

Abstract
In 1959, Dave Brubeck and Paul Desmond revolutionized modern jazz music by composing their unforgettable Take Five in 5/4, one of the most defiant time signatures in all music. Of similar revolutionary importance for biomedical and basic biochemical research is the identification of the minimal set of genes required to obtain a deadly time bomb ticking in all of us: Alzheimer's disease. It now appears that one needs to Take Five genes to produce a deadly peptide by a proteolytic mechanism, which paradoxically is otherwise of pivotal importance for development and cell fate decisions.
Keywords: Alzheimer's disease, amyloid β-peptide, processing, secretases
A 100 years anniversary of Alzheimer's disease research
It is almost precisely 100 years ago that Auguste D. reported to a German psychiatrist in Frankfurt with the words: ‘I lost myself'. The psychiatrist was non other than Alois Alzheimer, and this day should mark the beginning of Alzheimer's disease (AD) research. By that time Auguste D. was the one and only patient identified—now we have millions worldwide. As the major risk factor for AD is aging, one can only expect a further sharp increase in the number of patients in the near future. Therapeutic treatment against this devastating disease is desperately required. Fortunately, major progress has been made in the last years, which has led to the first human trials with drugs aimed to lower the burden of the major culprit, the neurotoxic amyloid β-peptide (Aβ) (Hock et al, 2003). Aβ is a highly hydrophobic peptide, which aggregates to form oligomers. If these oligomers aggregate further, they start forming fibers, which eventually precipitate and accumulate in the disease defining amyloid plaques (Figure 1). While the fibers are deposited in the brain parenchyma, additional toxic aggregation events appear to be induced within the cell. Tau, a protein that stabilizes microtubules, dissociates from the cytoskeleton and aggregates within the soma of neurons to form the intracellular tangles (Figure 1) (Hardy and Selkoe, 2002). This process may then initiate a variety of less understood toxic insults, ultimately leading to the tremendous neuronal loss observed in AD brains. This process, which appears to be initiated by the oligomerization of Aβ, is now known as the amyloid cascade (Hardy and Selkoe, 2002).
Figure 1.
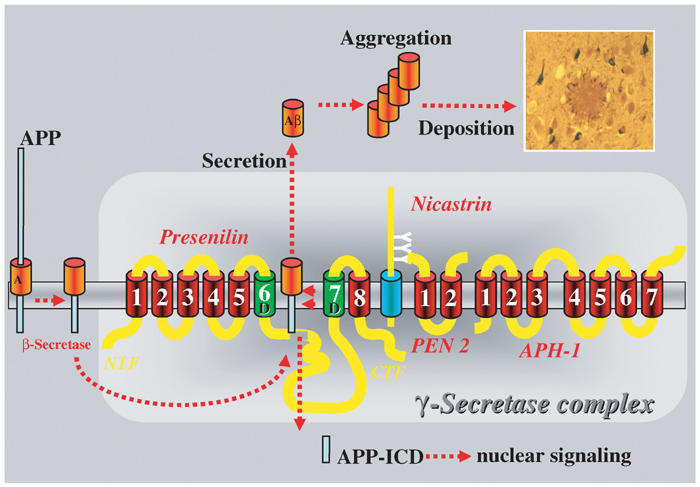
Generation of Aβ from APP via proteolytic processing by β- and γ-secretase (for details, see text). Aβ aggregates and finally precipitates in amyloid plaques. This event initiates the amyloid cascade resulting in additional intracellular aggregations of the tau protein, which then form tangles (the black structures surrounding the amyloid plaque).
A deadly time bomb
Surprisingly and against all predictions, Aβ was found in the early 1990s to be a physiologically normal metabolite generated in healthy persons (Hardy and Selkoe, 2002). Subtle changes in Aβ generation, or its metabolism, can occur as a result of aging, which leads to the accumulation and deposition of Aβ. Accordingly, in the rare familial AD (FAD) cases, mutations cause increased production of the two-amino-acid longer Aβ42 peptide, at the expense of the rather benign Aβ40. Aβ42 aggregates much faster, and consequently causes a much earlier onset of the disease (Hardy and Selkoe, 2002).
Generation of Aβ occurs by processing of the β-amyloid precursor protein (APP) via proteases called secretases. Three secretases are known, α-, β-, and γ-secretase. While β- and γ-secretase mediate the amyloidogenic cleavage events (Figure 1), α-secretase on the contrary prevents Aβ generation by cleaving APP in the middle of the Aβ domain (Figures 1 and 2).
Figure 2.

Cleavage sites of α-, β-, and γ-secretase. The Aβ domain is shown in red, the TMD is underlined, and the APP-ICD is shown in blue.
β-Secretase is an aspartyl protease of the pepsin family
β-Secretase (also called BACE-1 for β-site APP-cleaving enzyme) was identified as a type 1 transmembrane protein containing aspartyl protease activity (Vassar, 2001). BACE belongs to the pepsin family of aspartyl proteases, but defines a novel subgroup of membrane-associated hydrolases. BACE-1 mediates the primary amyloidogenic cleavage of APP and generates a membrane-bound APP C-terminal fragment (APP CTFβ), which is the immediate precursor for the intramembraneous γ-secretase cleavage (Figure 1). BACE-1 is clearly the only protease with a well-defined β-secretase activity. This was unambiguously shown by the homozygous knockout of the BACE-1 gene, which does not allow any Aβ generation (Cai et al, 2001; Luo et al, 2001). A close homolog of BACE-1, BACE-2, was identified as well (Vassar, 2001); however, BACE-2 exhibits an α-secretase-like activity (Figure 2), which cleaves APP in the middle of the Aβ domain at amino acids 19 and 20 (Farzan et al, 2000). Thus, BACE-2 does not contribute to the amyloidogenic processing of APP, which is consistent with the complete lack of Aβ generation in a BACE-1 knockout.
BACE-1 is generated as a preproenzyme. Upon removal of the signal peptide, the prodomain is cleaved by a furin-like protease (Bennett et al, 2000). BACE-1 is then targeted through the secretory pathway to the plasma membrane and clusters within lipid rafts (Ehehalt et al, 2003). APP processing by BACE-1 also occurs preferentially within lipid rafts (Ehehalt et al, 2003). Consistent with the finding of BACE-1 in lipid rafts, BACE-1 is sorted apically in polarized cells (Capell et al, 2002), the side where rafts are thought to accumulate. At the plasma membrane, BACE-1 (like APP) can be internalized to endosomes (Huse et al, 2000; Walter et al, 2001). Thus BACE-1 and APP follow similar trafficking routes and meet within endosomes, which may be the preferential site of BACE-1 activity due to its acidic pH optimum (Vassar, 2001).
Little is known about the physiological substrates of BACE-1. As described above, BACE-1 has only limited access to basolaterally sorted APP in polarized cells, thus suggesting that APP is not the main substrate. This is supported by the finding that APP does not bear the optimal cleavage site for BACE-1 (Liu et al, 2002). In fact, one of the FAD-associated mutations in APP (the so-called Swedish mutation) strongly enhances BACE-1 cleavage of APP simply by creating an ‘optimized' cleavage site (Citron et al, 1992). Besides APP, additional candidate BACE-1 substrates were identified recently; these include P-selectin glycoprotein ligand-1 (Lichtenthaler et al, 2003) and the sialyl-transferase ST6Gal I (Kitazume et al, 2001). However, the biological function of BACE-1 remains unclear, and may be difficult to address since no phenotype is observed upon removal of BACE-1 from the mouse genome.
Most interestingly, evidence exists that BACE-1 expression is significantly enhanced in brains derived from patients with sporadic AD (Yang et al, 2003). The reasons for the enhanced BACE-1 expression are currently unclear. However, in this regard, it is tempting to speculate that AD-associated stressors such as oxidative stress, radicals, unfolded proteins, head trauma, and others may induce BACE-1 transcription and/or expression/activity during aging.
The γ-secretase quartet
After BACE-1 has done its job to initiate Aβ generation, γ-secretase comes into play. γ-Secretase is required for the rather unusual intramembrane cleavage event (Figures 1 and 2), a process initially thought to be biochemically impossible. However, recent evidence now suggests the convergent evolution of several polytopic proteases, which are all able to cleave proteins inside the hydrophobic environment of a lipid bilayer. These include the signal peptide peptidases and their homologs, the type 4 prepilin peptidases (although these may cleave just outside of the membrane), rhomboid, S2P, and the γ-secretase itself (Haass and Steiner, 2002; Weihofen and Martoglio, 2003). Whereas S2P, rhomboid, and type 4 prepilins work by themselves, a set of four proteins is required to build up a γ-secretase complex (De Strooper, 2003).
Presenilins
The two homologous presenilins, PS1 and PS2, are apparently of exceptional importance for the γ-secretase cleavage event. This became obvious many years ago when the genetic linkage of PSs to numerous familial AD cases was discovered (Sherrington et al, 1995). More than 100 autosomal dominant PS point mutations have now been identified, which all cause aggressive early-onset AD by the same mechanism. These mutations seem to influence the γ-secretase cleavage event by simply shifting it two amino acids to the C-terminus (Hardy and Selkoe, 2002). The subsequent increase of the Aβ42 to Aβ40 ratio results in the enhanced oligomerization of the highly amyloidogenic Aβ42. This already suggests that PSs must be directly involved in the γ-secretase cleavage event or even be the γ-secretase itself. The latter is supported by the pivotal observation of De Strooper et al (1998), who found that a PS1 knockout severely reduced Aβ generation. When the second PS gene (PS2) was eliminated as well, no Aβ generation was observed at all (Herreman et al, 2000; Zhang et al, 2000). Direct evidence for PS presenting the catalytic core of γ-secretase came from the finding that all PSs contain two functionally important and highly conserved aspartate residues within transmembrane domains (TMDs) 6 and 7 (Wolfe et al, 1999). This was of great interest, since biochemical evidence suggested that γ-secretase belongs to the aspartyl protease family. Do these aspartates build up the catalytic center of the γ-secretase complex? When either of the two aspartates was mutagenized, Aβ generation was indeed abolished (Wolfe et al, 1999). Moreover, PSs are cleaved by endoproteolysis within the large cytoplasmic loop (Figure 1) (Haass and Steiner, 2002). This cleavage results in two fragments (the N-terminal (NTF) and the C-terminal (CTF) fragment), which remain bound to each other (Capell et al, 1998). Upon mutagenesis of either one of the two critical aspartates, this cleavage event does not occur anymore and PS accumulated as a holoprotein (Wolfe et al, 1999). This phenomenon would be consistent with the holoprotein being a zymogene, which is activated by autoproteolyis (Haass and Steiner, 2002).
Another piece of evidence that PSs provide the catalytic core for the aspartyl protease activity of the γ-secretase complex is that γ-secretase inhibitors can be directly crosslinked to PS (Esler et al, 2000; Li et al, 2000). However, in contrast to BACE-1 and all other conventional aspartyl proteases, PSs lack the D(T/S)G(T/S) aspartyl protease active site; instead, they contain a GxGD motif around the critical asparate in TMD7 (Steiner et al, 2000; Haass and Steiner, 2002). Most interestingly, this motif is fully conserved between presenilins, the type-4 prepilin peptidases (Steiner et al, 2000), and the signal peptide peptidases (Weihofen and Martoglio, 2003). This suggests convergent evolution of a novel active site for a common problem: intramembrane proteolysis.
Obviously, PSs did not evolve to produce the deleterious Aβ peptide. In fact, Aβ generation seems to be a rather unfortunate by-product of an otherwise highly important biological function of PSs. This became evident when PSs were eliminated in mice. A PS1 knockout resulted in a phenotype that closely resembled that of a Notch knockout (Selkoe and Kopan, 2003). A number of additional substrates of presenilins have also been identified besides APP and Notch (De Strooper, 2003). These include the APP homologs APLP-1 and -2, ErbB-4, E-cadherin, N-cadherin, LRP, Nectin-1-α, the Notch ligands Delta and Jagged, and CD44. In the case of Notch, the function of intramembraneous cleavage is well understood. Intramembraneous cleavage of Notch is required to liberate the Notch intracellular domain (ICD) for nuclear signaling and subsequent regulation of target gene transcription (summarized by Selkoe and Kopan, 2003). However, intramembraneous cleavage can also negatively regulate nuclear signaling. Robakis and co-workers (Marambaud et al, 2003) demonstrated that the ICD generated from N-cadherin functions as a potent repressor of CBP/CREB-mediated transcription. Moreover, generation of the ICD was reduced by FAD mutations. Interestingly, there is now evidence that the APP intracellular domain interacts with the nuclear adaptor protein Fe65 and with the histone acetyltransferase Tip60 to form a transcriptionally active complex (Cao and Südhof, 2001). Similar to the effects of FAD mutants on ICD generation of N-cadherin, some FAD mutations affect the generation of the APP and the Notch intracellular domain (Moehlmann et al, 2002). This suggests a loss of function (in signaling) in addition to the pathological gain of function related to Aβ42 generation.
Two intramembraneous cleavage events are required to liberate ICDs (Figure 2) and their corresponding Aβ or Aβ-like peptide (Sastre et al, 2001; Okochi et al, 2002). One cleavage event occurs within the middle of the TMDs (at amino acid 40 or 42). Another one occurs close to the cytoplasmic border of the TMD and is required to liberate ICD. It is possible that hydrophobic TMDs prevent efficient liberation of ICDs generated by the cleavage in the middle of the membrane, and thus require a second cleavage event to remove this ‘sticky' domain. Both cleavage events appear to occur simultaneously and are fully PS dependent (Sastre et al, 2001).
Nct, APH-1, and PEN-2
PSs are not sufficient for γ-secretase activity, but rather require additional cofactors to form a multiprotein high-molecular-weight complex (De Strooper, 2003). Biochemical purification of this complex led to the identification of nicastrin (Nct) (Yu et al, 2000). The remaining components of the complex were isolated by genetic screens for enhancers of a PS-dependent Notch-deficient phenotype in Caenorhabditis elegans. This led to the identification of two additional components: APH-1 (anterior pharynx-defective phenotype) and PEN-2 (PS-enhancer) (Francis et al, 2002; Goutte et al, 2002; De Strooper, 2003). When all four components were expressed together in Saccharomyces cerevisiae, an organism that lacks any endogenous γ-secretase activity, fully active γ-secretase was reconstituted (Edbauer et al, 2003).
PS expression and γ-secretase activity are regulated in a coordinated manner. Because overexpression of PS does not lead to the expression of higher levels of PS fragments (Haass and Steiner, 2002), nor to enhanced γ-secretase activity, a limiting factor regulating γ-secretase activity was postulated. Maybe there is no defined limiting factor; instead, the least abundant γ-secretase complex component, be it PS, PEN-2, APH-1, or Nct, may become limiting. If there is a disequilibrium of any of these components, excess amounts are destabilized or, in the case of Nct, fail to maturate (De Strooper, 2003).
Although the composition and the biological function of the γ-secretase complex seem to be largely solved, little is known about the function of the individual components. This is due to the difficulties in analyzing these components individually in the absence of the other partners.
Since the complete (Shirotani et al, 2003) luminal domain of Nct bound to the membrane (Morais et al, 2003) is required for γ-secretase activity within the complex, it is tempting to speculate that this globular, tightly folded ectodomain is responsible for measuring the length of the membrane-retained stub-like substrates. Such a function must be postulated for one of the γ-secretase complex components, because substrates are only recognized and proteolyzed if their ectodomain was previously truncated (Struhl and Adachi, 2000).
In the absence of PEN-2, PS is stabilized as an uncleaved holoprotein probably with the help of APH-1 and Nct (Luo et al, 2003; Takasugi et al, 2003). Thus APH-1 may be a stabilizer of the PS holoprotein, whereas PEN-2 is apparently required to initiate endoproteolysis of PS (Hu and Fortini, 2003; Luo et al, 2003; Takasugi et al, 2003). These findings may give some insights into the assembly of the γ-secretase complex. Since Nct is fairly stable in the ER, it is likely that it may be the scaffold for a second binding partner. APH-1 would then be recruited to Nct, as evidenced by the identification of a putative Nct/Aph-1 precomplex (LaVoie et al, 2003; Morais et al, 2003). Binding of PS to the APH-1/Nct complex would then result in the formation of a trimeric complex (Takasugi et al, 2003). Finally, PEN-2 joins the complex, thus allowing PS endoproteolysis and the eventual activation of the γ-secretase activity (Hu and Fortini, 2003; Takasugi et al, 2003). However, this is all still quite speculative, since it has so far proved impossible to monitor the assembly of the complex in vivo.
Developing drugs against AD
Having identified all the components required for γ-secretase function, it should be rather straightforward to interfere with Aβ production with the help of selective inhibitors. Several high-affinity inhibitors are indeed available. However, one concern remains. γ-Secretase is required for Notch signaling (and most likely for several other signaling pathways as well). Accordingly, deleterious side effects have been observed in vivo (Geling et al, 2002). However, as for any other novel drug, one needs to define a therapeutic window, which allows some γ-secretase activity for signaling but reduces Aβ generation significantly enough to slow aggregation and deposition.
BACE-1 is another obvious target for Aβ-lowering drugs. In contrast to γ-secretase, BACE-1 can be fully ablated in mice without causing any deleterious phenotype. Thus, interfering with BACE-1 is unlikely to result in unwanted side effects. Moreover, the three-dimensional structure of BACE-1 has been solved (Hong et al, 2000), and peptidomimetic and nonpeptidomimetic inhibitors have been generated (Hong et al, 2000; Vassar, 2001). These need to be developed further to improve their affinity with the rather large active site cleft of BACE-1 before they can be used in human trials. However, highly sensitive inhibitors will certainly be available in the near future.
AD futurology
Besides the obvious development of secretase inhibitors, other options remain as well. In fact, in my opinion, anti-Aβ vaccination will have the highest chance to be the first ‘true anti-AD' drug on the market, since first and very hopeful results from a small cohort of patients have already been published (Hock et al, 2003), and further trials are on the way. What these data may also tell us is that the amyloid cascade hypothesis may in fact indeed be true. Moreover, independent treatments may be important to circumvent the above-described side effects. Finally, additional options for anti-AD drugs remain, such as the nonsteroidal anti-inflammatory drugs (Weggen et al, 2001), and compounds aimed to prevent toxicity of Aβ and tau.
However, AD research is far from coming to an end. The secrets of the secretases are not all discovered yet. We still do not know the function of BACE or any of the γ-secretase complex components other than PS. The biggest challenge in that regard will be the discovery of the three-dimensional structure of the entire γ-secretase complex, maybe even co-crystallized with its substrate and/or inhibitors. Mechanisms of cellular toxicity are far from being understood, and finally and most embarrassingly we do not even understand the biological function of APP, the longest known player in AD pathology.
Conclusion
Thus after almost precisely 100 years, AD research came to a point where direct interference with deadly mechanisms in patients is possible and human trials with secretase inhibitors are on the way. As an important by-product, research in this field led to the elucidation of novel signaling pathways involved in cell fate decisions and shed light on completely unexpected mechanisms of intramembrane proteolysis.
Acknowledgments
I thank Drs Steiner and Lichtenthaler for critical comments and the Deutsche Forschungsgemeinschaft (DFG), the European Union (DIADEM), the American Health Assistance Foundation (AHAF), the National Genome Research Network (NGFN), and the Hans und Ilse Breuer Foundation for funding.
References
- Bennett BD, Denis P, Haniu M, Teplow DB, Kahn S, Louis JC, Citron M, Vassar R (2000) A furin-like convertase mediates propeptide cleavage of BACE, the Alzheimer's beta-secretase. J Biol Chem 275: 37712–37717 [DOI] [PubMed] [Google Scholar]
- Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, Wong PC (2001) BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat Neurosci 4: 233–234 [DOI] [PubMed] [Google Scholar]
- Cao X, Südhof TC (2001) A transcriptionally active complex of APP with Fe65 and histone acetyltransferase Tip60. Science 293: 115–120 [DOI] [PubMed] [Google Scholar]
- Capell A, Grünberg J, Pesold B, Diehlmann A, Citron M, Nixon R, Beyreuther K, Selkoe DJ, Haass C (1998) The proteolytic fragments of the Alzheimer's disease-associated presenilin-1 form heterodimers and occur as a 100–150-kDa molecular mass complex. J Biol Chem 273: 3205–3211 [DOI] [PubMed] [Google Scholar]
- Capell A, Meyn L, Fluhrer R, Teplow DB, Walter J, Haass C (2002) Apical sorting of beta-secretase limits amyloid beta-peptide production. J Biol Chem 277: 5637–5643 [DOI] [PubMed] [Google Scholar]
- Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, Vigo-Pelfrey C, Lieberburg I, Selkoe DJ (1992) Mutation of the β-amyloid precursor protein in familial Alzheimer's disease increases β-protein production. Nature 360: 672–674 [DOI] [PubMed] [Google Scholar]
- De Strooper B (2003) Aph-1, Pen-2, and nicastrin with presenilin generate an active γ-secretase complex. Neuron 38: 9–12 [DOI] [PubMed] [Google Scholar]
- De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F (1998) Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 391: 387–390 [DOI] [PubMed] [Google Scholar]
- Edbauer D, Winkler E, Regula JT, Pesold B, Steiner H, Haass C (2003) Reconstitution of γ-secretase activity. Nat Cell Biol 5: 486–488 [DOI] [PubMed] [Google Scholar]
- Ehehalt R, Keller P, Haass C, Thiele C, Simons K (2003) Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J Cell Biol 160: 113–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esler WP, Kimberly WT, Ostaszewski BL, Diehl TS, Moore CL, Tsai J-Y, Rahmati T, Xia W, Selkoe DJ, Wolfe MS (2000) Transition-state analogue inhibitors of γ-secretase bind directly to presenilin-1. Nat Cell Biol 2: 428–433 [DOI] [PubMed] [Google Scholar]
- Farzan M, Schnitzler CE, Vasilieva N, Leung D, Choe H (2000) BACE2, a beta-secretase homolog, cleaves at the beta site and within the amyloid-beta region of the amyloid-beta precursor protein. Proc Natl Acad Sci USA 97: 9712–9717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis R, McGrath G, Zhang J, Ruddy DA, Sym M, Apfeld J, Nicoll M, Maxwell M, Hai B, Ellis MC, Parks AL, Xu W, Li J, Gurney M, Myers RL, Himes CS, Hiebsch RD, Ruble C, Nye JS, Curtis D (2002) aph-1 and pen-2 are required for Notch pathway signaling, γ-secretase cleavage of βAPP, and presenilin protein accumulation. Dev Cell 3: 85–97 [DOI] [PubMed] [Google Scholar]
- Geling A, Steiner H, Willem M, Bally-Cuif L, Haass C (2002) A γ-secretase inhibitor blocks Notch signaling in vivo and causes a severe neurogenic phenotype in zebrafish. EMBO Rep 3: 688–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goutte C, Tsunozaki M, Hale VA, Priess JR (2002) APH-1 is a multipass membrane protein essential for the Notch signaling pathway in Caenorhabditis elegans embryos. Proc Natl Acad Sci USA 99: 775–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, Steiner H (2002) Alzheimer disease γ-secretase: a complex story of GxGD-type presenilin proteases. Trends Cell Biol 12: 556–562 [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297: 353–356 [DOI] [PubMed] [Google Scholar]
- Herreman A, Serneels L, Annaert W, Collen D, Schoonjans L, De Strooper B (2000) Total inactivation of γ-secretase activity in presenilin-deficient embryonic stem cells. Nat Cell Biol 2: 461–462 [DOI] [PubMed] [Google Scholar]
- Hock C, Konietzko U, Streffer JR, Tracy J, Signorell A, Muller-Tillmanns B, Lemke U, Henke K, Moritz E, Garcia E, Wollmer MA, Umbricht D, de Quervain DJ, Hofmann M, Maddalena A, Papassotiropoulos A, Nitsch RM (2003) Antibodies against beta-amyloid slow cognitive decline in Alzheimer's disease. Neuron 38: 547–554 [DOI] [PubMed] [Google Scholar]
- Hong L, Koelsch G, Lin X, Wu S, Terzyan S, Ghosh AK, Zhang XC, Tang J (2000) Structure of the protease domain of memapsin 2 (beta-secretase) complexed with inhibitor. Science 290: 150–153 [DOI] [PubMed] [Google Scholar]
- Hu Y, Fortini ME (2003) Different cofactor activities in γ-secretase assembly: evidence for a nicastrin-Aph-1 subcomplex. J Cell Biol 161: 685–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse JT, Pijak DS, Leslie GJ, Lee VM, Doms RW (2000) Maturation and endosomal targeting of beta-site amyloid precursor protein-cleaving enzyme. The Alzheimer's disease beta-secretase. J Biol Chem 275: 33729–33737 [DOI] [PubMed] [Google Scholar]
- Kitazume S, Tachida Y, Oka R, Shirotani K, Saido TC, Hashimoto Y (2001) Alzheimer's beta-secretase, beta-site amyloid precursor protein-cleaving enzyme, is responsible for cleavage secretion of a Golgi-resident sialyltransferase. Proc Natl Acad Sci USA 98: 13554–13559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaVoie MJ, Fraering PC, Ostaszewski BL, Ye W, Kimberly WT, Wolfe MS, Selkoe DJ (2003) Assembly of the gamma-secretase complex involves early formation of an intermediate subcomplex of Aph-1 and nicastrin. J Biol Chem 278: 37213–37222 [DOI] [PubMed] [Google Scholar]
- Li YM, Xu M, Lai MT, Huang Q, Castro JL, DiMuzio-Mower J, Harrison T, Lellis C, Nadin A, Neduvelli JG, Register RB, Sardana MK, Shearman MS, Smith AL, Shi XP, Yin KC, Shafer JA, Gardell SJ (2000) Photoactivated γ-secretase inhibitors directed to the active site covalently label presenilin 1. Nature 405: 689–694 [DOI] [PubMed] [Google Scholar]
- Lichtenthaler SF, Dominguez DI, Westmeyer GG, Reiss K, Haass C, Saftig P, De Strooper B, Seed B (2003) The cell adhesion protein P-selectin glycoprotein ligand-1 is a substrate for the aspartyl protease BACE1. J Biol Chem 278: 48713–48719 [DOI] [PubMed] [Google Scholar]
- Liu K, Doms RW, Lee VM (2002) Glu11 site cleavage and N-terminally truncated A beta production upon BACE overexpression. Biochemistry 41: 3128–3136 [DOI] [PubMed] [Google Scholar]
- Luo WJ, Wang H, Li H, Kim BS, Shah S, Lee HJ, Thinakaran G, Kim TW, Yu G, Xu H (2003) PEN-2 and APH-1 coordinately regulate proteolytic processing of presenilin 1. J Biol Chem 278: 7850–7854 [DOI] [PubMed] [Google Scholar]
- Luo Y, Bolon B, Kahn S, Bennett BD, Babu-Khan S, Denis P, Fan W, Kha H, Zhang J, Gong Y, Martin L, Louis JC, Yan Q, Richards WG, Citron M, Vassar R (2001) Mice deficient in BACE1, the Alzheimer's beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat Neurosci 4: 231–232 [DOI] [PubMed] [Google Scholar]
- Marambaud P, Wen PH, Dutt A, Shioi J, Takashima A, Siman R, Robakis NK (2003) A CBP binding transcriptional repressor produced by the PS1/epsilon-cleavage of N-cadherin is inhibited by PS1 FAD mutations. Cell 114: 635–645 [DOI] [PubMed] [Google Scholar]
- Moehlmann T, Winkler E, Xia X, Edbauer D, Murrell J, Capell A, Kaether C, Zheng H, Ghetti B, Haass C, Steiner H (2002) Presenilin-1 mutations of leucine 166 equally affect the generation of the Notch and APP intracellular domains independent of their effect on Aβ 42 production. Proc Natl Acad Sci USA 99: 8025–8030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morais VA, Crystal AS, Pijak DS, Carlin D, Costa J, Lee VM, Doms RW (2003) The transmembrane domain region of nicastrin mediates direct interactions with APH-1 and the gamma-secretase complex. J Biol Chem 278: 43284–43291 [DOI] [PubMed] [Google Scholar]
- Okochi M, Steiner H, Fukumori A, Tanii H, Tomita T, Tanaka T, Iwatsubo T, Kudo T, Takeda M, Haass C (2002) Presenilins mediate a dual intramembraneous γ-secretase cleavage of Notch-1. EMBO J 21: 5408–5416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastre M, Steiner H, Fuchs K, Capell A, Multhaup G, Condron MM, Teplow DB, Haass C (2001) Presenilin-dependent γ-secretase processing of β-amyloid precursor protein at a site corresponding to the S3 cleavage of Notch. EMBO Rep 2: 835–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe D, Kopan R (2003) Notch and presenilin: regulated intramembrane proteolysis links development and degeneration. Annu Rev Neurosci 26: 565–597 [DOI] [PubMed] [Google Scholar]
- Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, Tsuda T, Mar L, Foncin JF, Bruni AC, Montesi MP, Sorbi S, Rainero I, Pinessi L, Nee L, Chumakov I, Pollen D, Brookes A, Sanseau P, Polinsky RJ, Wasco W, Dasilva HAR, Haines JL, Pericakvance MA, Tanzi RE, Roses AD, Fraser PE, Rommens JM, Stgeorgehyslop PH (1995) Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature 375: 754–760 [DOI] [PubMed] [Google Scholar]
- Shirotani K, Edbauer D, Capell A, Schmitz J, Steiner H, Haass C (2003) Secretase activity is associated with a conformational change of nicastrin. J Biol Chem 278: 16474–16477 [DOI] [PubMed] [Google Scholar]
- Steiner H, Kostka M, Romig H, Basset G, Pesold B, Hardy J, Capell A, Meyn L, Grim MG, Baumeister R, Fechteler K, Haass C (2000) Glycine 384 is required for presenilin-1 function and is conserved in polytopic bacterial aspartyl proteases. Nat Cell Biol 2: 848–851 [DOI] [PubMed] [Google Scholar]
- Struhl G, Adachi A (2000) Requirements for presenilin-dependent cleavage of Notch and other transmembrane proteins. Mol Cell 6: 625–636 [DOI] [PubMed] [Google Scholar]
- Takasugi N, Tomita T, Hayashi I, Tsuruoka M, Niimura M, Takahashi Y, Thinakaran G, Iwatsubo T (2003) The role of presenilin cofactors in the γ-secretase complex. Nature 422: 438–441 [DOI] [PubMed] [Google Scholar]
- Vassar R (2001) The β-secretase, BACE. A prime target for Alzheimeŕs disease. J Mol Neurosci 17: 157–170 [DOI] [PubMed] [Google Scholar]
- Walter J, Fluhrer R, Hartung B, Willem M, Kaether C, Capell A, Lammich S, Multhaup G, Haass C (2001) Phosphorylation regulates intracellular trafficking of beta-secretase. J Biol Chem 276: 14634–14641 [DOI] [PubMed] [Google Scholar]
- Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, Findlay KA, Smith TE, Murphy MP, Bulter T, Kang DE, Marquez-Sterling N, Golde TE, Koo EH (2001) A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity. Nature 414: 212–216 [DOI] [PubMed] [Google Scholar]
- Weihofen A, Martoglio B (2003) Intramembrane-cleaving proteases: controlled liberation of functional proteins and peptides from membranes. Trends Cell Biol 13: 71–78 [DOI] [PubMed] [Google Scholar]
- Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ (1999) Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and γ-secretase activity. Nature 398: 513–517 [DOI] [PubMed] [Google Scholar]
- Yang LB, Lindholm K, Yan R, Citron M, Xia W, Yang XL, Beach T, Sue L, Wong P, Price D, Li R, Shen Y (2003) Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat Med 9: 3–4 [DOI] [PubMed] [Google Scholar]
- Yu G, Nishimura M, Arawaka S, Levitan D, Zhang L, Tandon A, Song YQ, Rogaeva E, Chen F, Kawarai T, Supala A, Levesque L, Yu H, Yang DS, Holmes E, Milman P, Liang Y, Zhang DM, Xu DH, Sato C, Rogaev E, Smith M, Janus C, Zhang Y, Aebersold R, Farrer LS, Sorbi S, Bruni A, Fraser P, St George-Hyslop P (2000) Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and βAPP processing. Nature 407: 48–54 [DOI] [PubMed] [Google Scholar]
- Zhang Z, Nadeau P, Song W, Donoviel D, Yuan M, Bernstein A, Yankner BA (2000) Presenilins are required for γ-secretase cleavage of βAPP and transmembrane cleavage of Notch-1. Nat Cell Biol 2: 463–465 [DOI] [PubMed] [Google Scholar]