

Abstract
Equilibrium models of isolation by distance predict an increase in genetic differentiation with geographic distance. Here we find a linear relationship between genetic and geographic distance in a worldwide sample of human populations, with major deviations from the fitted line explicable by admixture or extreme isolation. A close relationship is shown to exist between the correlation of geographic distance and genetic differentiation (as measured by FST) and the geographic pattern of heterozygosity across populations. Considering a worldwide set of geographic locations as possible sources of the human expansion, we find that heterozygosities in the globally distributed populations of the data set are best explained by an expansion originating in Africa and that no geographic origin outside of Africa accounts as well for the observed patterns of genetic diversity. Although the relationship between FST and geographic distance has been interpreted in the past as the result of an equilibrium model of drift and dispersal, simulation shows that the geographic pattern of heterozygosities in this data set is consistent with a model of a serial founder effect starting at a single origin. Given this serial-founder scenario, the relationship between genetic and geographic distance allows us to derive bounds for the effects of drift and natural selection on human genetic variation.
Keywords: genetic distance, genetic drift, HGDP-CEPH, human origins, microsatellites
A regular decrease of genetic similarity with increasing geographic distance has been predicted by the theory of isolation by distance (1) and by the stepping-stone model (2), under the assumption that movement connected with mating is usually restricted to short distances (3, 4). Data on genetic polymorphisms have confirmed a strong association between genetic and geographic distance; early studies were generally limited to short geographic ranges and within-regional analyses (5, 6), but later studies have been extended to wider areas (7-9). Here, we regress a measure of genetic differentiation on geographic distance at the global level using 783 microsatellite loci from the Human Genome Diversity Project-Centre d'Etude du Polymorphisme Humain (HGDP-CEPH) worldwide sample of populations (10, 11). We then use simulations to examine a serial founder effect scenario as a possible explanation for the observed relationship between genetic and geographic distance.
Materials and Methods
Data. The data set that we analyzed consists of 1,027 individuals from the HGDP-CEPH Human Genome Diversity Cell Line Panel (10). Several individuals from the collection of 1,056 individuals studied by Rosenberg et al. (11) were excluded from the present analysis. These included the following: (i) no. 1026, who was studied by Rosenberg et al. (11) but who was not in the HGDP-CEPH panel; (ii) nos. 770 and 980, who were identified by Rosenberg et al. (11) as likely labeling errors; (iii) nos. 589, 652, 659, 826, 979, 981, 1022, 1025, 1087, 1092, 1154, and 1235, each of whom was identified by Mountain and Ramakrishnan (12) as a duplicate sample of another individual included in the panel; (iv) nos. 111 and 220, who were identified by Mountain and Ramakrishnan (12) as duplicates of each other but whose population labels differed; and (v) 21 individuals from the Surui population, an extreme outlier in a variety of previous analyses (11, 13, 14). Individuals not studied by Rosenberg et al. (11) but analyzed here included the following: (i) no. 1331, whose genotypes had been unavailable at the time of the Rosenberg et al. (11) study; (ii) nos. 993, 994, 1028, 1030, 1031, 1033, 1034, and 1035, who were previously excluded as members of populations with small sample sizes but who were grouped for the present analysis into Southwestern Bantu (individuals no. 1028, 1031, and 1035) and Southeastern Bantu (individuals no. 993, 994, 1030, 1033, and 1034) populations. Thus, the present data set includes two additional populations along with all populations studied by Rosenberg et al. (11) except Surui for a total of 53 populations.
Each of the 1,027 individuals was genotyped for 783 autosomal microsatellite loci, which included the 377 loci from Marshfield Screening Set no. 10 that were previously studied by Rosenberg et al. (11), as well as 406 additional loci from Marshfield Screening Sets no. 13 and 52. The complete data set used in this study is available from the authors upon request.
Geographic locations of the samples were reported by Cann et al. (10). For populations where ranges of coordinates were provided, the mean of the latitudes and the mean of the longitudes of the reported region were used to characterize the population's location. For the Northern Han of East Asia, the coordinate pair used was (39N, 114E); 39N is the northern extreme of locations where Han individuals were sampled, whereas 114E fell in the middle of the interval of longitudes at which Han individuals were sampled.
Genetic Distance. genetic data analysis (gda) (15) was used to compute pairwise genetic distances, as measured by FST (16), for all pairs of populations. We refer to FST as a “genetic distance,” although strictly speaking it does not satisfy the triangle inequality (17). A pairwise matrix of RST values (18) also was computed, as were matrices for several other genetic distances. All distances were found to be highly correlated (Table 1), and, consequently, only FST was used in further analysis.
Table 1.
Mantel correlations between various distances
References for measures of genetic differentiation are as follows: D1 (19), (δμ)2 (20), FST (16), GST (21), proportion of shared alleles (PSA) (22), and RST (18).

Geographic Distance. For each pair of populations, we calculated geographic distance in kilometers based on great circle distances using the haversine (23), according to which the distance D between two points specified by (latitude, longitude) coordinates (α1, δ1) and (α2, δ2), with a central angle of θ between the two points is
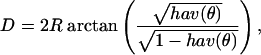 |
[1] |
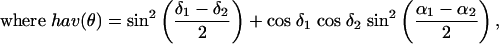 |
[2] |
and R is the radius of the Earth, which we assume to be 6,371 km.
In addition to great circle geographic distances, we also calculated pairwise geographic distances using five obligatory waypoints. Waypoints were used to make our between-continent distance estimates more reflective of human migration patterns, taking into account the belief that until recently humans did not generally cross large bodies of water while migrating. These waypoints were as follows: Anadyr, Russia (64N, 177E); Cairo, Egypt (30N, 31E); Istanbul, Turkey (41N, 28E); Phnom Penh, Cambodia (11N, 104E); and Prince Rupert, Canada (54N, 130W). The distance between two points is then the sum of the great circle distances between the points and the waypoint(s) in the path connecting them, plus the great circle distance(s) between waypoints if two or more waypoints are needed. Including the waypoints in between-continent distance calculations forced movement, for example, to Oceania via Southeast Asia, and to America via the Bering Strait and western coast of North America (see Fig. 6, which is published as supporting information on the PNAS web site).
Because there may have been an important expansion route along the south Asian coast (24), we also considered a waypoint at the southern part of the Red Sea. However, changing the waypoint from the north to the south of the Red Sea or using two waypoints at the Red Sea does not substantially change the quantitative results (results not shown).
Jackknifing over Populations. To determine which populations were most influential in the linear regression, we jackknifed over each of the 53 populations and fitted a new regression line with the remaining 52 populations and their pairwise comparisons. For each pair (i, j), we then calculated the deleted residual for eliminated population i with population j, di,j
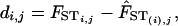 |
where FSTi,j is the observed genetic distance between populations i and j and  is the predicted FST between populations i and j using the regression line generated when population i is eliminated from the data set.
is the predicted FST between populations i and j using the regression line generated when population i is eliminated from the data set.
This process allows us to compute 52 deleted residuals for each population; the sorted averages of those residuals are reported in Table 2, which is published as supporting information on the PNAS web site.
Principal Coordinates. Principal coordinates were calculated on both the genetic (FST) and geographic distance matrices (calculated using the five waypoints) by using routines in the matlab language from the res5 library (25). The calculation of principal coordinates involves converting a distance matrix into Gower's centered matrix, which is decomposed into its eigenvalues and eigenvectors (26). Each eigenvector is then divided by the square root of its corresponding eigenvalue to yield principal coordinate scores for each population in the distance matrix (26). Each coordinate was converted to standardized scores (such that each had mean 0 and SD 1) independently within each type of data (genetic and geographic). Because the sign of a principal coordinate is arbitrary, we adjusted the first principal coordinate of the genetic distance matrix by multiplying by -1 so that projection on a common set of coordinates would better visually reflect the patterns of geographic association.
Origin of the Human Expansion. Regressions on geographic distance from a center were performed by using each of 4,210 centers drawn from the surface of the earth as follows. By using a lattice of 200 longitudes and 79 latitudes constructed so that each lattice point represented an equal area, 4,210 lattice points on land were identified (excluding Antarctica and islands farther south than the southern tip of South America). Rivers and all lakes other than Huron, Michigan, Superior, Victoria, and the Caspian and Aral Seas were treated as land.
The Relationship Between FST and Heterozygosities. Taking equation 5.12 from Weir (16), if ui denotes allele u in population i, l is the locus under consideration, and p̃lui is the frequency at locus l of allele u in population i, then 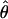 (the estimator for FST) is
(the estimator for FST) is
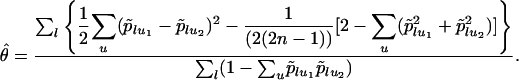 |
[3] |
Restricting our computations to one locus (removing l from Eq. 3), we obtain for a sample of size n
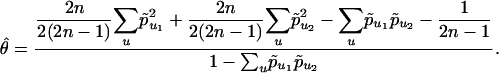 |
[4] |
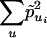 is the homozygosity in population i and is therefore equal to 1 - Hi, where Hi is the heterozygosity in population i. Assuming 2n/(2[2n - 1]) ≈ 1/2, then Eq. 4 reduces to
is the homozygosity in population i and is therefore equal to 1 - Hi, where Hi is the heterozygosity in population i. Assuming 2n/(2[2n - 1]) ≈ 1/2, then Eq. 4 reduces to
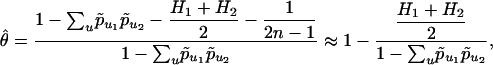 |
[5] |
assuming (1/[2n - 1])/(1 - ∑up̃u1 p̃u2) is small. If we fix population 1 as Africa and denote it by α, and if we write
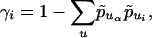 |
[6] |
then equating FST = a + b × (geographic distance) with Eq. 5 it follows that an estimate for the heterozygosity in population i is
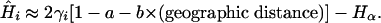 |
[7] |
It is only because geographic distance is a good predictor of FST that this calculation can be made.
Results
Fig. 1A shows a scatterplot of pairwise genetic distances (as measured by FST) against great circle geographic distances. Fitting a linear regression of FST on geographic distance produces R2 = 0.5882. Incorporating waypoints to account for more likely paths of past migrations increases R2 for the regression to 0.7834 (Fig. 1B).
Fig. 1.

Scatterplot of FST and geographic distance. Red dots denote within-region comparisons, green triangles indicate comparisons between populations in Africa and Eurasia, and blue diamonds represent comparisons with America and Oceania. (A) The relationship between FST and geographic distance computed using great circle distances. R2 for the linear regression of genetic distance on geographic distance is 0.5882. (B) The correction for large bodies of water produces a different scatterplot (R2 = 0.7835). The regression line fitted to the data [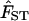 = 4.35 × 10-3 + (6.28 × 10-6) × (geographic distance in kilometers)] is drawn in black.
= 4.35 × 10-3 + (6.28 × 10-6) × (geographic distance in kilometers)] is drawn in black.
The Mantel correlation between FST and pairwise geographic distance incorporating waypoints is 0.8851 (p < 10-4). The correlations of other measures of genetic differentiation with geographic distance are also high (Table 1). Table 1 shows that the Mantel correlation between genetic distance and geographic distance is almost as high as those between any two different estimates of genetic distance calculated from the data set.
Fig. 2 highlights comparisons of those populations that had the most influence on the regression (see Materials and Methods) and shows the strong contribution of the American populations to the relationship between geographic and genetic distance at a large scale (Fig. 2A). The deviation of the Maya (labeled as 2 in Fig. 2A) from the regression line is possibly a result of admixture between Europeans and the Maya during colonization. To some extent, this relationship was observed earlier (11), and it has the effect here of lowering the Maya's genetic distance from Eurasians (Fig. 3). The Old World deviations from the linear regression of FST on geographic distance can be explained by genetic isolation; the Kalash, Mbuti Pygmies, and San (Fig. 2B) are each more highly differentiated genetically than is predicted based on the regression. An earlier study (29) of correlations between genetic and geographic distance showed an asymptote at high geographic distances within each continent; this asymptotic relationship is not observed with the present microsatellite data, although the sampling of populations within continents here is not dense in any particular continent (10).
Fig. 2.

Populations influencing the linear regression. The two plots are identical except that different features are highlighted in A and B. The number representing each population is the rank of its influence on the regression, with 1 indicating the population whose removal from the data alters the regression by the greatest amount (see Materials and Methods and Table 2). All other points not involving comparisons with the populations of greatest influence are in gray. (A) Red 1 denotes comparisons including Karitiana; green 2, Maya; navy blue 3, Pima; and purple 4, Colombia. Black squares show comparisons between the American populations. Comparisons involving the Maya (labeled as 2) tend to produce smaller FST values than are predicted by the regression line, and excluding the Maya from analysis increases R2 to 0.8183. The slight increase in the error sum of squares of the regression when the Maya are included in the data set shows that they have little influence on the observed pattern. (B) Orange 5 denotes comparisons including Kalash; brown 6, San; and blue 7, Mbuti Pygmy. The black circle is the comparison between the San and Mbuti Pygmies. The black triangles are comparisons of the Kalash to the San and Mbuti. The Kalash have been identified as a genetic isolate (11) from the rest of Pakistan; here, comparisons of the Kalash with other Central/South Asian and East Asian populations produce large residuals, whereas comparisons with European and Middle Eastern groups do not, consistent with the closer relationships of the Kalash to groups in these regions than to groups in East Asia or to other groups in Pakistan (11, 27). The high FST values observed in comparisons with the Mbuti Pygmies or the San, both hunter-gatherer populations, are likely to be a consequence of the deep genetic structure believed to exist in Africa and of the amount of genetic isolation these groups have experienced from other African populations (8, 28).
Fig. 3.

Standardized principal coordinates of both the geographic and genetic distance matrices of all pairwise comparisons, superimposed on a common set of axes. Scale on axes indicates SDs from the mean of each respective coordinate. Each population is represented by two points joined by a line: its geographic standardized principal coordinate score is shown by an open circle, and its genetic standardized principal coordinate score is marked by an open diamond (except in the case of the labeled populations, which are indicated by crosses). Regions of the world and certain populations of interest are labeled. The first three principal coordinates from the genetic distance matrix explain 50.8%, 16.1%, and 8.1% of the variation of genetic distance across populations, respectively, and the first three principal coordinates of the geographic distance matrix explain 73.2%, 21.3%, and 2.7% of the variation of geographic distance across populations.
The observed relationship of genetic and geographic distance should not be interpreted simply as following from theories of isolation by distance (1, 2), which are valid only at equilibrium between migration, mutation, and drift. There clearly has not been time to reach equilibrium between the extremes of man's inhabited range, or even within continents, in the very short evolutionary history of modern humans (29). An expansion of modern humans outward from a single center is an alternative way of producing a global correlation between geographic and genetic distances. Geographical expansion events may have happened in many small steps, with each such migration involving a sampling from the previous subset of the original population. This sampling would have led to a stepwise increase in genetic drift and a concomitant decrease in genetic diversity: a serial founder effect (30, 31).
Genetic data are found to be in strong agreement with this expansion model. The rank order of continents by genetic diversity for Y-chromosomal and chromosome-21 polymorphisms correlates with the archaeologically estimated order in which modern humans entered into continents (32, 33), and expected heterozygosity (calculated by using 377 loci from the HGDP-CEPH data set) has been found to decrease linearly with distance from a possible site for the geographic origin of modern humans in East Africa (34). We have confirmed the latter observation by augmenting the 377 loci previously studied with 406 additional microsatellites from the same individuals (Fig. 4A).
Fig. 4.

The decay of heterozygosity plotted against geographic distance between populations and a possible origin of expansion. (A) Heterozygosity in the HGDP-CEPH populations against distance from Addis Ababa, Ethiopia (9N, 38E). Distances were corrected for large bodies of water. The equation of the regression line is heterozygosity = 0.7682 - (6.52 × 10-6) × (distance from Addis Ababa). R2 = 0.7630. (B) Simulation results of the decay of heterozygosity with distance using a model of a serial founder effect. The simulation is based exclusively on mutation at a realistic rate and drift, as described in more detail in Supporting Text. The parameter values generating the simulation were chosen so as to fit the observed ΔH of A. The number of bottlenecks is n = 100, and the number of founders per bottleneck, Nb, is 250, which approximates the effective population size of a population of hunter-gatherers (35, 36). Other pairs of values of n and Nb in the same ratio would fit the data equally well, because their ratio is the main quantity affecting the slope. The equation of the fitted line is heterozygosity = 0.8761 - 0.0012 × (distance from the parental colony). R2 = 0.8587.
Assuming that there was an initial site from which the human expansion occurred, Fig. 5 shows that the pattern of expected heterozygosities in the data set is best explained by an expansion originating in Africa. For each of 4,210 points on a lattice of latitudes and longitudes (see Materials and Methods), we regressed expected heterozygosity in the HGDP-CEPH populations on geographic distance to the lattice point (Fig. 5). The 936 locations in Africa used as origins resulted in R2 values ranging from 0.757 to 0.870 (the SD of R2 within Africa was 0.017), whereas R2 using the 3,274 non-African locations as origins ranged from 1.67 × 10-7 to 0.744 (the SD of R2 outside of Africa was 0.245). Thus, no origin outside of Africa had the explanatory power of an origin anywhere in Africa (see also ref. 37). Because sampling was not very dense in Africa, especially in Eastern and Northern Africa, a larger sample might enable this approach to further localize the specific origin of the expansion.
Fig. 5.

The origin of the human expansion. The color or shade of each of the 4,210 locations (shown as dots) indicates either a correlation coefficient r or an R2 value for the regression of expected heterozygosities in 53 HGDP-CEPH populations on geographic distance (corrected for large bodies of water) to the location displayed. Note that, for a simple linear regression, r2 = R2. Grayscale points indicate R2 values, as shown by the gradient on the right, and correlation coefficients r are displayed in Africa and South America to reflect the sign of the relationship between heterozygosity and geographic distance to locations in these continents. R2 values range from 0.757 to 0.870 in Africa and from 0.519 to 0.659 in South America. The maximum value of r (≈0.812) is observed when the origin is (30S, 50.2W); the minimum value of r (approximately -0.933) is observed when the origin is (4.3N, 12.8E).
Regressions based on origins in South America had the highest R2 values of the non-African locations, but the correlation of expected heterozygosities with geographic distance to South America is positive, indicating that whereas heterozygosity decreases linearly with distance from Africa, it increases with distance from South America. These observations, together with the high genetic diversity in Africa and low diversity in the Americas, are consistent with an expansion from Africa, with South America being among the last places reached by migrating populations.
The linear relationships observed in Figs 4A and 1B are different depictions of the same phenomenon because pairwise FST is directly related to the homozygosities of each population in the comparison (see Eq. 7) and is therefore inversely related to the populations' heterozygosities (38). Suppose i is any non-African population and α is a fixed African population. We can regard γi (see Eq. 6) as an index of the similarity of alleles between populations i and α where puα is the frequency of allele u in the fixed African population α, pui is the frequency of the allele u in population i, and the sum is taken over all alleles u.
Pooling all of the sub-Saharan African populations in the data set and averaging γi across loci between Africa (α) and each non-African population (i) in the sample, we find that the mean of γi is 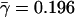 with a coefficient of variation of 1%. Substituting
with a coefficient of variation of 1%. Substituting 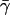 and the values of the slope b and the intercept a from the regression of FST on geographic distance from Africa into Eq. 7, the estimate of the expected heterozygosities of all populations in the sample is within 3% of the observed values; the difference between the estimate
and the values of the slope b and the intercept a from the regression of FST on geographic distance from Africa into Eq. 7, the estimate of the expected heterozygosities of all populations in the sample is within 3% of the observed values; the difference between the estimate 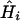 and the observed value has a SD of 0.0207. Thus, we can “transform” Figs. 4A and 1B into each other almost without loss of information, as is reflected by the similar explanatory power of the linear regressions of FST (R2 = 0.78) and of expected heterozygosity (R2 = 0.76) on geographic distance.
and the observed value has a SD of 0.0207. Thus, we can “transform” Figs. 4A and 1B into each other almost without loss of information, as is reflected by the similar explanatory power of the linear regressions of FST (R2 = 0.78) and of expected heterozygosity (R2 = 0.76) on geographic distance.
Testing whether a serial founder effect could give rise to the decay of expected heterozygosity with distance observed in Fig. 4A requires appropriate demographic models for calculating the effect of drift. We performed simulations of evolutionary processes to assess whether we could recover a similar pattern to what was computed from the data as shown in Fig. 4A (37). Assume for simplicity that we begin with a parental population, and there are n serial bottleneck episodes starting at the origin (the location of the parental population). In each bottleneck, a sample of individuals of size Nb founds the next colony, which is established at some distance from the previous colony and which remains isolated from all other colonies. This subsampling generates a succession of colonies in time, each of which grows to a large size K before generating the next colony in the chain. Each bottleneck episode decreases expected heterozygosity in the new colony by a factor of 1 - 1/(2Nb) (39). To be precise, this computation includes the drift effect only of the first generation after the bottleneck.
Based on this simple model of n bottlenecks with Nb founders at each bottleneck, an approximation for the total loss of expected heterozygosity from the beginning to the end of the expansion from the parental population due to the sequence of bottlenecks alone will be
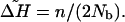 |
[8] |
Regressing heterozygosity on distance from the parental colony, we can estimate ΔH by calculating the difference between the intercept of the regression line and the fitted value for the last population in the expansion (the furthest population from the origin). In Fig. 4A, the observed ΔH is 0.12. Because n and Nb are unknown, Eq. 8 only allows the estimation of their ratio. Moreover, this simple model assumes no intermigration among colonies after their founding; it only accounts for genetic drift that occurs as a result of the bottlenecks in the serial founder effect, ignoring genetic drift (i) during the growth period where the founding population increases in size to carrying capacity and (ii) while the population stays at carrying capacity as the subsequent colonies are formed. These components will increase the amount of drift experienced by populations over that which would ensue from a population of constant size K.
Simulation enables the evaluation of these components of the evolutionary process by using estimable quantities, such as the mutation rate of microsatellites and the sizes of populations (see Supporting Text, which is published as supporting information on the PNAS web site, for more discussion). Fig. 4B shows that simulation can produce heterozygosity values similar to those observed in the data set, giving a simulated value for ΔH of 0.12, very close to the observed value. ΔHsim will differ from 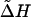 in Eq. 8 (see Supporting Text). The main assumption in the simulation (Fig. 4B) is that Nb, the number of founders at each bottleneck, is of the order of a hunter-gatherer tribe (35, 36).
in Eq. 8 (see Supporting Text). The main assumption in the simulation (Fig. 4B) is that Nb, the number of founders at each bottleneck, is of the order of a hunter-gatherer tribe (35, 36).
Discussion
Geographic distance is a good predictor of genetic distance on a global scale (Fig. 1). The pattern's robustness is indicated by our ability to reasonably explain anomalies (Fig. 2) based on what is generally believed to have occurred during the past 100,000 years of modern human history (29). We also find a close relationship between the correlation of FST and geographic distance (Fig. 1) and the geographic pattern of heterozygosity across populations (Fig. 4A). An increase in genetic distance with geographic distance has been observed in the past and has been attributed to equilibrium models of isolation by distance, but simulation results show that the geographic pattern of heterozygosities in the HGDP-CEPH populations is consistent with a serial founder effect starting at a single origin. Further, the observed pattern of within-population diversity is best explained by an origin in Africa (Fig. 5).
By studying the relationship between genetic and geographic distance, we can assess the relative importance of genetic drift and natural selection in determining the genetic variation observed among human populations. The average contribution of drift generated by the serial founder effect might be estimated from the properties of the regression in Figs. 1B and 4A. Because our regressions explain 76-78% of the observed genetic variation, this quantity is therefore an estimate of the minimum influence that drift, due to the serial founder effect, has on the total variation observed. In other words, the fraction of the variation in heterozygosity across human populations that is explained by drift is at least 76-78%. If stabilizing selection has been a major force in human evolution, then the decrease of average heterozygosity would be reduced, and the slope in Fig. 4A would be less negative (by an unknown amount).
The residual 22-24% of genetic variation not explained by the regression is generated by population-specific selection, drift, and mutational histories. The deviation from the regression of each individual population (Fig. 4A) or of each population pair (Fig. 2) is a consequence of each population's particular demographic history (40). But it is clear that part of these deviations also may be due to different selective conditions met by these populations in the different environments to which they have been exposed. Therefore, we estimate that 76-78% can be considered a lower bound on the effect of drift, and 22-24% an upper bound on the effect of selection, in the genetic differentiation of human populations.
Supplementary Material
Acknowledgments
We thank Saurabh Mahajan for bioinformatics assistance and Lynn Jorde and Montgomery Slatkin for helpful comments on the manuscript. This work was supported by National Institutes of Health Grants GM28106 and GM28428. S.R. is supported by a National Defense Science and Engineering Graduate fellowship. C.C.R. is supported by a fellowship from the Morrison Institute for Population and Resource Studies. N.A.R. is supported by a Burroughs Wellcome Fund Career Award in the Biomedical Sciences.
Author contributions: S.R., M.W.F., and L.L.C.-S. designed research; S.R., O.D., and C.C.R. performed research; S.R., C.C.R., and N.A.R. analyzed data; and S.R., O.D., C.C.R., N.A.R., M.W.F., and L.L.C.-S. wrote the paper.
Abbreviation: HGDP-CEPH, Human Genome Diversity Project-Centre d'Etude du Polymorphisme Humain.
References
- 1.Malécot, G. (1991) The Mathematics of Heredity (Freeman, San Francisco).
- 2.Kimura, M. & Weiss, G. H. (1964) Genetics 49, 561-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cavalli-Sforza, L. L., Barrai, I. & Edwards, A. W. F. (1964) Cold Spring Harbor Symp. Quant. Biol. 29, 9-20. [DOI] [PubMed] [Google Scholar]
- 4.Wijsman, E. M. & Cavalli-Sforza, L. L. (1984) Annu. Rev. Ecol. Syst. 15, 279-301. [Google Scholar]
- 5.Morton, N. E. (1973) in Genetic Structure of Populations, ed. Morton, N. E. (Univ. Press of Hawaii, Honolulu), pp. 76-79.
- 6.Jorde, L. B. (1980) in Current Developments in Anthropological Genetics, eds. Mielke, J. H. & Crawford, M. H. (Plenum, New York), Vol. 1, pp. 135-208. [Google Scholar]
- 7.Cavalli-Sforza, L. L., Menozzi, P. & Piazza, A. (1994) The History and Geography of Human Genes (Princeton Univ. Press, Princeton).
- 8.Eller, E. (1999) Am. J. Phys. Anthropol. 108, 147-159. [DOI] [PubMed] [Google Scholar]
- 9.Relethford, J. H. (2001) (2001) Hum. Biol. 73, 629-636. [DOI] [PubMed] [Google Scholar]
- 10.Cann, H. M., de Toma, C., Cazes, L., Legrand, M. F., Morel, V., Piouffre, L., Bodmer, J., Bodmer, W. F., Bonne-Tamir, B., Cambon-Thomasen, A., et al. (2002) Science 296, 261-262. [DOI] [PubMed] [Google Scholar]
- 11.Rosenberg, N. A., Pritchard, J. K., Weber, J. L., Cann, H. M., Kidd, K. K., Zhivotovsky, L. A. & Feldman, M. W. (2002) Science 298, 2381-2385. [DOI] [PubMed] [Google Scholar]
- 12.Mountain, J. L. & Ramakrishnan, U. (2005) Hum. Genomics 2, 4-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramachandran, S., Rosenberg, N. A., Zhivotovsky, L. A. & Feldman, M. W. (2004) Hum. Genomics 1, 87-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhivotovsky, L. A., Rosenberg, N. A. & Feldman, M. W. (2003) Am. J. Hum. Genet. 72, 1171-1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lewis, P. O. & Zaykin, D. (2001) gda (genetic data analysis): Computer Program for the Analysis of Allelic Data (Univ. of Connecticut, Storrs, CT), Version 1.0 d16c. Available at: http://hydrodictyon.eeb.uconn.edu/people/plewis/software.php.
- 16.Weir, B. (1996) genetic data analysis ii (Sinauer, Sunderland, MA).
- 17.Wright, S. (1978) Evolution and the Genetics of Populations (University of Chicago, Chicago), Vol. IV, p. 89. [Google Scholar]
- 18.Slatkin, M. (1995) Genetics 139, 457-462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goldstein, D. B., Ruiz-Linares, A., Cavalli-Sforza, L. L. & Feldman, M. W. (1995) Genetics 139, 463-471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldstein, D. B., Ruiz-Linares, A., Cavalli-Sforza, L. L. & Feldman, M. W. (1995) Proc. Natl. Acad. Sci. USA 92, 6723-6727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nei, M. (1972) Am. Nat. 106, 283-292. [Google Scholar]
- 22.Mountain, J. L. & Cavalli-Sforza, L. L. (1997) Am. J. Hum. Genet. 61, 705-718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sinnott, R. W. S. (1984) Sky Telescope 68, 159. [Google Scholar]
- 24.Stringer, C. (2000) Nature 405, 24-27. [DOI] [PubMed] [Google Scholar]
- 25.Strauss, R. E. (2002) res5 (MathWorks, Natick, MA). Available at www.biol.ttu.edu/Strauss/Matlab/matlab.htm.
- 26.Gower, J. C. (1966) Biometrika 53, 325-338. [Google Scholar]
- 27.Qamar, R., Ayub, Q., Mohyuddin, A., Helgason, A., Mazhar, K., Mansoor, A., Zerjal, T., Tyler-Smith, C. & Mehdi, S. Q. (2002) Am. J. Hum. Genet. 70, 1107-1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tishkoff, S. A. & Williams, S. M. (2002) Nat. Rev. Genet. 3, 611-621. [DOI] [PubMed] [Google Scholar]
- 29.Cavalli-Sforza, L. L. & Feldman, M. W. (2003) Nat. Genet. 33, 266-275. [DOI] [PubMed] [Google Scholar]
- 30.Harpending, H. & Rogers, A. (2000) Annu. Rev. Genomics Hum. Genet. 1, 361-385. [DOI] [PubMed] [Google Scholar]
- 31.Eswaran, V. (2002) Curr. Anthropol. 43, 749-774. [Google Scholar]
- 32.Underhill, P. A., Passarino, G., Lin, A. A., Shen, P., Mirazón Lahr, M., Foley, R. A., Oefner, P. J. & Cavalli-Sforza, L. L. (2001) Ann. Hum. Genet. 65, 43-62. [DOI] [PubMed] [Google Scholar]
- 33.Jin, L., Underhill, P. A., Doctor, V., Davis, R. W., Shen, P. D., Cavalli-Sforza, L. L. & Oefner, P. J. (1999) Proc. Natl. Acad. Sci. USA 93, 3796-3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prugnolle, F., Manica, A. & Balloux, F. (2005) Curr. Biol. 15, 159-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee, R. B. & DeVore, I., eds. (1968) Man the Hunter (Aldine, Chicago).
- 36.Cavalli-Sforza, L. L. (2004) in Examining the Farming/Language Dispersal Hypothesis, eds. Bellwood, P. & Renfrew, C. (McDonald Institute Monographs, Cambridge, U.K.).
- 37.Ray, N., Currat, M., Berthier, P. & Excoffier, L. (2005) Genome Res. 15, 1161-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Charlesworth, B. (1998) Mol. Biol. Evol. 15, 538-543. [DOI] [PubMed] [Google Scholar]
- 39.Hartl, D. L. & Clark, A. G. (1997) Principles of Population Genetics (Sinauer, Sunderland, MA), 3rd Ed., p. 172.
- 40.Cavalli-Sforza, L. L., ed. (1986) African Pygmies (Academic, New York).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.