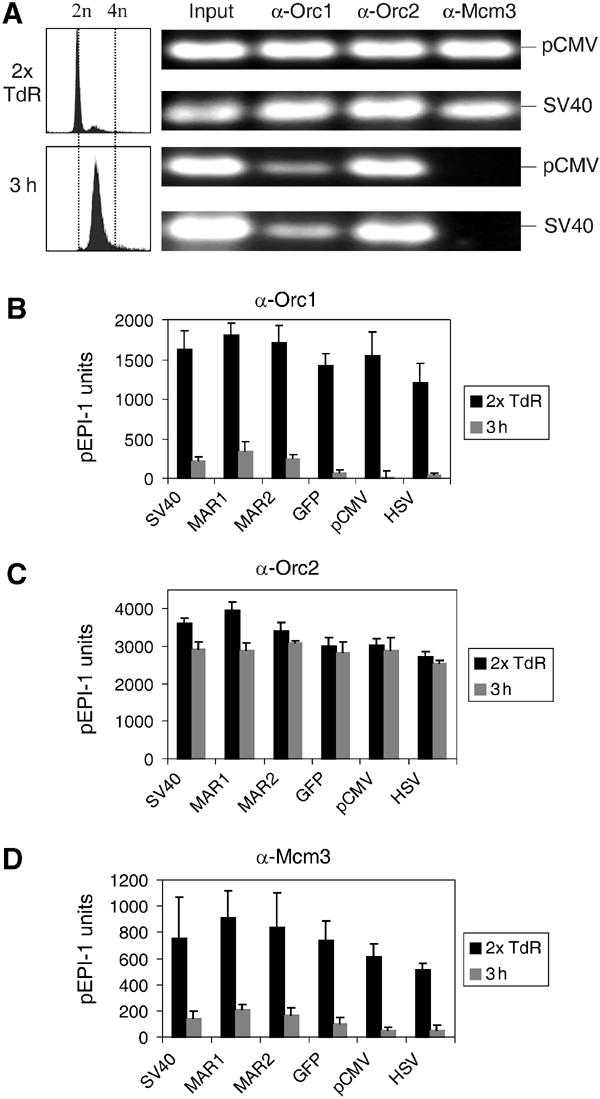
Orc1p is released from pEPI-1 during the S phase in HeLa cells. A HeLa cell clone containing episomal pEPI-1 was investigated (see Figure 1B). (A) Left, FACS analysis showing the passage of cells through the S phase. Right, conventional PCR using DNA from crosslinked chromatin. PCR was performed with 5 ng nucleoprotein before immunoprecipitation (Input) or 1/100th of the recovered DNA after immunoprecipitation with Orc1p (α-Orc1), Orc2p (α-Orc2) and MCM3p (α-Mcm3) antibodies using primer pairs SV40 and pCMV with 20/23 cycles. (B–D) Quantitative real-time PCR were performed with six different primer pairs on DNA templates extracted from chromatin precipitated with antibodies against Orc1p, Orc2p or Mcm3p.
The conclusion is that pre-RCs on the extrachromosomal replicon partially disassemble during the S phase like they do on cellular replicons, and this may be one of the control mechanisms that underlies the once-per-cell-cycle replication of pEPI-1 as documented in Figure 2.
No preference for a cellular ORC-binding site
As shown above, pre-RC proteins, Orc1p and Orc2p as well as Mcm3p, are bound to many sites on the extrachromosomal plasmid. We were concerned though that their promiscuous binding may occur by default, that is, because the plasmid lacks a strong ORC-binding site as present in the cellular genome. To address this point, we inserted into the multiple cloning site of pEPI-1 an established ORC-binding region, the upstream promoter region of the human MCM4 gene (UPR). The control was a sequence in exon IX (EX9), about 6 kb in distance, which has never been shown to carry ORC in vivo (Ladenburger et al, 2002). We transfected these constructs, pEPI-UPR and pEPI-EX9 (Figure 7A), into HeLa cells, and showed that both plasmids were transmitted as low-copy-number extrachromosomal elements over many cell generations without selection exactly as the parent plasmid pEPI-1 (not shown). Proliferating HeLa cells, carrying pEPI-UPR or pEPI-EX9, were processed for ChIP as described above. DNA in the immunoprecipitates from Orc2 and Mcm3 antibodies was analyzed by real-time PCR with the six primer pairs described and an additional primer pair designed to amplify the two inserts (Figure 7A). The results of these experiments indicate that Orc2p and Mcm3p dispersed over the entire DNA circle without a preference for the inserted chromosomal DNA sequences. In fact, the MCM4 upstream promoter region (with its mapped ORC-binding site) and the exon-IX region (which does not serve as an ORC site in vivo) were as abundant in the chromatin immunoprecipitates as the other plasmid sequences (Figure 7B; the difference in the calculated numbers between the pEPI-UPR and the pEPI-EX9 experiment was due to the fact that the pEPI-UPR input contained four–six plasmid copies per cell and the pEPI-EX9 input contained only two–three copies per cell). We also performed nascent strand abundance assays and could show that sequences corresponding to all regions of the two plasmids were enriched by factors of 1.2–1.8 over the HSV region (Figure 7C).
Figure 7.
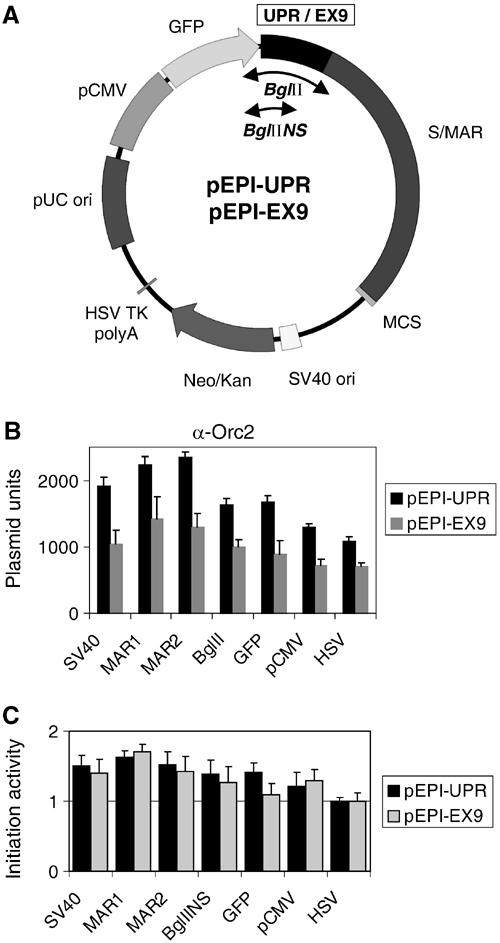
Orc2p binds sequence-independently to plasmid DNA in vivo. (A) Maps of pEPI-UPR and pEPI-EX9. The UPR and the EX9 sequence of the human MCM4 gene were cloned into the BglII site of the vector pEPI-1. The divergent arrowheads indicate the location of the regions analyzed by PCR amplification with the additional primer pairs BglII and BglIINS. ChIP assays with antibodies against Orc2p (B) and nascent strand abundance assays (C) were performed with asynchronously growing HeLa clones containing either episomal pEPI-UPR (black bars) or pEPI-EX9 (gray bars).
Discussion
We have shown here that a plasmid, pEPI-1, replicates in hamster CHO and in human HeLa cells as an extrachromosomal replicon in a once-per-cell-cycle manner and segregates faithfully over many cell generations in the absence of selection. We have taken advantage of this system and investigated the in vivo binding sites for two Orc proteins, Orc1p and Orc2p, and for another member of the pre-RC, Mcm3p. We could show that these proteins, and by implication other components of the pre-RC, bind to multiple sites on the extrachromosomal replicon. We present evidence that the pre-RCs on pEPI-1 are functional because they partially disassemble in the S phase exactly as known for cellular pre-RCs. In addition, replication can be initiated at various regions of pEPI-1. The important conclusion is that the chromatin-binding sites for ORC and therefore for other components of the pre-RC are not much, if at all, determined by DNA sequence elements.
An extrachromosomal replicon in mammalian cells
Viral systems provide useful models for the study of extrachromosomal replicons in mammalian systems. The appropriate model is not so much the run-away replication of viral DNA in lytically infected cells that depends on a highly specific interaction between an initiator protein, T antigen in the case of SV40, and a circumscribed origin sequence. Rather, it is the once-per-cell-cycle replication of papilloma virus and herpes virus genomes in latently infected cells. In the case of EBV, viral DNA replication appears to initiate in a region of the viral genome at or around the viral origin of replication that includes the binding sites for a viral protein, the Epstein–Barr Virus Nuclear Antigen 1 (EBNA-1) (Collins and Medveczky, 2002). EBNA-1 appears to interact with ORC, which in turn recruits other pre-RC proteins to the viral replication start site (Chaudhuri et al, 2001; Schepers et al, 2001). Together, these proteins are responsible for the coordination of viral DNA replication in the mammalian cell cycle (Dhar et al, 2001). However, EBNA-1 has a second function, namely the (direct or indirect) tethering of the viral episomes to chromosomes. This interaction provides the basis for the successful segregation of replicated episomes at mitosis (Kapoor and Frappier, 2003). Thus, the two essential functions for the extrachromosomal maintenance of viral episomes, replication and segregation, appear to be determined by virally encoded factors such as EBNA-1.
Does the research on EBV episomes help to understand how pEPI-1 is maintained as an extrachromosomal element? Experiments performed over many years in several laboratories suggest that transfected plasmid DNA of essentially any sequence can replicate in mammalian cells. However, Price et al. (2003) have recently suggested that a loosely defined 36-bp sequence seems to be required for at least a few rounds of semiconservative plasmid DNA replication in transfected mammalian cells. Allowing three mismatches, this 36-bp sequence occurs six times in the pEPI-1 sequence. We believe, however, that the presence of an active strong promoter, pCMV, is a more important requirement for the extrachromosomal maintenance of pEPI-1 because its deletion abolishes extrachromosomal replication (Stehle et al, 2003). Our present model is that transcription from this strong eukaryotic promoter affects the structure of the chromatin such that Orc proteins and other initiator proteins gain access to their binding sites on pEPI-1. Previous studies had shown that ORC and replication start sites are located in the upstream promoter regions of several constitutively transcribed genes (Keller et al, 2002). We had therefore expected to find a preferred ORC-binding site and a replication start site in the vicinity of the CMV promoter in pEPI-1. We found, however, that ORC assembles at many sites on the plasmid circle. Whether this implies that active transcription alters the chromatin structure around the entire plasmid circle, or whether other mechanisms are involved, is presently under investigation.
Replication is one reaction required for the maintenance of the extrachromosomal state another reaction is the segregation of replicated plasmids at mitosis. It appears to be most likely that the essential element here is S/MAR. These elements are not necessarily preferred sites for replication initiation in mammalian DNA (Keller et al, 2002; Mesner et al, 2003), but they are believed to be important for the organization of chromatin loops and of chromosomes in mitosis. Indeed, extrachromosomal pEPI-1 is found in association with mitotic chromosomes (Baiker et al, 2000), and this could well be mediated by S/MAR because the scaffold attachment factor A (SAF-A), a major component of cellular chromatin and chromosomes (Kipp et al, 2000), associates with pEPI-1 due to its high affinity for S/MAR (Jenke et al, 2002). Thus, in contrast to viral episomes which encode their own tethering factors, pEPI-1 contains a DNA element, S/MAR, that interacts with a chromosomal factor, SAF-A, provided by the host cell.
In conclusion, much like ARS-bearing plasmids in yeast cells, pEPI-1 appears to be a useful model as an extrachromosomal replicon in mammalian cells. The practical side is that pEPI-1 could serve as a valuable vector for the expression of foreign proteins in mammalian cells such as required in gene therapy (Lipps et al, 2003).
Sites of ORC binding
We have used a ChIP approach to determine the distribution of ORC proteins on the extrachromosomal DNA circle. This procedure involves in vivo crosslinking of chromatin proteins to DNA, immunoprecipitations of the resulting nucleoprotein fragments and the identification of enriched specific DNA sequences in the immunoprecipitates. The procedure has been successfully used to identify unique ORC-binding sites within several >100-kb-long sections of human chromatin (Keller et al, 2002) and in episomal herpesviral genomes (Schepers et al, 2001). By the same experimental approach, we now show that Orc1p and Orc2p, and by implication other components of ORC, bind to multiple sites on the plasmid DNA circle. Similarly, Mcm3p, a component of the six-membered Mcm protein complex, also occurs at multiple sites on pre-S-phase chromatin as expected, since bound ORC is known to recruit Mcm proteins to complete the formation of pre-RCs.
We recall that we have measured large plasmid populations, not individual plasmids, and it is quite possible that each plasmid DNA circle carries just one or only a few pre-RCs. However, this point has yet to be investigated. In either case, evidence indicates that pre-RCs on pEPI-1 are functional because they undergo the same transitions during the S phase as pre-RCs on cellular chromatin involving the dissociation of Orc1p and Mcm proteins. In addition, replicative nascent DNA strands correspond in sequence to many regions of pEPI-1, just as the ORC-binding sites.
A second point that needs further attention is whether ORC prefers particular sequences within the regions identified by PCR. If it does, the similarities between these sequences must be low, because we detect no obvious sequence similarities in pairwise comparisons, except perhaps for elements like the Price et al. sequence (see above), which is, however, rather loosely defined. Indeed, a sequence, UPR of human MCM4 gene that is known to be a preferred ORC-binding site in the context of the cellular genome, attracts ORC to pEPI-UPR with the same efficiency as a control target sequence in exon IX (EX9), which has never been shown to harbor ORC in vivo (Figure 7). In addition, nascent strand analysis indicates that replication can be initiated at various regions of both constructs. Related observations have been reported for yeast where replication initiation was found to occur at a fixed chromosomal site, but at many more sites when the same sequence was present in an autonomously replicating episome (Bielinsky and Gerbi, 1999).
The results described here have major implications for our thinking on the organization of genome replication in mammalian cells. They suggest that specific DNA sequences are not the major determinant for the assembly of ORC, although we cannot completely dismiss the possibility that AT-rich elements, DNA unwinding elements (DUEs) and other sequences that have previously been discussed as components of mammalian origins may facilitate the binding of ORC (DePamphilis, 1999). In fact, pEPI-1 has a high overall AT content (56%) with a maximum of 69% in S/MAR. This could have modulating effects on ORC binding. However, the major mechanism for the selection of ORC sites must be epigenetic in nature. For episomal EBV it is EBNA-1 which that ORC to the viral origin region, and in cellular chromatin it could be that transcription factors or other DNA-binding proteins determine the site of ORC binding. More generally, we expect that all mechanisms that hold chromatin in an extended confirmation or otherwise facilitate the access of proteins to DNA may in principle be involved in the localization of ORC (Gilbert, 2001; Mechali, 2001). This explains why origins can either be strictly localized to defined DNA sequences (Abdurashidova et al, 2000) or widely distributed over long chromatin regions (Dijkwel et al, 2002). In the former case, specifically bound transcription factors may direct ORC to precise sites, whereas, in the case of wide open chromatin regions, ORC can choose among a much larger set of sequences.
In summary, what we have presented above may be considered to be an in vivo version of an ORC-binding experiment—a foreign DNA is introduced into cultured mammalian cells and recruits ORC which binds to multiple sites as a requirement for ordered replication. Vashee et al. (2003) have recently published corresponding in vitro ORC-binding experiments and arrive at the same conclusion, namely that the binding of ORC and the initiation of replication do not depend on the underlying DNA sequence.
Materials and methods
Cell culture and synchronization
Available as Supplementary data at The EMBO Journal Online.
Plasmids and transfection
The plasmids used were pEPI-1 (Piechaczek et al, 1999), pGFP-C1 (Clontech), pEPI-UPR and pEPI-EX9. The latter two plasmids were constructed by cloning PCR-amplified UPR- and EX9-sequences (see Table I) into the BglII restriction site of pEPI-1 using the TOPO TA cloning kit (Invitrogen).
Table 1.
Sequences and amplification conditions for primers
| Primer | Sequence (5′ to 3′) | Size (bp) of PCR product | Annealing temp. (°C) | |
|---|---|---|---|---|
| pEPI | ||||
| SV40-F | CCCCTAACTCCGCCCAGT | |||
| SV40-R | TGTGCCCAGTCATAGCCGAA | 254 | 60 | |
| MAR1-F | AAGGTAATGCTGGCCATA | |||
| MAR1-R | AGGTTCAGGGGGAGGT | 239 | 68 | |
| MAR2-F | TGGCATTTTACAATGGG | |||
| MAR2-R | TCATGATGGCATGCTTCT | 194 | 60 | |
| GFP-F | CATTGAAGATGGAAGCGTTC | |||
| GFP-R | GTCCGGACTTGTATAGGTC | 211 | 60 | |
| pCMV-F | CCAAGTCTCCACCCCATTG | |||
| pCMV-R | GAATTGGGACAACTCCAGTG | 231 | 60 | |
| HSV-F | AATAAAACGCACGGTCGTTGG | |||
| HSV-R | CAAAAAGGATCTTCACCTAGA | 277 | 62 | |
| BglII-F | TGGGATTACACATGGCAT | 629 (pEPI-UPR) | ||
| BglII-R | TCTCTCCGTTTCTGCATT | 533 (pEPI-EX9) | 64 | |
| BglIINS-F | CCTTTTACCAGACAACCA | |||
| BglIINS-UPR-R | AGCCAACGCTAGAGGAACA | 279 | 60 | |
| BglIINS-EX9-R | ATGCAGATTTTGTCCCCCT | 264 | 60 | |
| DHFR locus | ||||
| DHFRex1-F | TGGCCTCCGATTCACAAGT | |||
| DHFRex1-R | TTCCCTAACCCAATCCAGCCAGTA | 217 | 60 | |
| DHFR3′-F | CCAAAGACATAGAGCCCCTGA | |||
| DHFR3′-R | TTTACCCATTTTCTGTCTGAGTGA | 210 | 60 | |
| Ori-β-F | GTCCCTGCCTCAAAACACAA | |||
| Ori-β-R | CCTTCATGCTGACATTTGTC | 278 | 62 | |
| Ori-β2.5-F | GGACACTAAGTCTAGGTACTACA | |||
| Ori-β2.5-R | GCTGGGATAAGTTGAAATCC | 259 | 58 | |
| MCM4 locus | ||||
| EX9-F | GTAGATCTATGTCTTCCGGAGACTCCTGAAGC | |||
| EX9-R | CTAGATCTGGCCTCCTATTCTCAGAATCATGC | 403 | 60 | |
| UPR-F | GTAGATCTAAACCAGAAGTAGGCCTCGCTCGG | |||
| UPR-R | CTAGATCTGTCTGACCTGCGGAGGTAGTTTGG | 499 | 68 |
Details of transfection, maintenance of transfected cells, fluctuation assay, rescue experiments and DNA extraction methods are described as Supplementary data.
Separation of BrdU-substituted DNA
Total DNA was isolated from transfected CHO cells cultivated for indicated times in BrdU-containing medium (30 μg/ml). After sonication and digestion with EcoRI, DNA was loaded onto density gradients (5 μg DNA/ml), the refractive index was adjusted to 1.4038, and the gradients were spun in a Beckman SW40 rotor first at 40 000 rpm for 24 h and then at 30 000 rpm another 24 h. Fractions of 250 μl were collected from the top of the gradient and their refractive index was determined. Samples were dialyzed against TE, precipitated and further analyzed by quantitative PCR.
PCR-based nascent strand abundance assay
Total genomic DNA was prepared from 108 exponentially growing cells. After denaturation at 85°C for 10 min followed by rapid cooling on ice, the single-stranded DNA was loaded on a 5–30% neutral sucrose gradient and centrifuged in a Beckman SW28 rotor for 20 h at 26 000 rpm.
Fractions of 1 ml were collected and the size of DNA in each fraction was determined by alkaline agarose gel electrophoresis. Fractions containing fragments from 0.8 to 1.4 kb were pooled, concentrated by ethanol precipitation, treated with polynucleotide kinase and λ-exonuclease as described previously (Gerbi and Bielinsky, 1997) and used for quantitative PCR analysis. For an input control, total genomic DNA was sheared to fragments 1–2 kb in size by sonication before loading on the gradient. The quality of the DNA strands was tested by PCR with specific primers for the DHFR origin (see below). To compare different experiments, the abundance of the target sequences for each primer pair was expressed in relation to the abundance of the target sequences of the HSV primer pair in the case of pEPI-1 and the DHFRex1 primer pair for the control in each experiment. These two were defined as 1 and the resulting ratio was termed ‘initiation activity'.
In vivo crosslinking, ChIP, conventional and quantitative PCR
These procedures were performed as described previously (Ladenburger et al, 2002) with modifications (see Supplementary data).
Supplementary Material
Supplementary Data
Acknowledgments
We thank V Ullrich for allowing us to use the Light Cycler instrument and M Kulartz for a critical reading. This work was supported by the Deutsche Forschungsgemeinschaft (DFG).
References
- Abdurashidova G, Deganuto M, Klima R, Riva S, Biamonti G, Giacca M, Falaschi A (2000) Start sites of bidirectional DNA synthesis at the human lamin B2 origin. Science 287: 2023–2026 [DOI] [PubMed] [Google Scholar]
- Alexandrow MG, Ritzi M, Pemov A, Hamlin JL (2002) A potential role for mini-chromosome maintenance (MCM) proteins in initiation at the dihydrofolate reductase replication origin. J Biol Chem 277: 2702–2708 [DOI] [PubMed] [Google Scholar]
- Altman AL, Fanning E (2001) The Chinese hamster dihydrofolate reductase replication origin beta is active at multiple ectopic chromosomal locations and requires specific DNA sequence elements for activity. Mol Cell Biol 21: 1098–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aparicio OM, Weinstein DM, Bell SP (1997) Components and dynamics of DNA replication complexes in S cerevisiae: redistribution of MCM proteins and Cdc45p during S phase. Cell 91: 59–69 [DOI] [PubMed] [Google Scholar]
- Baiker A, Maercker C, Piechaczek C, Schmidt SB, Bode J, Benham C, Lipps HJ (2000) Mitotic stability of an episomal vector containing a human scaffold/matrix-attached region is provided by association with nuclear matrix. Nat Cell Biol 2: 182–184 [DOI] [PubMed] [Google Scholar]
- Bell SP, Dutta A (2002) DNA replication in eukaryotic cells. Annu Rev Biochem 71: 333–374 [DOI] [PubMed] [Google Scholar]
- Bell SP, Stillman B (1992) ATP-dependent recognition of eukaryotic origins of DNA replication by a multiprotein complex [see comments]. Nature 357: 128–134 [DOI] [PubMed] [Google Scholar]
- Bielinsky AK, Gerbi SA (1999) Chromosomal ARS1 has a single leading strand start site. Mol Cell 3: 477–486 [DOI] [PubMed] [Google Scholar]
- Bielinsky AK, Gerbi SA (2001) Where it all starts: eukaryotic origins of DNA replication. J Cell Sci 114: 643–651 [DOI] [PubMed] [Google Scholar]
- Blow JJ, Hodgson B (2002) Replication licensing—defining the proliferative state? Trends Cell Biol 12: 72–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode J, Kohwi Y, Dickinson L, Joh T, Klehr D, Mielke C, Kohwi-Shigematsu T (1992) Biological significance of unwinding capability of nuclear matrix-associating DNAs. Science 255: 195–197 [DOI] [PubMed] [Google Scholar]
- Caddle MS, Calos MP (1992) Analysis of the autonomous replication behavior in human cells of the dehydrofolate reductase putative chromosomal origin of replication. Nucleic Acids Res 20: 5971–5978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri B, Xu H, Todorov I, Dutta A, Yates JL (2001) Human DNA replication initiation factors, ORC and MCM, associate with oriP of Epstein–Barr virus. Proc Natl Acad Sci USA 98: 10085–10089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang RY, Kelly TJ (1999) The fission yeast homologue of Orc4p binds to replication origin DNA via multiple AT-hooks. Proc Natl Acad Sci USA 96: 2656–2661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins CM, Medveczky PG (2002) Genetic requirements for the episomal maintenance of oncogenic herpesvirus genomes. Adv Cancer Res 84: 155–174 [DOI] [PubMed] [Google Scholar]
- Coverley D, Laskey RA (1994) Regulation of eukaryotic DNA replication. Annu Rev Biochem 63: 745–776 [DOI] [PubMed] [Google Scholar]
- DePamphilis ML (1999) Replication origins in metazoan chromosomes: fact or fiction? Bioessays 21: 5–16 [DOI] [PubMed] [Google Scholar]
- DePamphilis ML (2003) The ‘ORC cycle': a novel pathway for regulating eukaryotic DNA replication. Gene 310: 1–15 [DOI] [PubMed] [Google Scholar]
- Dhar SK, Yoshida K, Machida Y, Khaira P, Chaudhuri B, Wohlschlegel JA, Leffak M, Yates J, Dutta A (2001) Replication from oriP of Epstein–Barr virus requires human ORC and is inhibited by geminin. Cell 106: 287–296 [DOI] [PubMed] [Google Scholar]
- Dijkwel PA, Hamlin JL (1992) Initiation of DNA replication in the dihydrofolate reductase locus is confined to the early S period in CHO cells synchronized with the plant amino acid mimosine. Mol Cell Biol 12: 3715–3722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkwel PA, Wang S, Hamlin JL (2002) Initiation sites are distributed at frequent intervals in the Chinese hamster dihydrofolate reductase origin of replication but are used with very different efficiencies. Mol Cell Biol 22: 3053–3065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerbi SA, Bielinsky AK (1997) Replication initiation point mapping. Methods 13: 271–280 [DOI] [PubMed] [Google Scholar]
- Giacca M, Pelizon C, Falaschi A (1997) Mapping replication origins by quantifying relative abundance of nascent DNA strands using competitive polymerase chain reaction. Methods 13: 301–312 [DOI] [PubMed] [Google Scholar]
- Gilbert DM (2001) Making sense of eukaryotic DNA replication origins. Science 294: 96–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert DM (2002) Replication timing and transcriptional control: beyond cause and effect. Curr Opin Cell Biol 14: 377–383 [DOI] [PubMed] [Google Scholar]
- Jenke BH, Fetzer CP, Stehle IM, Jonsson F, Fackelmayer FO, Conradt H, Bode J, Lipps HJ (2002) An episomally replicating vector binds to the nuclear matrix protein SAF-A in vivo. EMBO Rep 3: 349–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor P, Frappier L (2003) EBNA1 partitions Epstein–Barr virus plasmids in yeast cells by attaching to human EBNA1-binding protein 2 on mitotic chromosomes. J Virol 77: 6946–6956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller C, Ladenburger EM, Kremer M, Knippers R (2002) The origin recognition complex marks a replication origin in the human TOP1 gene promoter. J Biol Chem 277: 31430–31440 [DOI] [PubMed] [Google Scholar]
- Kipp M, Gohring F, Ostendorp T, van Drunen CM, van Driel R, Przybylski M, Fackelmayer FO (2000) SAF-Box, a conserved protein domain that specifically recognizes scaffold attachment region DNA. Mol Cell Biol 20: 7480–7489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Rein T, DePamphilis ML (1998) Identification of primary initiation sites for DNA replication in the hamster dihydrofolate reductase gene initiation zone. Mol Cell Biol 18: 3266–3277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitz S, Ritzi M, Baack M, Knippers R (2001) The human origin recognition complex protein 1 dissociates from chromatin during S phase in HeLa cells. J Biol Chem 276: 6337–6342 [DOI] [PubMed] [Google Scholar]
- Krysan PJ, Smith JG, Calos MP (1993) Autonomous replication in human cells of multimers of specific human and bacterial DNA sequences. Mol Cell Biol 13: 2688–2696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labib K, Tercero JA, Diffley JF (2000) Uninterrupted MCM2-7 function required for DNA replication fork progression. Science 288: 1643–1647 [DOI] [PubMed] [Google Scholar]
- Ladenburger EM, Keller C, Knippers R (2002) Identification of a binding region for human origin recognition complex proteins 1 and 2 that coincides with an origin of DNA replication. Mol Cell Biol 22: 1036–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DG, Bell SP (1997) Architecture of the yeast origin recognition complex bound to origins of DNA replication. Mol Cell Biol 17: 7159–7168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CM, Fu H, Martinovsky M, Bouhassira E, Aladjem MI (2003) Dynamic alterations of replication timing in mammalian cells. Curr Biol 13: 1019–1028 [DOI] [PubMed] [Google Scholar]
- Lipps HJ, Jenke AC, Nehlsen K, Scinteie MF, Stehle IM, Bode J (2003) Chromosome-based vectors for gene therapy. Gene 304: 23–33 [DOI] [PubMed] [Google Scholar]
- Liu G, Malott M, Leffak M (2003) Multiple functional elements comprise a mammalian chromosomal replicator. Mol Cell Biol 23: 1832–1842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWhinney C, Leffak M (1990) Autonomous replication of a DNA fragment containing the chromosomal replication origin of the human c-myc gene. Nucleic Acids Res 18: 1233–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechali M (2001) DNA replication origins: from sequence specificity to epigenetics. Nat Rev Genet 2: 640–645 [DOI] [PubMed] [Google Scholar]
- Mesner LD, Li X, Dijkwel PA, Hamlin JL (2003) The dihydrofolate reductase origin of replication does not contain any nonredundant genetic elements required for origin activity. Mol Cell Biol 23: 804–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piechaczek C, Fetzer C, Baiker A, Bode J, Lipps HJ (1999) A vector based on the SV40 origin of replication and chromosomal S/MARs replicates episomally in CHO cells. Nucleic Acids Res 27: 426–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price GB, Allarakhia M, Cossons N, Nielsen T, Diaz-Perez M, Friedlander P, Tao L, Zannis-Hadjopoulos M (2003) Identification of a cis-element that determines autonomous DNA replication in eukaryotic cells. J Biol Chem 278: 19649–19659 [DOI] [PubMed] [Google Scholar]
- Ritzi M, Baack M, Musahl C, Romanowski P, Laskey RA, Knippers R (1998) Human minichromosome maintenance proteins and human origin recognition complex 2 protein on chromatin. J Biol Chem 273: 24543–24549 [DOI] [PubMed] [Google Scholar]
- Schaarschmidt D, Ladenburger EM, Keller C, Knippers R (2002) Human Mcm proteins at a replication origin during the G1 to S phase transition. Nucleic Acids Res 30: 4176–4185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schepers A, Ritzi M, Bousset K, Kremmer E, Yates JL, Harwood J, Diffley JF, Hammerschmidt W (2001) Human origin recognition complex binds to the region of the latent origin of DNA replication of Epstein–Barr virus. Embo J 20: 4588–4602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons DT (2000) SV40 large T antigen functions in DNA replication and transformation. Adv Virus Res 55: 75–134 [DOI] [PubMed] [Google Scholar]
- Smith JG, Calos MP (1995) Autonomous replication in Drosophila melanogaster tissue culture cells. Chromosoma 103: 597–605 [DOI] [PubMed] [Google Scholar]
- Stehle IM, Scinteie MF, Baiker A, Jenke AC, Lipps HJ (2003) Expoiting a minimal system to study the epigenetic control of DNA replication: the interplay between transcription and replication. Chromosome Res 11: 413–421 [DOI] [PubMed] [Google Scholar]
- Tye BK (1999) MCM proteins in DNA replication. Annu Rev Biochem 68: 649–686 [DOI] [PubMed] [Google Scholar]
- Vashee S, Cvetic C, Lu W, Simancek P, Kelly TJ, Walter JC (2003) Sequence-independent DNA binding and replication initiation by the human origin recognition complex. Genes Dev 17: 1894–1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Z, Komamura Y, Ishimi Y (1999) Biochemical analysis of the intrinsic Mcm4–Mcm6–Mcm7 DNA helicase activity. Mol Cell Biol 19: 8003–8015 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data