

Abstract
In various human diseases, altered gene expression patterns are often the result of deregulated gene-specific transcription factor activity. To further understand disease on a molecular basis, the comprehensive analysis of transcription factor signaling networks is required. We developed an experimental approach, combining chromatin immunoprecipitation (ChIP) with a yeast-based assay, to screen the genome for transcription factor binding sites that link to transcriptionally regulated target genes. We used the tumor suppressor p53 to demonstrate the effectiveness of the method. Using primary and immortalized, nontransformed cultures of human mammary epithelial cells, we isolated over 100 genomic DNA fragments that contain novel p53 binding sites. This approach led to the identification and validation of novel p53 target genes involved in diverse signaling pathways, including growth factor signaling, protein kinase/phosphatase signaling, and RNA binding. Our results yield a more complete understanding of p53-regulated signaling pathways, and this approach could be applied to any number of transcription factors to further elucidate complex transcriptional networks.
Regulation of gene expression is required for the maintenance of cellular homeostasis. Deregulation of transcription factor (TF) activities is directly linked to many human diseases, including cancer. It has been estimated that there are ∼1,850 TFs encoded within the human genome, and the comprehensive examination of these proteins and their associated regulatory pathways is required to further understand the molecular basis of disease (71). The elucidation of complex transcriptional cellular networks (the identification of TFs, binding sites within the genome, and regulated genes) is an area of intense research focus in the post-human genome-sequencing era.
Many techniques such as subtractive hybridization, differential display, microarrays, and serial analysis of gene expression have been employed to monitor gene expression changes on a genome-wide level. However, a limitation of these methods is that the observed gene expression changes may be due to direct or indirect regulation, making it challenging and time-consuming to delineate the primary gene targets. To this end, chromatin immunoprecipitation (ChIP), the localization of genomic TF binding sites through the cross-linking and immunoprecipitation of an endogenous TF from cells, is a valuable tool used to identify regulatory regions directly bound by a TF in the context of the native chromatin structure. A method widely used to study TF binding is ChIP followed by microarray analysis (ChIP-chip); this method often involves microarray hybridization of the ChIP DNA fragments to specific, known promoter regions or to genomic CpG island regions (35, 41, 47, 55). One critical limitation of some ChIP-chip analyses is that only segments of promoter regions are analyzed. Thus, other genomic regions that may be bound by TFs (distal regions and enhancers) are excluded from these analyses. By using ChIP and high-density oligonucleotide arrays to analyze Sp1, c-Myc, and p53 binding on chromosomes 21 and 22, investigators found that a majority of the binding sites for these transcription factors do not reside within 1 kb of a CpG island or the 5′ exon of a gene (8) and, thus, would have been overlooked using limited promoter fragments on a microarray chip. ChIP-chip involving high-density oligonucleotide arrays is beneficial for whole-genome profiling of TF binding, but currently only single chromosomes are being analyzed, limiting global pathway analyses (19, 48). To circumvent some of these issues, we developed an experimental approach to assay the genome for TF binding sites that link to transcriptionally regulated target genes and used the TF p53 to demonstrate the effectiveness of the method.
The tumor suppressor p53 binds DNA with high affinity in a sequence-specific manner (40) and regulates multiple signaling pathways involved primarily in growth arrest, DNA repair, and apoptosis in response to various cellular stresses (64). To date, more than 100 genes directly regulated by p53 have been identified, leading to a better understanding of the mechanism of p53 tumor suppression (14, 30, 50). However, it is estimated that there are ∼1,600 sites to which p53 can bind in the human genome (8). Thus, it seems the genome contains many unidentified p53 target genes, and the identification of these genes will give further insight to complex p53 signaling networks.
We used ChIP to capture p53 binding to genomic DNA on a global level after damage, and the resulting DNA fragments were screened for functional p53 binding and transactivation of a reporter gene using a modified yeast one-hybrid system (69). Our screen yielded not only valuable information regarding the sequence and characteristics of functional p53 binding sites within the genome but also novel target genes and would be a valuable approach to pursue for any number of TFs.
MATERIALS AND METHODS
Cell culture, treatments, and adenovirus infection.
The MCF-10A cells were obtained from the American Type Culture Collection (Manassas, VA), and primary human mammary epithelial cells (HMEC) were purified from normal breast tissue obtained from the Vanderbilt-Ingram Cancer Center Human Tissue Acquisition and Pathology Shared Resource (63); both were cultured as previously described (3). Second-passage primary human epidermal keratinocyte (HK) cells, obtained from the Vanderbilt Skin Disease Research Core, were isolated and grown as previously described (23). The human colorectal carcinoma HCT116 cell lines (6) were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin. The HIp53 ponasterone A-inducible p53 cell lines (24) were cultured in DMEM supplemented with 10% fetal bovine serum, 1% penicillin-streptomycin, 600 μg/ml G418 (Mediatech, Herndon, VA), and 400 μg/ml Zeocin (Cayla, Toulouse, France). All cells were grown at 37°C with 5% CO2 in a humidified incubator. The MCF-10A cells, HMEC, and HCT116 cells were treated with 350 nM adriamycin (ADR); the HIp53 cells were treated with 10 μM ponasterone A; and the HK cells were infected with adenovirus expressing green fluorescent protein (GFP) or p53 (32).
Western analyses.
Western analysis was performed as described previously (72) using anti-MDM2 SMP14, anti-p53 PAb1801 (Santa Cruz Biotechnology, Santa Cruz, CA), and anti-p21Waf1/Cip1 Ab-1 (Oncogene Research Products, Cambridge, MA) antibodies.
Formaldehyde cross-linking and chromatin immunoprecipitation.
Cells were formaldehyde cross-linked in a 1.6% solution for 10 min as described previously (66). For every 2 mg of protein extract processed for ChIP, 10 μg of mouse immunoglobulin G bound to protein A-Sepharose (Pharmacia Biotech, Piscataway, NJ) was used to preclear for 1 h with rocking at 4°C. The extracts were immunoprecipitated with 1 μg of anti-p53 PAb1801 (Santa Cruz) and 1 μg of anti-p53 PAb421 (Oncogene Research Products, Cambridge, MA) or 2 μg of anti-cyclin B1 GNS1 antibodies (Santa Cruz) by rocking overnight at 4°C. For the library generation, a total of 75 mg of MCF-10A cell or HMEC protein lysates was immunoprecipitated, and for the ChIP analyses shown in Fig. 3 and 4, 2 mg of protein lysates was immunoprecipitated from the MCF-10A cells, HMEC, or HK cells. Immunocomplexes were washed eight times as described previously (66), and the protein was degraded in digestion buffer (120 μg/ml proteinase K, 10 mM Tris [pH 7.5], 5 mM EDTA, and 0.5% sodium dodecyl sulfate) at 56°C overnight and then incubated at 65°C for 30 min. The DNA was phenol-chloroform extracted, and ethanol precipitated.
FIG. 3.

Analysis of p53 in vivo binding to consensus binding sites. Three sets each of MCF-10A cells, HMEC, and HK cells were identically processed: one set was treated with ADR (350 nM for 5 h) and formaldehyde cross-linked (ADR +, X-L +), another set was not treated with ADR and was formaldehyde cross-linked (ADR −, X-L +), and a final set was treated with ADR and not formaldehyde cross-linked (ADR +, X-L −). The DNA for PCRs was derived from p53-specific and cyclin B1-specific immunoprecipitations (IP) and amplified using primers flanking the p53 response elements in genes encoding the indicated proteins. PCRs were resolved with polyacrylamide gel electrophoresis, and the gels were stained with ethidium bromide. The cyclin B1-specific IPs were included to assess any DNA fragments purified from cross-linked lysates nonspecifically. Input +, genomic input; input −, water control. PCR results with primers directed to the coding region of GAPDH serve as a control for nonspecific DNA IP by p53-specific antibodies.
FIG. 4.

Comparative analysis of p53 binding to sites in promoter regions of known and candidate target genes. In the left panels, four sets of HMEC were processed as follows: one set was treated with ADR (350 nM for 5 h), formaldehyde cross-linked, and immunoprecipitated with a p53 antibody (solid bars); another set was treated with ADR, formaldehyde cross-linked, and immunoprecipitated with a cyclin B1 antibody (open bars); a third set was treated with ADR, not formaldehyde cross-linked, and immunoprecipitated with a p53 antibody (dotted bars); and a final set was formaldehyde cross-linked but not treated with ADR and then immunoprecipitated with a p53 antibody (gray bars). Quantitative real-time PCR was performed, and each sample was normalized to the same genomic DNA that was isolated from cells that were cross-linked and processed the same with the exception that the immunoprecipitation step was not performed. The binding sites shown are those that were recovered from the library screen (LS), those that were previously reported in the literature (RS), and those that were potential binding sites found by gene analysis using the p53MH algorithm (PS). The base pair match of the binding site to the p53 consensus is shown in parentheses. The results, shown as percentages of input DNA, are from at least three independent experiments, with the error bars representing standard deviations. The right panels show schematics of the genomic structure and localization of known and putative p53 binding sites analyzed. The bar shading indicates species conservation as indicated. Exons are indicated with an E followed by the exon number in either an open box or a shaded box (representing the terminal exon). The sequences of the binding sites present in the regions analyzed are shown, and in parentheses the distances of the binding sites from the start of exon 1 are given.
Yeast selection system.
The immunoprecipitated genomic DNA was processed using a PCR polishing kit (Pfu based; Stratagene, La Jolla, CA), ligated into the HIS3 reporter plasmid, pBM947 (generously provided by M. Johnston; [73]), and amplified by growth in TransforMax EPI300 electrocompetent Escherichia coli (Epicenter, Madison, WI). The Saccharomyces cerevisiae yeast strain YPH681 containing the pRS314SN vector (51) was transformed as described previously (25) with 100 μg of each pBM947-based library DNA. The cells were plated onto SG media lacking Trp, Ura, and His (SG-Trp-Ura-His) and incubated at 30°C, and colonies were replica plated onto SD-Trp-Ura-His media to screen for false positives.
The pBM947-based library DNA fragments were amplified from the yeast that grew in a p53-dependent manner using a modified yeast colony PCR technique (7). Briefly, 50 cycles of PCR were performed after an initial 4 min at 95°C; each cycle consisted of 1 min at 95°C, 1 min at 56°C, and 1.5 min at 72°C, before a final elongation step of 10 min at 72°C. PCR DNA products were resolved in a 1% agarose gel, stained with ethidium bromide, purified, and sequenced.
RNA preparation and reverse transcription.
mRNA was harvested from the HIp53 and HK cells as previously described (66). Total RNA was harvested from the HCT116 cells using the Aurum total RNA minikit (Bio-Rad Laboratories, Hercules, CA). Reverse transcription of 100 ng of poly(A) RNA from the HIp53 and HK cells and 250 ng of total RNA from the HCT116 cells was performed using the TaqMan reverse transcription reagent kit (Applied Biosystems, Foster City, CA).
PCR amplification.
The ChIP PCR amplifications for CDKN1A (p21) site 1 and DDB2 (p48) were performed in 16.6 mM (NH4)2SO4, 0.67 mM Tris (pH 8.8), 6.7 mM MgCl2, 10 mM β-mercaptoethanol, 10% dimethyl sulfoxide, 1.5 mM nucleotides, and 1.25 U of Taq polymerase (Fig. 3). A total of 175 ng of each primer was used per 25-μl reaction mixture. The ChIP PCR amplifications for EDN2, PPM1J, RPS27L, UBTD1, PDGFC, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were performed in 10 mM Tris (pH 9.0), 50 mM KCl, 0.1% Triton X-100, 0.5 mM MgCl2, 0.25 mM nucleotides, and 1.25 U of Taq polymerase (Fig. 3). Each primer was used at 0.4 μM per 25-μl PCR mixture. Primer sequences and PCR conditions are provided in Table S1 in the supplemental material.
Quantitative real-time PCR (Q-RT-PCR) was performed for p53 binding analyses (Fig. 4) using chromatin-immunoprecipitated DNA (described above) and normalized to a genomic template that was cross-linked and sonicated, but not immunoprecipitated. Q-RT-PCR was performed for the gene expression analyses (Fig. 5) using 2 ng of reverse-transcribed mRNA or 5 ng of reverse-transcribed total RNA template. Q-RT-PCR SYBR-Green Supermix (Bio-Rad) reaction mixtures contained 200 nM of each primer and were run on an iCycler thermal cycler (Bio-Rad). Primers were designed using the Beacon Designer software (Bio-Rad). Target gene expression was normalized to GAPDH, and changes were calculated relative to control samples. For primer sequences and PCR conditions see Table S1 in the supplemental material.
FIG. 5.

p53-dependent regulation of representative candidate target gene expression. The isogenic pair of HCT116 p53+/+ and p53−/− cells were treated with ADR (350 nM) for 0, 6, 12, and 24 h; the HIp53 cells (p53) and corresponding vector control cell line (Ø) were treated with ponasterone A (10 μM) for 24 h, and the HK cells were infected with a GFP- or p53-expressing adenovirus for 30 h. Total RNA from HCT116 cells and mRNA from HIp53 and HK cells was purified and reverse transcribed and quantitative real-time PCR performed. The samples were normalized to GAPDH, and the results are presented as changes relative to either the 0-h HCT116 p53+/+ sample (left panel), the HIp53 vector control cells treated with ponasterone (middle panel), or the HK cells infected with a GFP-expressing adenovirus (right panel). The results are the means of three independent experiments (MCF-10A cells and HMEC) or duplicate experiments (HK cells), with error bars representing the standard deviations. Note that the y axes are set to 7.0 with the exceptions of the left and middle panels for the CDKN1A gene and the left and right panels for the EDN-2 gene.
RESULTS
Library generation and yeast screen.
We used primary HMEC and the immortal, nontransformed MCF-10A human mammary epithelial cell line to generate libraries of DNA fragments that p53 bound after the cells were treated with the genotoxic agent ADR. We selected immortal, nontransformed and primary cells to avoid genetic and epigenetic chromatin alterations observed in transformed cells. To verify p53 signaling in these systems, cells were treated with ADR (350 nM) for 8 h and analyzed for changes in p53, MDM2, and p21 protein levels. ADR treatment led to increased p53 levels in HME and MCF-10A cells as well as elevation of the downstream targets p21 and MDM2 (Fig. 1). Of note, under this treatment condition, both the HMEC and the MCF-10A cells underwent cell cycle arrest as opposed to apoptosis (data not shown).
FIG. 1.
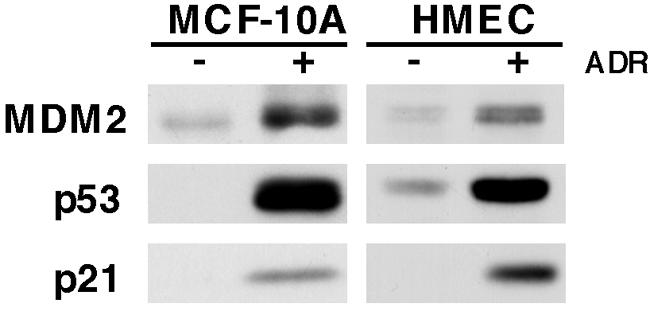
Analysis of p53, MDM2, and p21 protein levels in MCF-10A cells and HMEC after ADR treatment. Shown is Western blot analysis of p53, MDM2, and p21 protein harvested from MCF-10A and primary HMEC that were not treated (−) or treated (+) with ADR (350 nM) for 8 h.
To isolate DNA fragments that p53 bound after ADR treatment, the MCF-10As were treated with ADR for 5 h and the HMEC for 4, 8, 16, and 24 h and both cell types were cross-linked with formaldehyde and lysed (the HMEC lysates were pooled). The lysates were sonicated to shear the chromatin into ∼600- to 1,000-bp fragments. The chromatin was immunoprecipitated using p53-specific antibodies, the cross-linking was reversed, and the DNA fragments were purified and cloned into the pBM947 yeast expression vector upstream of a minimal promoter that controls the expression of the HIS3 gene. The pBM947-based library of putative p53-binding DNA fragments was transformed into an auxotrophic histidine-deficient yeast strain with a galactose-inducible yeast expression vector containing the human p53 gene (pRS314SN). Yeast transformants containing both the pRS314SN and pBM947 vectors were able to grow on galactose-containing, histidine-deficient media if the expressed p53 bound to the human genomic DNA fragment in the pBM947 vector and activated transcription of the HIS3 gene. To rule out false-positive clones that grew in a p53-independent manner, we also replica plated all clones on glucose-containing, histidine-deficient media. The clones that grew in the presence of glucose and, thus, in the absence of p53, were considered false positives and not further analyzed (Fig. 2).
FIG. 2.

Yeast selection system. The candidate upstream activating sequences (UAS) recovered from ChIP were cloned into the pBM947 reporter vector containing the HIS3 gene under the control of a basal GAL1 promoter and a URA1 marker. The pBM947-based library was transformed into an auxotrophic His-deficient yeast strain containing the pRS314SN vector, which expresses a galactose-inducible human wild-type p53 and a TRP1 marker. Yeasts containing both the vectors were grown on galactose-containing, histidine-deficient media (SG-Trp-Ura-His) to assay for the ability of p53 to bind to the potential UAS in the pBM947 vector and activate transcription of the HIS3 gene. Replica plating of all clones on glucose-containing, histidine-deficient media (SD-Trp-Ura-His) was performed to rule out false-positive clones. The clones that grew in the presence of glucose were considered false positive, and only the clones that grew on galactose, and presumably in a p53-dependent manner, were analyzed further. A clone containing a fragment of the p21 promoter encompassing site 1 is indicated as an example of a positive result.
For the MCF-10A library, ∼3.2 × 106 yeast transformants were screened, and 187 clones grew in a p53-dependent manner. After sequencing, 51 nonredundant sequences were identified. For the HMEC library, ∼1.7 × 106 yeast transformants were screened, resulting in 74 nonredundant positive clones that grew in a p53-dependent manner. A control library generated from immunoprecipitation of identically processed lysates using an isotype-matched antibody (cyclin B1) was also screened to determine the level of nonspecifically bound DNA fragments that contained sequences conforming to the p53 consensus. There was a sevenfold reduction in the number of colonies from the control library that grew in a p53-dependent manner; of those only four clones had putative p53 binding sites as identified by the p53MH algorithm (34). Thus, the majority of the DNA fragments present in the experimental libraries resulted from specific interactions with p53.
Analysis of genomic DNA fragments for p53 consensus binding sites and the identification of candidate target genes.
The DNA fragments isolated from the combined screening of the HMEC and MCF-10A cell libraries were analyzed for the presence of p53 consensus DNA binding sites using the p53MH algorithm (34). The canonical p53 binding site consists of two repeats of the 10-bp sequence PPPCWWGYYY (where P = A/G, W = A/T, and Y = T/C) separated by a 0- to 13-bp spacer region, (15). Of the fragments isolated from the HMEC and MCF-10A cell libraries, 99% contained putative p53 binding sites. A representative list is shown in Table 1; for the additional sites see Table S2 in the supplemental material.
TABLE 1.
Representative p53 consensus binding sites and candidate target genes
| Gene or DNA fragmenta | GenBank accession no. | Description | p53 binding siteb | Matchc | Locationd | Distance from exon 1 (kb) | Chromosome locationf |
|---|---|---|---|---|---|---|---|
| PPPCWWGYYY-x-PPPCWWGYYYe | |||||||
| CDKN1A | NM_078467 | Cyclin dependent kinase inhibitor 1A (p21, Cip1) | cAACATGTTg-0-GGACATGTTC | 18 | 5′ | 2.2 | 6p21.2 |
| CDKN1A | NM_078467 | Cyclin dependent kinase inhibitor 1A (p21, Cip1) | AGACTgGgCa-0-tGtCTgGgCa | 12 | 5′ | 1.3 | 6p21.2 |
| MDM2 | NM_002392 | Murine double minute 2, p53 binding protein | GGtCAAGTTC-0-AGACAcGTTC | 18 | Intron 1 | <1 | 12q14.3-15 |
| DDB2 | NM_000107 | Damage-specific DNA binding protein 2 (p48) | GAACAAGCCC-1-GGGCATGTTT | 20 | Exon 1 | <1 | 11p12-p11 |
| AKAP6 | NM_004274 | A kinase anchor protein 6 | AGACATGCCC-0-AAACATGTCT | 20 | Intron 1 | 16 | 14q12 |
| FLJ32800† | NM_152647 | Predicted | AGGCATGTCT-0-GGACATGTTT | 20 | 5′ | 4 | 15q21.1-.2 |
| MDS009† | NM_020234 | DTWD1 (DTW domain containing 1) | Intron 2 | 2.5 | 15q21.2 | ||
| H955 | NAg | NA | GGACATGCCC-0-GGACAAGCCT | 20 | NA | NA | 13q14.3 |
| H1160 | NA | NA | AGACATGCCC-0-GGGCATGCCT | 20 | NA | NA | 1q25.3 |
| RHOC‡ | NM_175744 | Ras homolog gene family product, member C | GAACATGCCT-0-GAGCAAGCCC | 20 | Intron 2 | 3 | 1p13.1 |
| MGC19531‡ | NM_005167 | PPM1J (protein phosphatase 1J, PP2C domain containing) | 3′ | 11 | 1p13.1 | ||
| PPP2R3A | NM_002718 | Protein phosphatase 2, regulatory subunit B, alpha | AGACATGTCT-2-AGACATGCCC | 20 | Intron 14 | 176 | 3q22.1 |
| TMEM30A | NM_018247 | Transmembrane protein 30A | GGACTTGCTT-0-GGGCTTGTCC | 20 | Intron 1 | 12 | 6q14.1 |
| UBTD1§ | NM_024954 | Ubiquitin domain containing 1 | tGACAAGTCT-0-GGGCTTGCTC | 19 | Intron 1 | 50 | 10q24.1-.2 |
| ANKRD2§ | NM_020349 | Ankyrin repeat domain 2 | 5′ | 22 | 10q24.2 | ||
| H1026 | NA | NA | GGGCATGTTg-0-GGGCATGTCC | 19 | NA | NA | 5p15.3 |
| LOC441968 | XM_497787 | Similar to extensin-like protein | GGGCATGCCa-0-GGACATGTCT | 19 | 5′ | 6 | 21q22.3 |
| PDGFC | NM_016205 | Platelet-derived growth factor, isoform C | GGtCATGTTC-0-AGACTTGCCC | 19 | Intron 4 | 200 | 4q32 |
| ANKRD11 | NM_013275 | Ankyrin repeat domain protein 11 | AGACATGaCC-0-tGGCATGTTC | 18 | Intron 1 | 50 | 16q24.3 |
| EDN-2 | NM_001956 | Endothelin-2 | ctGCAAGCCC-0-GGGCATGCCC | 18 | Intron 3 | 2 | 1p34 |
| FLJ12484¶ | NM_022767 | ISG20L1 (interferon-stimulated exonuclease 20 kDa-like 1) | 5′ | <1 | 15q26.1 | ||
| ISG20¶ | NM_002201 | Interferon-stimulated exonuclease, 20 kDa | ccACATGCCC-0-GGGCAAGCCC | 18 | 5′ | 17 | 15q26 |
| LOC388171¶ | XM_373647 | Predicted | 3′ | 17 | 15q26.1 | ||
| PTPN14 | NM_005401 | Protein tyrosine phosphatase, nonreceptor type 14 | AAACATGTTg-0-cAACATGTCC | 18 | Intron 3 | 115 | 1q32.2 |
| VTI1A | NM_145206 | Vesicle transport through interaction with t-SNARE homolog 1A | cAACATGTCT-0-tGGCATGTTC | 18 | Intron 7 | 270 | 10q25.2 |
| H1634 | NA | NA | tGGCATGTCC-11-cAACATGCCT | 18 | NA | NA | 12q23.3 |
| C9ORF88 | XM_497077 | Chromosome 9 open reading frame 88 product | tGGCAAGCag-0-AGGCTTGTTT | 17 | Intron 1 | 3 | 9q34.11 |
| RBM19 | NM_016196 | RNA binding motif protein 19 | GtACATGTCa-0-GGGCATGTTg | 17 | Intron 24 | 127 | 12q24.13-21 |
| RPS27L | NM_015920 | Ribosomal protein S27-like (40S ribosomal protein S27 isoform) | GGGCATGTag-0-tGACTTGCCC | 17 | Intron 1 | 2 | 15q22.2 |
| SMARCC1 | NM_003074 | SWI/SNF-related, matrix-associated actin-dependent regulator of chromatin | GGGCATGaTg-4-AtGCcTGTaa | 14 | Intron 7 | 65 | 3p23-21 |
The four items above the space are known p53 target genes; those below the space are products of candidate genes, except for H955, H1160, H1026, and H1634, which are DNA fragments that aligned to areas of the genome that did not contain any candidate genes within the criterion range. Items with the same symbol (†, ‡, §, or ¶) are products of multiple candidate genes identified within the criterion range of a single binding site.
Lowercase letters indicate consensus binding site mismatches.
Number of base pairs (out of 20) in the putative p53 binding site that match the consensus binding site.
Location of the p53 binding site in relation to the known/candidate target gene.
p53 consensus binding site, where P = purine (A/G), W = A/T, Y = pyrimidine (C/T), and x = 0- to 13-bp spacer region.
Location of the candidate target gene or binding site (if a candidate gene was not identified).
NA, not applicable.
The sequences of the library DNA fragments were aligned with the human genome using the BLAST search of the Human Genome Resources Database from the National Center for Biotechnology Information (NCBI) website, and candidate target genes were selected for validation. The criterion for selection was any known or predicted gene with the start of exon 1 within 20 kb upstream or downstream of where the sequence of the DNA fragment aligned. This criterion was chosen because approximately 99% of known p53 binding sites occur within 20 kb of the regulated gene. However, if the library DNA fragment aligned to an intron of a gene, regardless of the distance between the alignment and the start of exon 1, the gene was included as a candidate. After analyzing the DNA fragments generated from both libraries, ∼100 candidate target genes were identified, including both known and predicted genes. The chromosomal location, the location of the binding site in relation to the known or candidate target gene, and the distance of the binding site from the beginning of exon 1 of the associated known or candidate gene are shown (Table 1; see Table S2 in the supplemental material). The genes identified from the screen encode proteins involved in numerous cellular pathways including protein kinase/phosphatase signaling, protein transport, growth factor signaling, RNA binding, and chromatin remodeling. Numerous predicted genes with unknown functions were also identified. Of note, less than 25% of the DNA fragments aligned to regions of the genome that did not contain known or predicted genes within the criterion range. We recovered binding sites that were both 5′ to the candidate target genes and intronic, and, interestingly, we also recovered p53 binding sites 3′ of putative target genes. In addition to novel genomic p53 binding sites, the library screen (LS) yielded multiple known p53 binding sites. For instance, both of the p53 binding sites from the CDKN1A (p21) promoter were isolated (17), as well as the p53 binding sites from the MDM2 (77) and DDB2 (68) genes. The recovery of known p53 response elements that regulate key p53 target genes validated our method and demonstrates that the screen selects for functionally relevant binding sites and physiologically important target genes.
Analysis of in vivo binding of p53 to novel consensus binding sites.
To further validate p53 binding to the novel binding sites recovered from the library screen, we generated pools of DNA fragments from ChIP of ADR-treated HMEC and MCF-10A cells and primary cultures of HK cells. The latter were included in our analyses to compare binding results across primary cell types from two human tissues. The ChIP DNA was used as a template for PCR amplification of the genomic regions containing response elements obtained from the library screen. All of the genomic binding sites shown in Table 1 were present in these replicate pools of DNA fragments, and representative ChIP-PCR results from the HMEC, MCF-10A cell, and HK cell lysates are shown in Fig. 3. The binding sites shown were chosen due to their differential locations with regard to exon 1 of the associated candidate gene, as well as the varied cellular functions of the target genes. Response elements in CDKN1A (p21) and DDB2 (p48) genes, known p53 target genes, as well as the binding sites associated with EDN-2, PPM1J, RPS27L, and PDGFC genes were bound by p53 before ADR treatment as well as after ADR treatment in all cell systems analyzed. The UBTD1 gene-associated p53 response element was bound by p53 before ADR treatment in the MCF-10A cells, but binding was not detectable in the HMEC and HK cells. However, binding of p53 to the UBTD1 gene response element was detected after ADR treatment in all cell lines analyzed.
To analyze the specificity of p53 binding to the sites that we recovered from the libraries, we performed Q-RT-PCR on ChIP DNA fragments using primers specific to regions containing the binding sites identified from our screen, as well as other putative binding sites within 20 kb upstream or downstream of the start of exon 1 identified by the p53MH algorithm (34), but not recovered from our screen (Fig. 4). Cells were treated as outlined in the Fig. 3 legend, and the ChIP DNA was analyzed by Q-RT-PCR. All samples were normalized to a sample of genomic DNA from treated, cross-linked, and sonicated lysates that were not chromatin immunoprecipitated, and the results are shown as percentages of that input. Schematics showing the structure of each gene and the locations of binding sites that were either identified through the library screen, previously reported, or identified using the p53MH algorithm are shown in the right panels of Fig. 4. Also shown for these sites is their conservation across species. For CDKN1A, four potential p53 binding sites were analyzed along with the two binding sites that were recovered from the library screen. These two binding sites are the two previously reported p53 binding sites for the CDKN1A gene (16). Significant binding of p53 was observed only at both of the previously reported sites. Of note, all of the algorithm-derived, potential binding sites matched the consensus to a higher degree than the RS/LS2 site (12 of 20); however, none of the potential sites exhibited more binding than this site. For EDN-2, five potential binding sites, including the one site isolated from our screen, were analyzed. Binding was greatest at the intronic site recovered from the library screen that fit the consensus to a high degree, 18 of 20 bp. Interestingly, this site was not one of the two sites localized nearest to exon 1. For the PPM1J gene, we analyzed the site recovered from our library as well as three other potential p53 binding sites. The only sample that showed appreciable binding was that isolated from the library screen. Interestingly, both the EDN-2 and PPM1J sites isolated from the library screen are not as well conserved (only in rats) as the other binding sites analyzed for these genes. For the RPS27L gene, two potential binding sites and the site isolated by the screen were analyzed, and for the PDGFC gene, three potential sites were analyzed as well as the library-generated site. In both genes, p53 binding was seen predominantly in the library-generated sites and was observed both before and after treatment with ADR. Although several primer sets were used, the UBTD1 gene could not be reliably analyzed by Q-RT-PCR.
p53-dependent regulation of candidate target gene mRNA.
To determine if p53 binding to response elements correlated with p53-dependent transcription for the genes shown in Fig. 3, we performed Q-RT-PCR analysis on RNA isolated from three cell model systems: (i) the isogenic pair of HCT116 p53+/+ and p53−/− cells (6) treated with ADR, (ii) an inducible H1299 cell line in which p53 expression was under the control of the ecdysone promoter and induced by ponasterone A addition (HIp53), and (iii) primary cultures of HK cells infected with a p53- or GFP-expressing adenovirus (Fig. 5).
p21 and p48 are encoded by known p53 target genes; they function in cell cycle arrest and DNA repair (64), respectively, and were included in the analysis for comparison of expression between novel and known targets. p21 was induced by p53 in all cell lines examined, and p48 was regulated in only two of the three cell lines. The EDN-2 gene, a gene encoding a secreted vasoactive peptide (ET-2) with a proposed role in cell survival after hypoxia in breast epithelial cells (2, 27), was elevated in a p53-dependent manner in all cell types analyzed. PPM1J, a member of the protein phosphatase type 2C family (52), was also induced in a p53-dependent manner in all cell types. As shown in Table 1, another gene, the RHOC (ras homolog gene family, member C protein [9]) gene, is located in close proximity to the PPM1J gene and the associated p53 binding site. However, p53-dependent regulation of the RHOC gene was not observed (data not shown). The RPS27L gene, a gene similar to the 40S ribosomal subunit S27 gene (65, 78), was also regulated in a p53-dependent manner in all three systems analyzed. Similar to the p48 gene, the UBTD1 gene (9, 65) was regulated by p53 in two of the three cell systems analyzed. Another gene, the ANKRD2 (ankyrin repeat domain 2 protein [39]) gene, is located in close proximity to the UTBD1 gene and the p53 binding site; however, we did not observe p53-dependent regulation of the ANKRD2 gene (data not shown). The gene encoding PDGFC, a member of the PDGF growth factor family (44), was not regulated in a p53-dependent manner in any of the cell systems used in this study. There were no open reading frames within 20 kb upstream or downstream of the binding site, thus ruling out the possibility that a neighboring gene within this distance was the target. Our analysis of PDGFC provides additional evidence that p53 binding does not necessarily equate to transcriptional regulation. In sum, the genes analyzed can be divided into three categories: those that were p53 regulated in all cell systems (CDKN1A, EDN-2, PPM1J, and RPS27L genes), those that were regulated in a subset (DDB2 and UBTD1 genes), and those that were bound but not regulated by p53 under the conditions analyzed in this study (PDGFC gene).
DISCUSSION
Many studies focusing on target gene identification have used techniques that only provide information about gene expression but do not yield information regarding the direct/indirect nature of gene regulation. Of those studies that analyze TF binding, (e.g., ChIP-chip), typically only promoter regions are used in the analyses, excluding other genomic regions potentially bound by TFs (distal regions and enhancers). One advantage of the screen used in the present study is the ability to identify functional TF binding sites throughout the genome, not only in promoter regions, similar to a recent ChIP-based screen performed by Impey et al. (36). Our analysis of binding site locations in the human genome in relation to putative target genes showed that a majority of p53 binding sites were localized to the introns of candidate target genes, not to 5′-proximal promoters. This coincides with the prevalence of intronic binding sites in many known p53 target genes, including the MDM2 and GADD45 genes (11, 77). This intronic-binding-site characteristic is not unique to p53. When Martone et al. studied NF-κB binding to the human genome using ChIP followed by a genomic microarray of human chromosome 22, they also found a significant proportion of binding occurred in the intronic areas (48). We also recovered a binding site that was 3′ of the candidate target PPM1J gene, the first report of a functional 3′ binding site for a putative p53 target gene. These data reiterate the importance of analyzing the whole genome, not only proximal promoters, when searching for functional TF regulatory regions. A further advantage of using the ChIP-based screen described herein over a screen of random genomic DNA is that the former identifies binding sites that a TF “is” bound to at a specific time under a certain physiological condition versus a screen of random genomic DNA that only identifies fragments of DNA that a TF “can” bind to, but may not under any physiologically relevant condition. Under the conditions used to generate the libraries of DNA fragments for our screen, ADR treatment of breast epithelial cells, we recovered three known p53 target genes, the functions of which are consistent with the biological end point we observed. It was not anticipated that the screen would identify all known p53 target genes given that the ChIP performed would select only for the genomic loci that p53 was bound to after ADR treatment.
After analyzing the characteristics of the binding sites recovered in this study, we found that, for the MCF-10A library, 45% of the putative binding sites matched the consensus site at greater than or equal to 17 of 20 bp, and, for those binding sites, 56% had spacer regions of 2 bp or less. For the HMEC library, 83% of the putative binding sites obtained from the positive clones matched the consensus site at greater than or equal to 17 of 20 bp, and for those binding sites, 83% had spacer regions of 2 bp or less. In addition, ∼25% of the isolated fragments had p53 binding sites that fit the p53 consensus perfectly (a match of 20 of 20 bp). Preceding this study only two p53 target genes had been identified with binding sites that fit the consensus at 20 of 20 bp, the DDB2 and type IV collagenase genes (4, 68). Interestingly, the binding site of one of the novel target genes identified by our screen, the PPM1J gene, is a 20- of 20-bp fit to the canonical binding sequence. Although a majority of the binding sites recovered were in close concordance with the consensus binding site, highly degenerate response elements were also identified. For example, the highly degenerate, but physiologically relevant, second p53 response element in p21 (17), which matches the consensus at only 12 of 20 bp, was isolated. In addition, we observed trends involving the lengths of the binding site spacer regions; a majority of the novel binding sites contained a 0- to 2-bp spacer, indicating that p53 binding to genomic DNA under the conditions assayed was more prevalent at sites that have short spacers or none at all. This observation is supported by a previous finding showing that artificially constructed binding sites containing a 4- or 14-bp spacer have greater-than-90%-reduced activity in a yeast reporter assay system than did the same binding site with a 0-bp spacer (69). Our data also correlate with the characteristics of known p53 target gene binding sites, ∼70% of which contain 0- to 2-bp spacers (54).
The in vivo specificity of the binding sites recovered from our screen was evidenced by the fact that p53 preferentially bound to these sites even though some of the p53MH algorithm-derived binding sites in the region matched the consensus to an equal or higher degree, were closer to exon 1, or were more conserved. Our results show that p53 binding is not solely dependent upon the fit of the binding site to the consensus, and matching the consensus is not sufficient for p53 binding. Other factors could also play a role in dictating binding site specificity, including the sequence context surrounding the binding site as well as access of p53 to the DNA due to chromatin structure. In addition, posttranslational modifications of the p53 protein, such as phosphorylation and acetylation, and cofactor interactions could affect its DNA binding characteristics (5, 12).
In our analyses, we highlighted four novel p53 target genes involved in diverse signaling pathways, each potentially contributing to our further understanding of p53 biology. EDN-2 is a member of the endothelin family, first discovered as potent vasoactive peptides (2). The protein ET-2 can function as an autocrine survival factor in response to hypoxia in breast cells (27) and as a macrophage cell chemoattractant (28). The identification of the EDN-2 gene as a p53 target gene, in combination with the findings that p53 can regulate other genes involved in growth factor and survival signaling, such as genes encoding HB-EGF (heparin-binding epidermal growth factor) (20), EGFR (epidermal growth factor receptor) (46), HGF (hepatocyte growth factor) (49), TGF-α (transforming growth factor alpha) (61), and, most recently, p53CSV (p53-inducible cell survival factor) (53), strongly suggests that, in addition to the well-defined role p53 plays in promoting cell viability through cell cycle arrest and DNA repair, the protein can also play an active role in initiating survival pathway signaling in the cell.
The identification of the gene encoding PPM1J, a PP2C family member, as a p53 target gene contributes to the growing list of phosphatase genes regulated by p53, including the PTEN (62), DUSP5 (70), PAC1 (75), and WIP-1 (21) genes. Interestingly, WIP-1, also known as PPM1D, is also a family member of the protein phosphatase 2C family and inhibits UV-induced apoptosis by negatively regulating the p38 mitogen-activated protein kinase-p53 signaling pathway (67). The exact role of PPM1J in human cells is not known, but its gene is conserved in mice and rats, and the murine homolog, PP2CZ, can associate with the ubiquitin-conjugating enzyme 9 (Ubc9). Ubc9 is involved in the sumoylation of protein substrates, and the PP2CZ-Ubc9 interaction is enhanced in the presence of the small ubiquitin-related modifier (SUMO) protein (38). These observations suggest that PPM1J may be involved in the regulation of sumoylation processes in the cell. Since sumoylation regulates the localization and activity of proteins involved in numerous cellular functions including transcription, DNA repair, and chromatin structure (26), the involvement of p53 in the regulation of the gene could dramatically impact cellular dynamics.
Another gene identified as a p53 target in this study was the RPS27L gene (65, 78). Although the function of human RPS27L is unknown, data show that ARS27A, one of the three ribosomal protein S27 homologues in Arabidopsis thaliana, is involved in genotoxic stress-dependent mRNA degradation. Loss of ARS27A gene expression in Arabidopsis thaliana leads to hypersensitivity to methyl methanesulfate treatment that is accompanied by inhibition of seedling growth and formation of tumor-like structures instead of auxiliary roots. The ARS27A protein is not required for translation; instead it is needed for the degradation of damaged mRNA in response to UV (56). It is possible that the RPS27L gene could play a similar role in humans. Recently, another p53 target gene, encoding MCG10, a KH-domain containing RNA-binding protein, has been identified (80). These genes link p53 to pathways involved in RNA binding and regulation, and this aspect of p53 function has yet to be explored.
As mentioned previously, we identified candidate target genes whose functions are currently unknown. One such example is the UBTD1 gene, a gene encoding a protein containing an ubiquitin domain (9, 65). p53 is well linked to the ubiquitination pathway through its transcriptional regulation of four ubiquitin ligases, MDM2 (31, 42), COP1 (13), Siah1b (22), and Pirh2 (43). However, based on the limited information available for UBTD1, we cannot speculate on the role its gene may play in p53 signaling. Also, the UBTD1 gene is regulated in only a subset of the cell model systems assayed. This example illustrates the discriminatory nature of p53 target gene selectivity, which is dependent on various factors including, but not limited to, the cell types analyzed and the specific signals leading to p53 activation.
A number of candidate target genes identified in our screen were not regulated in a p53-dependent manner, although we validated binding to DNA regions adjacent to the genes through additional ChIP analyses (the PDGFC gene being the example shown). There are many potential reasons for this observed lack of transcriptional regulation. For instance, it is likely that p53-dependent expression of these genes could be tissue/cell type specific and/or specific to the type of cellular stress activating p53, which has been reported from other studies analyzing the regulation of p53-inducible genes (76, 79). In addition, it is possible that p53 regulates antisense or noncoding RNAs not analyzed in this study. The transcriptome consists of an RNA population that is larger than what would be expected if only taking into account the protein-coding transcripts (10, 37, 57, 60). By analyzing a subset of the novel transcripts in the transcriptome, it was concluded that they represent RNA that is most likely not protein encoding, and some of the transcripts even had an orientation that was antisense to a known protein-coding region (37). In addition, when Cawley et al. examined TF (Sp1, cMyc, and p53) binding on chromosomes 21 and 22 using high-density oligonucleotide arrays, they found that many binding sites were significantly correlated with noncoding RNAs. They concluded that there are about the same number of protein-coding and noncoding genes in the transcriptome and that these two types of genes can be bound and regulated by common TFs (8). Alternatively, p53 occupancy at certain response elements (without measurable gene regulation) may affect chromatin remodeling in select regions of the genome. It is well established that p53 is required for global genomic repair in the nucleotide excision repair pathway following DNA damage (29). Further, p53-dependent global chromatin relaxation occurs in response to low doses of UV that are not high enough to activate the protein's activity as a transcription factor (45, 58, 59, 74). Also, p53 can impact histone H3 posttranslational modifications under normal cellular conditions (1). These data suggest that p53 can play a role in chromatin relaxation and remodeling.
The lack of identification of p53-repressed genes was not surprising, given that the majority of p53-repressed genes lack a consensus p53 binding site (33) and thus would not have been isolated. Also, we did not identify DNA fragments adjacent to genes involved in apoptotic signaling, consistent with our finding that ADR (350 nM) does not induce apoptosis in HMEC and MCF-10A cells and the findings of other studies showing lack of constitutive p53 binding to proapoptotic genes (18, 66).
The screen presented herein can be used to directly identify novel genes regulated by other TFs of interest (that maintain activity in yeast) on a genome-wide scale. Comparative analyses of functional binding sites occupied by transcriptional activators under various cellular conditions can be performed with this type of screen. As more genomic, proteomic, and bioinformatic advances are made, the hope is that ChIP-based methods will allow further characterization of protein complexes and chromatin structure at binding sites identified by methods such as those described in this study.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health grants (CA70856, CA105436 [J.A.P], ES00267, and CA68485 [Core Services]), The Komen Foundation, and a U.S. Army grant (DAMD17-02-1-0605 [J.M.H.]).
We thank the members of the Pietenpol laboratory for helpful discussions and critical reading of the manuscript.
Footnotes
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Allison, S. J., and J. Milner. 2003. Loss of p53 has site-specific effects on histone H3 modification, including serine 10 phosphorylation important for maintenance of ploidy. Cancer Res. 63:6674-6679. [PubMed] [Google Scholar]
- 2.Bagnato, A., and P. G. Natali. 2004. Targeting endothelin axis in cancer. Cancer Treat. Res. 119:293-314. [DOI] [PubMed] [Google Scholar]
- 3.Band, V., and R. Sager. 1989. Distinctive traits of normal and tumor-derived human mammary epithelial cells expressed in a medium that supports long-term growth of both cell types. Proc. Natl. Acad. Sci. USA 86:1249-1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bian, J., and Y. Sun. 1997. Transcriptional activation by p53 of the human type IV collagenase (gelatinase A or matrix metalloproteinase 2) promoter. Mol. Cell. Biol. 17:6330-6338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bode, A. M., and Z. Dong. 2004. Post-translational modification of p53 in tumorigenesis. Nat. Rev. Cancer 4:793-805. [DOI] [PubMed] [Google Scholar]
- 6.Bunz, F., P. M. Hwang, C. Torrance, T. Waldman, Y. Zhang, L. Dillehay, J. Williams, C. Lengauer, K. W. Kinzler, and B. Vogelstein. 1999. Disruption of p53 in human cancer cells alters the responses to therapeutic agents. J. Clin. Investig. 104:263-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burke, D., D. Dawson, and T. Stearns. 2000. Yeast colony PCR protocol, p. 141-142. In D. Burke, D. Dawson, and T. Stearns (ed.), Methods in yeast genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 8.Cawley, S., S. Bekiranov, H. H. Ng, P. Kapranov, E. A. Sekinger, D. Kampa, A. Piccolboni, V. Sementchenko, J. Cheng, A. J. Williams, R. Wheeler, B. Wong, J. Drenkow, M. Yamanaka, S. Patel, S. Brubaker, H. Tammana, G. Helt, K. Struhl, and T. R. Gingeras. 2004. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell 116:499-509. [DOI] [PubMed] [Google Scholar]
- 9.Chardin, P., P. Madaule, and A. Tavitian. 1988. Coding sequence of human rho cDNAs clone 6 and clone 9. Nucleic Acids Res. 16:2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen, J., M. Sun, S. Lee, G. Zhou, J. D. Rowley, and S. M. Wang. 2002. Identifying novel transcripts and novel genes in the human genome by using novel SAGE tags. Proc. Natl. Acad. Sci. USA 99:12257-12262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chin, P. L., J. Momand, and G. P. Pfeifer. 1997. In vivo evidence for binding of p53 to consensus binding sites in the p21 and GADD45 genes in response to ionizing radiation. Oncogene 15:87-99. [DOI] [PubMed] [Google Scholar]
- 12.Coutts, A. S., and N. B. La Thangue. 2005. The p53 response: emerging levels of co-factor complexity. Biochem. Biophys. Res. Commun. 331:778-785. [DOI] [PubMed] [Google Scholar]
- 13.Dornan, D., I. Wertz, H. Shimizu, D. Arnott, G. D. Frantz, P. Dowd, K. O'Rourke, H. Koeppen, and V. M. Dixit. 2004. The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature 429:86-92. [DOI] [PubMed] [Google Scholar]
- 14.El-Deiry, W. S. 1998. Regulation of p53 downstream genes. Cancer Biol. 8:345-357. [DOI] [PubMed] [Google Scholar]
- 15.el-Deiry, W. S., S. E. Kern, J. A. Pietenpol, K. W. Kinzler, and B. Vogelstein. 1992. Definition of a consensus binding site for p53. Nat. Genet. 1:45-49. [DOI] [PubMed] [Google Scholar]
- 16.El-Deiry, W. S., T. Tokino, V. E. Velculescu, D. B. Levy, R. Parsons, J. M. Trent, D. Lin, W. E. Mercer, K. W. Kinzler, and B. Vogelstein. 1993. WAF1, a potential mediator of p53 tumor suppression. Cell 75:817-825. [DOI] [PubMed] [Google Scholar]
- 17.el-Deiry, W. S., T. Tokino, T. Waldman, J. D. Oliner, V. E. Velculescu, M. Burrell, D. E. Hill, E. Healy, J. L. Rees, S. R. Hamilton, et al. 1995. Topological control of p21WAF1/CIP1 expression in normal and neoplastic tissues. Cancer Res. 55:2910-2919. [PubMed] [Google Scholar]
- 18.Espinosa, J. M., R. E. Verdun, and B. M. Emerson. 2003. p53 functions through stress- and promoter-specific recruitment of transcription initiation components before and after DNA damage. Mol. Cell 12:1015-1027. [DOI] [PubMed] [Google Scholar]
- 19.Euskirchen, G., T. E. Royce, P. Bertone, R. Martone, J. L. Rinn, F. K. Nelson, F. Sayward, N. M. Luscombe, P. Miller, M. Gerstein, S. Weissman, and M. Snyder. 2004. CREB binds to multiple loci on human chromosome 22. Mol. Cell. Biol. 24:3804-3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fang, L., G. Li, G. Liu, S. W. Lee, and S. A. Aaronson. 2001. p53 induction of heparin-binding EGF-like growth factor counteracts p53 growth suppression through activation of MAPK and PI3K/Akt signaling cascades. EMBO J. 20:1931-1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fiscella, M., H. Zhang, S. Fan, K. Sakaguchi, S. Shen, W. E. Mercer, G. F. Vande Woude, P. M. O'Connor, and E. Appella. 1997. Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. Proc. Natl. Acad. Sci. USA 94:6048-6053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fiucci, G., S. Beaucourt, D. Duflaut, A. Lespagnol, P. Stumptner-Cuvelette, A. Geant, G. Buchwalter, M. Tuynder, L. Susini, J. M. Lassalle, C. Wasylyk, B. Wasylyk, M. Oren, R. Amson, and A. Telerman. 2004. Siah-1b is a direct transcriptional target of p53: identification of the functional p53 responsive element in the siah-1b promoter. Proc. Natl. Acad. Sci. USA 101:3510-3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flatt, P. M., J. O. Price, A. Shaw, and J. A. Pietenpol. 1998. Differential cell cycle checkpoint response in normal human keratinocytes and fibroblasts. Cell Growth Differ. 9:535-543. [PubMed] [Google Scholar]
- 24.Flatt, P. M., L. J. Tang, C. D. Scatena, S. T. Szak, and J. A. Pietenpol. 2000. p53 regulation of G2 checkpoint is retinoblastoma protein dependent. Mol. Cell Biol. 20:4210-4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gietz, R. D., and R. A. Woods. 2002. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 350:87-96. [DOI] [PubMed] [Google Scholar]
- 26.Gill, G. 2004. SUMO and ubiquitin in the nucleus: different functions, similar mechanisms? Genes Dev. 18:2046-2059. [DOI] [PubMed] [Google Scholar]
- 27.Grimshaw, M. J., S. Naylor, and F. R. Balkwill. 2002. Endothelin-2 is a hypoxia-induced autocrine survival factor for breast tumor cells. Mol. Cancer Ther. 1:1273-1281. [PubMed] [Google Scholar]
- 28.Grimshaw, M. J., J. L. Wilson, and F. R. Balkwill. 2002. Endothelin-2 is a macrophage chemoattractant: implications for macrophage distribution in tumors. Eur. J. Immunol. 32:2393-2400. [DOI] [PubMed] [Google Scholar]
- 29.Hanawalt, P. C. 2001. Controlling the efficiency of excision repair. Mutat. Res. 485:3-13. [DOI] [PubMed] [Google Scholar]
- 30.Harms, K., S. Nozell, and X. Chen. 2004. The common and distinct target genes of the p53 family transcription factors. Cell Mol. Life Sci. 61:822-842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haupt, Y., R. Maya, A. Kazaz, and M. Oren. 1997. Mdm2 promotes the rapid degradation of p53. Nature 387:296-299. [DOI] [PubMed] [Google Scholar]
- 32.He, T. C., S. Zhou, L. T. da Costa, J. Yu, K. W. Kinzler, and B. Vogelstein. 1998. A simplified system for generating recombinant adenoviruses. Proc. Natl. Acad. Sci. USA 95:2509-2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ho, J., and S. Benchimol. 2003. Transcriptional repression mediated by the p53 tumour suppressor. Cell Death Differ. 10:404-408. [DOI] [PubMed] [Google Scholar]
- 34.Hoh, J., S. Jin, T. Parrado, J. Edington, A. J. Levine, and J. Ott. 2002. The p53MH algorithm and its application in detecting p53-responsive genes. Proc. Natl. Acad. Sci. USA 99:8467-8472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Horak, C. E., M. C. Mahajan, N. M. Luscombe, M. Gerstein, S. M. Weissman, and M. Snyder. 2002. GATA-1 binding sites mapped in the beta-globin locus by using mammalian chIp-chip analysis. Proc. Natl. Acad. Sci. USA 99:2924-2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Impey, S., S. R. McCorkle, H. Cha-Molstad, J. M. Dwyer, G. S. Yochum, J. M. Boss, S. McWeeney, J. J. Dunn, G. Mandel, and R. H. Goodman. 2004. Defining the CREB regulon: a genome-wide analysis of transcription factor regulatory regions. Cell 119:1041-1054. [DOI] [PubMed] [Google Scholar]
- 37.Kapranov, P., S. E. Cawley, J. Drenkow, S. Bekiranov, R. L. Strausberg, S. P. Fodor, and T. R. Gingeras. 2002. Large-scale transcriptional activity in chromosomes 21 and 22. Science 296:916-919. [DOI] [PubMed] [Google Scholar]
- 38.Kashiwaba, M., K. Katsura, M. Ohnishi, M. Sasaki, H. Tanaka, Y. Nishimune, T. Kobayashi, and S. Tamura. 2003. A novel protein phosphatase 2C family member (PP2Czeta) is able to associate with ubiquitin conjugating enzyme 9. FEBS Lett. 538:197-202. [DOI] [PubMed] [Google Scholar]
- 39.Kemp, T. J., T. J. Sadusky, F. Saltisi, N. Carey, J. Moss, S. Y. Yang, D. A. Sassoon, G. Goldspink, and G. R. Coulton. 2000. Identification of Ankrd2, a novel skeletal muscle gene coding for a stretch-responsive ankyrin-repeat protein. Genomics 66:229-241. [DOI] [PubMed] [Google Scholar]
- 40.Kern, S. E., K. W. Kinzler, A. Bruskin, D. Jarosz, P. Friedman, C. Prives, and B. Vogelstein. 1991. Identification of p53 as a sequence-specific DNA-binding protein. Science 252:1708-1710. [DOI] [PubMed] [Google Scholar]
- 41.Kirmizis, A., and P. J. Farnham. 2004. Genomic approaches that aid in the identification of transcription factor target genes. Exp. Biol Med. (Maywood). 229:705-721. [DOI] [PubMed] [Google Scholar]
- 42.Kubbutat, M. H. G., S. N. Jones, and K. H. Vousden. 1997. Regulation of p53 stability by Mdm2. Nature 387:299-303. [DOI] [PubMed] [Google Scholar]
- 43.Leng, R. P., Y. Lin, W. Ma, H. Wu, B. Lemmers, S. Chung, J. M. Parant, G. Lozano, R. Hakem, and S. Benchimol. 2003. Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell 112:779-791. [DOI] [PubMed] [Google Scholar]
- 44.Li, X., A. Ponten, K. Aase, L. Karlsson, A. Abramsson, M. Uutela, G. Backstrom, M. Hellstrom, H. Bostrom, H. Li, P. Soriano, C. Betsholtz, C. H. Heldin, K. Alitalo, A. Ostman, and U. Eriksson. 2000. PDGF-C is a new protease-activated ligand for the PDGF alpha-receptor. Nat. Cell Biol. 2:302-309. [DOI] [PubMed] [Google Scholar]
- 45.Ljungman, M., and F. Zhang. 1996. Blockage of RNA polymerase as a possible trigger for u.v. light-induced apoptosis. Oncogene 13:823-831. [PubMed] [Google Scholar]
- 46.Ludes-Meyers, J. H., M. A. Subler, C. V. Shivakumar, R. M. Munoz, P. Jiang, J. E. Bigger, D. R. Brown, S. P. Deb, and S. Deb. 1996. Transcriptional activation of the human epidermal growth factor receptor promoter by human p53. Mol. Cell. Biol. 16:6009-6019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mao, D. Y., J. D. Watson, P. S. Yan, D. Barsyte-Lovejoy, F. Khosravi, W. W. Wong, P. J. Farnham, T. H. Huang, and L. Z. Penn. 2003. Analysis of Myc bound loci identified by CpG island arrays shows that Max is essential for Myc-dependent repression. Curr. Biol. 13:882-886. [DOI] [PubMed] [Google Scholar]
- 48.Martone, R., G. Euskirchen, P. Bertone, S. Hartman, T. E. Royce, N. M. Luscombe, J. L. Rinn, F. K. Nelson, P. Miller, M. Gerstein, S. Weissman, and M. Snyder. 2003. Distribution of NF-κB-binding sites across human chromosome 22. Proc. Natl. Acad. Sci. USA 100:12247-12252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Metcalfe, A. M., R. M. Dixon, and G. K. Radda. 1997. Wild-type but not mutant p53 activates the hepatocyte growth factor/scatter factor promoter. Nucleic Acids Res. 25:983-986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nakamura, Y. 2004. Isolation of p53-target genes and their functional analysis. Cancer Sci. 95:7-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nigro, J. M., R. Sikorski, S. I. Reed, and B. Vogelstein. 1992. Human p53 and CDC2Hs genes combine to inhibit the proliferation of Saccharomyces cerevisiae. Mol. Cell. Biol. 12:1357-1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ota, T., Y. Suzuki, T. Nishikawa, T. Otsuki, T. Sugiyama, R. Irie, A. Wakamatsu, K. Hayashi, H. Sato, K. Nagai, K. Kimura, H. Makita, M. Sekine, M. Obayashi, T. Nishi, T. Shibahara, T. Tanaka, S. Ishii, J. Yamamoto, K. Saito, Y. Kawai, Y. Isono, Y. Nakamura, K. Nagahari, K. Murakami, T. Yasuda, T. Iwayanagi, M. Wagatsuma, A. Shiratori, H. Sudo, T. Hosoiri, Y. Kaku, H. Kodaira, H. Kondo, M. Sugawara, M. Takahashi, K. Kanda, T. Yokoi, T. Furuya, E. Kikkawa, Y. Omura, K. Abe, K. Kamihara, N. Katsuta, K. Sato, M. Tanikawa, M. Yamazaki, K. Ninomiya, T. Ishibashi, H. Yamashita, K. Murakawa, K. Fujimori, H. Tanai, M. Kimata, M. Watanabe, S. Hiraoka, Y. Chiba, S. Ishida, Y. Ono, S. Takiguchi, S. Watanabe, M. Yosida, T. Hotuta, J. Kusano, K. Kanehori, A. Takahashi-Fujii, H. Hara, T. O. Tanase, Y. Nomura, S. Togiya, F. Komai, R. Hara, K. Takeuchi, M. Arita, N. Imose, K. Musashino, H. Yuuki, A. Oshima, N. Sasaki, S. Aotsuka, Y. Yoshikawa, H. Matsunawa, T. Ichihara, N. Shiohata, S. Sano, S. Moriya, H. Momiyama, N. Satoh, S. Takami, Y. Terashima, O. Suzuki, S. Nakagawa, A. Senoh, H. Mizoguchi, Y. Goto, F. Shimizu, H. Wakebe, H. Hishigaki, T. Watanabe, A. Sugiyama, et al. 2004. Complete sequencing and characterization of 21,243 full-length human cDNAs. Nat. Genet. 36:40-45. [DOI] [PubMed] [Google Scholar]
- 53.Park, W. R., and Y. Nakamura. 2005. p53CSV, a novel p53-inducible gene involved in the p53-dependent cell-survival pathway. Cancer Res. 65:1197-1206. [DOI] [PubMed] [Google Scholar]
- 54.Qian, H., T. Wang, L. Naumovski, C. D. Lopez, and R. K. Brachmann. 2002. Groups of p53 target genes involved in specific p53 downstream effects cluster into different classes of DNA binding sites. Oncogene 21:7901-7911. [DOI] [PubMed] [Google Scholar]
- 55.Ren, B., H. Cam, Y. Takahashi, T. Volkert, J. Terragni, R. A. Young, and B. D. Dynlacht. 2002. E2F integrates cell cycle progression with DNA repair, replication, and G2/M checkpoints. Genes Dev. 16:245-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Revenkova, E., J. Masson, C. Koncz, K. Afsar, L. Jakovleva, and J. Paszkowski. 1999. Involvement of Arabidopsis thaliana ribosomal protein S27 in mRNA degradation triggered by genotoxic stress. EMBO J. 18: 490-499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rinn, J. L., G. Euskirchen, P. Bertone, R. Martone, N. M. Luscombe, S. Hartman, P. M. Harrison, F. K. Nelson, P. Miller, M. Gerstein, S. Weissman, and M. Snyder. 2003. The transcriptional activity of human chromosome 22. Genes Dev. 17:529-540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rubbi, C. P., and J. Milner. 2003. Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. EMBO J. 22:6068-6077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rubbi, C. P., and J. Milner. 2003. p53 is a chromatin accessibility factor for nucleotide excision repair of DNA damage. EMBO J. 22:975-986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Saha, S., A. B. Sparks, C. Rago, V. Akmaev, C. J. Wang, B. Vogelstein, K. W. Kinzler, and V. E. Velculescu. 2002. Using the transcriptome to annotate the genome. Nat. Biotechnol. 20:508-512. [DOI] [PubMed] [Google Scholar]
- 61.Shin, T. H., A. J. Paterson, and J. E. Kudlow. 1995. p53 stimulates transcription from the human transforming growth factor alpha promoter: a potential growth-stimulatory role for p53. Mol. Cell. Biol. 15:4694-4701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stambolic, V., D. MacPherson, D. Sas, Y. Lin, B. Snow, Y. Jang, S. Benchimol, and T. W. Mak. 2001. Regulation of PTEN transcription by p53. Mol. Cell 8: 317-325. [DOI] [PubMed] [Google Scholar]
- 63.Stampfer, M., P. Yaswen, and J. Taylor-Papadimitriou. 2002. Culture of human mammary epithelial cells, p. 109-114. In R. I. Freshney and M. G. Freshney (ed.), Culture of epithelial cells, 2nd ed. Wiley-Liss, Inc., New York, N.Y.
- 64.Stewart, Z. A., and J. A. Pietenpol. 2001. p53 signaling and cell cycle checkpoints. Chem. Res. Toxicol. 14:243-263. [DOI] [PubMed] [Google Scholar]
- 65.Strausberg, R. L., E. A. Feingold, L. H. Grouse, J. G. Derge, R. D. Klausner, F. S. Collins, L. Wagner, C. M. Shenmen, G. D. Schuler, S. F. Altschul, B. Zeeberg, K. H. Buetow, C. F. Schaefer, N. K. Bhat, R. F. Hopkins, H. Jordan, T. Moore, S. I. Max, J. Wang, F. Hsieh, L. Diatchenko, K. Marusina, A. A. Farmer, G. M. Rubin, L. Hong, M. Stapleton, M. B. Soares, M. F. Bonaldo, T. L. Casavant, T. E. Scheetz, M. J. Brownstein, T. B. Usdin, S. Toshiyuki, P. Carninci, C. Prange, S. S. Raha, N. A. Loquellano, G. J. Peters, R. D. Abramson, S. J. Mullahy, S. A. Bosak, P. J. McEwan, K. J. McKernan, J. A. Malek, P. H. Gunaratne, S. Richards, K. C. Worley, S. Hale, A. M. Garcia, L. J. Gay, S. W. Hulyk, D. K. Villalon, D. M. Muzny, E. J. Sodergren, X. Lu, R. A. Gibbs, J. Fahey, E. Helton, M. Ketteman, A. Madan, S. Rodrigues, A. Sanchez, M. Whiting, A. C. Young, Y. Shevchenko, G. G. Bouffard, R. W. Blakesley, J. W. Touchman, E. D. Green, M. C. Dickson, A. C. Rodriguez, J. Grimwood, J. Schmutz, R. M. Myers, Y. S. Butterfield, M. I. Krzywinski, U. Skalska, D. E. Smailus, A. Schnerch, J. E. Schein, S. J. Jones, and M. A. Marra. 2002. Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proc. Natl. Acad. Sci. USA 99:16899-16903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Szak, S. T., D. Mays, and J. A. Pietenpol. 2001. Kinetics of p53 binding to promoter sites in vivo. Mol. Cell. Biol. 21:3375-3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Takekawa, M., M. Adachi, A. Nakahata, I. Nakayama, F. Itoh, H. Tsukuda, Y. Taya, and K. Imai. 2000. p53-inducible wip1 phosphatase mediates a negative feedback regulation of p38 MAPK-p53 signaling in response to UV radiation. EMBO J. 19:6517-6526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tan, T., and G. Chu. 2002. p53 binds and activates the xeroderma pigmentosum DDB2 gene in humans but not mice. Mol. Cell. Biol. 22:3247-3254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tokino, T., S. Thiagalingam, W. S. El-Deiry, T. Waldmann, K. W. Kinzler, and B. Vogelstein. 1994. p53 tagged sites from human genomic DNA. Hum. Mol. Genet. 3:1537-1542. [DOI] [PubMed] [Google Scholar]
- 70.Ueda, K., H. Arakawa, and Y. Nakamura. 2003. Dual-specificity phosphatase 5 (DUSP5) as a direct transcriptional target of tumor suppressor p53. Oncogene 22:5586-5591. [DOI] [PubMed] [Google Scholar]
- 71.Venter, J. C., M. D. Adams, E. W. Myers, P. W. Li, R. J. Mural, G. G. Sutton, H. O. Smith, M. Yandell, C. A. Evans, R. A. Holt, J. D. Gocayne, P. Amanati des, R. M. Ballew, D. H. Huson, J. R. Wortman, Q. Zhang, C. D. Kodira, X. H. Zheng, L. Chen, M. Skupski, G. Subramanian, P. D. Thomas, J. Zhang, G. L. Gabor Miklos, C. Nelson, S. Broder, A. G. Clark, J. Nadeau, V. A. McKusick, N. Zinder, A. J. Levine, R. J. Roberts, M. Simon, C. Slayman, M. Hunkapiller, R. Bolanos, A. Delcher, I. Dew, D. Fasulo, M. Flanigan, L. Florea, A. Halpern, S. Hannenhalli, S. Kravitz, S. Levy, C. Mobarry, K. Reinert, K. Remington, J. Abu-Threideh, E. Beasley, K. Biddick, V. Bonazzi, R. Brandon, M. Cargill, I. Chandramouliswaran, R. Charlab, K. Chaturvedi, Z. Deng, V. Di Francesco, P. Dunn, K. Eilbeck, C. Evangelista, A. E. Gabrielian, W. Gan, W. Ge, F. Gong, Z. Gu, P. Guan, T. J. Heiman, M. E. Higgins, R. R. Ji, Z. Ke, K. A. Ketchum, Z. Lai, Y. Lei, Z. Li, J. Li, Y. Liang, X. Lin, F. Lu, G. V. Merkulov, N. Milshina, H. M. Moore, A. K. Naik, V. A. Narayan, B. Neelam, D. Nusskern, D. B. Rusch, S. Salzberg, W. Shao, B. Shue, J. Sun, Z. Wang, A. Wang, X. Wang, J. Wang, M. Wei, R. Wides, C. Xiao, C. Yan, et al. 2001. The sequence of the human genome. Science 291:1304-1351. [DOI] [PubMed] [Google Scholar]
- 72.Westfall, M. D., D. J. Mays, J. C. Sniezek, and J. A. Pietenpol. 2003. The ΔNp63α phosphoprotein binds the p21 and 14-3-3σ promoters in vivo and has transcriptional repressor activity that is reduced by Hay-Wells syndrome-derived mutations. Mol. Cell. Biol. 23:2264-2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wilson, T. E., T. J. Fahrner, M. Johnston, and J. Milbrandt. 1991. Identification of the DNA binding site for NGFI-B by genetic selection in yeast. Science 252:1296-1300. [DOI] [PubMed] [Google Scholar]
- 74.Yamaizumi, M., and T. Sugano. 1994. u.v.-induced nuclear accumulation of p53 is evoked through DNA damage of actively transcribed genes independent of the cell cycle. Oncogene 9:2775-2784. [PubMed] [Google Scholar]
- 75.Yin, Y., Y. X. Liu, Y. J. Jin, E. J. Hall, and J. C. Barrett. 2003. PAC1 phosphatase is a transcription target of p53 in signalling apoptosis and growth suppression. Nature 422:527-531. [DOI] [PubMed] [Google Scholar]
- 76.Yu, J., L. Zhang, P. M. Hwang, C. Rago, K. W. Kinzler, and B. Vogelstein. 1999. Identification and classification of p53-regulated genes. Proc. Natl. Acad. Sci. USA 96:14517-14522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zauberman, A., D. Flusberg, Y. Haupt, Y. Barak, and M. Oren. 1995. A functional p53-responsive intronic promoter is contained within the human mdm2 gene. Nucleic Acids Res. 23:2584-2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang, Q. H., M. Ye, X. Y. Wu, S. X. Ren, M. Zhao, C. J. Zhao, G. Fu, Y. Shen, H. Y. Fan, G. Lu, M. Zhong, X. R. Xu, Z. G. Han, J. W. Zhang, J. Tao, Q. H. Huang, J. Zhou, G. X. Hu, J. Gu, S. J. Chen, and Z. Chen. 2000. Cloning and functional analysis of cDNAs with open reading frames for 300 previously undefined genes expressed in CD34+ hematopoietic stem/progenitor cells. Genome Res. 10:1546-1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhao, R. B., K. Gish, M. Murphy, Y. X. Yin, D. Notterman, W. H. Hoffman, E. Tom, D. H. Mack, and A. J. Levine. 2000. Analysis of p53-regulated gene expression patterns using oligonucleotide arrays. Genes Dev. 14:981-993. [PMC free article] [PubMed] [Google Scholar]
- 80.Zhu, J., and X. Chen. 2000. MCG10, a novel p53 target gene that encodes a KH domain RNA-binding protein, is capable of inducing apoptosis and cell cycle arrest in G2-M. Mol. Cell. Biol. 20:5602-5618. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.