

Abstract
By means of in vivo selection, transcriptomic analysis, functional verification and clinical validation, here we identify a set of genes that marks and mediates breast cancer metastasis to the lungs. Some of these genes serve dual functions, providing growth advantages both in the primary tumour and in the lung microenvironment. Others contribute to aggressive growth selectively in the lung. Many encode extracellular proteins and are of previously unknown relevance to cancer metastasis.
Metastasis is frequently a final and fatal step in the progression of solid malignancies. Tumour cell intravasation, survival in circulation, extravasation into a distant organ, angiogenesis and uninhibited growth constitute the metastatic process1. The molecular requirements for some of these steps may be tissue specific. Indeed, the proclivity that tumours have for specific organs, such as breast carcinomas for bone and lung, was noted more than a century ago2.
The identity and time of onset of the changes that endow tumour cells with these metastatic functions are largely unknown and are a subject of debate. It is believed that genomic instability generates large-scale cellular heterogeneity within tumour populations, from which rare cellular variants with augmented metastatic abilities evolve through a darwinian selection process2,3. Work on experimental metastasis with tumour cell lines has demonstrated that reinjection of metastatic cell populations can lead to enrichment in the metastatic phenotype4–6. Recently, however, the existence of genes expressed by rare cellular variants that specifically mediate metastasis has been challenged7. Transcriptomic profiling of primary human carcinomas has identified gene expression patterns that, when present in the bulk primary tumour population, predict a poor prognosis for patients8–10. The existence of such signatures has been interpreted to mean that genetic lesions acquired early in tumor-igenesis are sufficient for the metastatic process, and that consequently no metastasis-specific genes exist. However, it is unclear whether these genes predicting metastatic recurrence are also functional mediators.
The lungs and bones are frequent sites of breast cancer metastasis, and metastases to these sites differ in terms of their evolution, treatment, morbidity and mortality11. Reasoning that each organ places different demands on circulating cancer cells for the establishment of metastases, we sought to identify genes expressed in breast cancer cells that selectively mediate lung metastasis and that are correlated with the propensity of primary human breast cancers to relapse to the lungs.
Selection of cells metastatic to the lungs
The cell line MDA-MB-231 was derived from the pleural effusion of a breast cancer patient suffering from widespread metastasis years after removal of her primary tumour12. Individual MDA-MB-231 cells grown and tested as single-cell-derived progenies (SCPs) have distinct metastatic abilities and tissue tropisms13 despite having similar expression levels of genes constituting a validated Rosetta-type poor prognosis signature9 (Supplementary Fig. S1). These different meta-static behaviours, including different tropisms to bone and lung, are associated with discrete variation in overall gene expression patterns (Supplementary Fig. S1; ref. 13). We therefore proposed that organ-specific metastasis must be determined by genes that are distinct from a Rosetta-type poor prognosis signature and are differentially expressed within the MDA-MB-231 population. Indeed, previous work has shown this to be true for most of the genes linked to the activity of bone metastatic subpopulations4,13.
To identify genes that mediate lung metastasis we tested parental MDA-MB-231 cells and the 1834 sub-line (an in vivo isolate with no enhancement in bone metastatic behaviour4; Fig. 1a) by injection into the tail vein of immunodeficient mice (Fig. 1b). Metastatic activity was assayed by bioluminescence imaging (BLI) of luciferase-transduced cells as well as gross examination of the lungs at necropsy. The 1834 cells exhibited limited but significant lung metastatic activity compared with the parental population (Fig. 1b). When 1834-derived lung lesions were expanded in culture and reinoculated into mice, these cells (denoted LM1 subpopulations; Fig. 1a) showed increased lung metastatic activity. Another round of selection in vivo yielded second-generation populations (denoted LM2) that were rapidly and efficiently metastatic to the lungs (Fig. 1b). Histological analysis confirmed that LM2 lesions replaced large areas of the lung parenchyma, whereas 1834 cells exhibited intravascular growth with less extensive extravasation and parenchymal involvement (Fig. 1c). Inoculation of as few as 2 × 103 LM2 cells was sufficient for the emergence of aggressive lung metastases, whereas inoculation of 2 × 105 parental cells left only a residual, indolent population in the lungs (Fig. 1d). Furthermore, the enhancement in lung meta-static activity was tissue specific. When LM2 populations were inoculated into the left cardiac ventricle to facilitate bone metastasis, their metastatic activity was comparable to that of the parental and 1834 populations, and it was markedly inferior to that of a previously described, highly aggressive bone metastatic population (Fig. 1b).
Figure 1. Selection of breast cancer cells metastatic to lung.
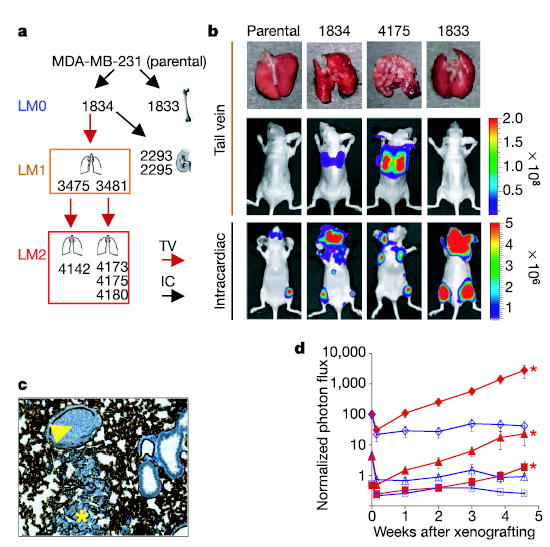
a , Flow chart of the selection of organ-specific metastatic subpopulations in vivo, indicating the organs from which these subpopulations were isolated. Each subsequent lung-metastatic generation is designated LM0, LM1 and LM2. The LM2 cells were further analysed for metastasis by either tail-vein (TV) or intracardiac (IC) xenografting. Metastatic propensities for all cell lines used in this study are listed in Supplementary Table 1. b, Representative lungs harvested at necropsy and BLI of the indicated cell lines are shown after tail-vein or intracardiac injection. The colour scale depicts the photon flux (photons per second) emitted from xenografted mice c, A representative image of haematoxylin staining of lung cryosections from mice injected with moderately metastatic 1834 cells showing an invading lesion (asterisk) and an embolus within the vascular space (arrowhead). Vascular walls are stained with the endothelial cell marker CD31. d, Parental cells (red) and 4175 (LM2) cells (blue) were tested for lung metastatic activity. Numbers of cells injected were as follows: diamonds, 2 × 105; triangles, 2 × 104; squares, 2 × 103. Plots show a quantification of the luminescence signal as a function of time. Results are means ± s.e.m. for each cohort. Asterisks, P < 0.05 with a one-sided rank test, compared with mice injected with an equivalent number of parental cells.
Establishing a lung metastasis signature
To identify patterns of gene expression associated with aggressive lung metastatic behaviour, we performed a transcriptomic micro-array analysis of the highly and weakly lung-metastatic cell populations. The gene list obtained from a class comparison between parental and LM2 populations was filtered to exclude genes that were expressed at low levels in a majority of samples and to ensure a threefold or higher change in expression level between the two groups. A total of 95 unique genes (113 probe sets) met these criteria: 48 were overexpressed and 47 underexpressed in cell populations most metastatic to the lungs (Fig. 2a and Supplementary Table 2). This gene set was largely distinct from the bone metastasis gene-expression signature previously identified in bone metastatic isolates derived from the same parental cell line4. In fact, only six genes overlapped with concordant expression patterns between the two groups (Supplementary Table 3).
Figure 2. Gene-expression signature associated with lung metastasis.
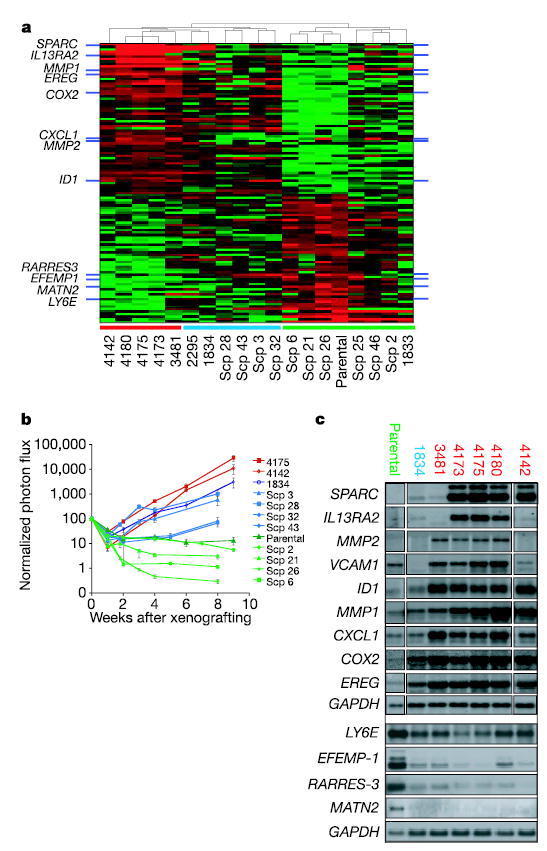
a, Comparison of gene expression profiles of LM2 populations with parental cells identifies 113 probe sets that are correlated with lung metastatic activity. This signature clusters populations selected in vivo and SCPs into groups that resemble the LM2 cell lines (red bar along the bottom), the parental MDA-MB-231 cell line (green bar) or an intermediate group (blue bar). b, LM2 populations 4175 and 4142 were assayed for lung metastatic activity as measured by BLI and were compared with parental populations and various SCPs13. Plots show a quantification of the luminescence signal as a function of time. Results are means ± s.e.m. for each cohort. Colour-coding is as in a. c, Northern blot analysis of parental, LM0, LM1 and LM2 cell lines with a set of nine lung metastasis genes selected for functional validation, as well as four genes underexpressed in the lung-metastatic populations.
Hierarchical clustering with the 95-gene list confirmed a robust relationship between this gene expression signature and the lung-specific metastatic activity of cell populations selected in vivo (Fig. 2a). In addition, this gene expression signature segregated the SCPs (which were not used in generating the gene list) into two major groups, one transcriptomically resembling the parental cells, the other more similar to the lung-metastatic populations selected in vivo. This latter group of SCPs was also more metastatic to lung than the former group (Fig. 2b). However, unlike the LM2 populations, none of the lung-metastatic SCPs concordantly expressed all of the genes in the lung metastasis signature (Fig. 2a). Consistent with this was our observation that the lung metastatic activity of the LM2 populations was about one order of magnitude greater than the most aggressive SCPs (Fig. 2b). We postulated that the subset of genes from the 95-gene signature that are uniformly expressed by all lung-metastatic SCPs and populations selected in vivo might confer baseline lung-metastatic functions, which we define as lung meta-stagenicity. Genes expressed exclusively in the most aggressive LM2 populations may serve specialized, lung-restricted functions, which we collectively describe as lung-metastatic virulence. A final list of 54 candidate lung metastagenicity and virulence genes was selected for further evaluation (Supplementary Methods and Supplementary Table 4).
Genes that mediate lung metastasis
A subset of biologically interesting genes overexpressed in the 54 gene list was selected for functional validation. These genes include those encoding the epidermal-growth-factor family member epiregulin (EREG), which is a broad-specificity ligand for the HER/ErbB family of receptors14,15, the chemokine GRO1/CXCL1 (ref. 16), the matrix metalloproteinases MMP1 (collagenase 1)17 and MMP2 (gelatinase A)18, the cell adhesion molecule SPARC19, the interleukin-13 decoy receptor IL13Rα2 (ref. 20) and the cell adhesion receptor VCAM1 (refs 21, 22) (Fig. 2a). These genes encode secretory or receptor proteins, indicating possible roles in the tumour cell microenvironment. In addition to these genes, we included the transcriptional inhibitor of cell differentiation and senescence ID1 (refs 23, 24) and the prostaglandin-endoperoxide synthase PTGS2/COX2 (ref. 25). Northern blot analysis of the various cell populations selected in vivo revealed expression patterns for these genes that were correlated with metastatic behaviour (Fig. 2c). SPARC, IL13RA2, VCAM1 and MMP2 belong to the subset of genes whose expression is generally restricted to aggressive lung-metastatic populations and are rarely expressed (less than 10% prevalence for VCAM1 and IL13Rα2, and less than 2% prevalence for SPARC and MMP2) in randomly picked SCPs (data not shown). In contrast, the expression of ID1, CXCL1, COX2, EREG and MMP1 is not restricted to aggressive lung metastasis populations but increases with lung metastatic ability. Analysis of protein expression for these genes confirmed that the differences in mRNA levels translated into significant alterations in protein levels (Supplementary Fig. S2).
To determine whether these genes have a causal function in lung metastasis, they were overexpressed by retroviral infection in the parental population either individually, in groups of three, or in groups of six (Supplementary Fig. S3). Only cells overexpressing ID1 alone were modestly more active at forming lung metastases than cells infected with vector controls (Fig. 3a). Consistent with the hypothesis that metastasis requires the concerted action of multiple effectors was our observation that combinations of these genes invariably led to more aggressive metastatic activity and that some combinations recapitulated the aggressiveness of the 4175 LM2 population (Fig. 3b). Triple combinations of lung metastasis genes in parental cells did not enhance bone metastatic activity (Supplementary Fig. S4), supporting their identity as tissue-specific mediators of metastasis. The requirement for some of these genes was tested by stably decreasing their expression in 4175 (LM2) cells with short-hairpin RNA-mediated interference (RNAi) vectors (Fig. 3c). A decrease in ID1, VCAM1 or IL13Rα2 levels decreased the lung metastatic activity of 4175 cells more than tenfold (Fig. 3d). These effects were not due to activation of the RNAi machinery, because efficient knockdown of another gene, ROBO1, did not inhibit lung metastasis formation (data not shown). Collectively, the results show that these nine genes are not only markers but also functional mediators of lung-specific metastasis.
Figure 3. Genes in the expression signature mediate lung metastasis.
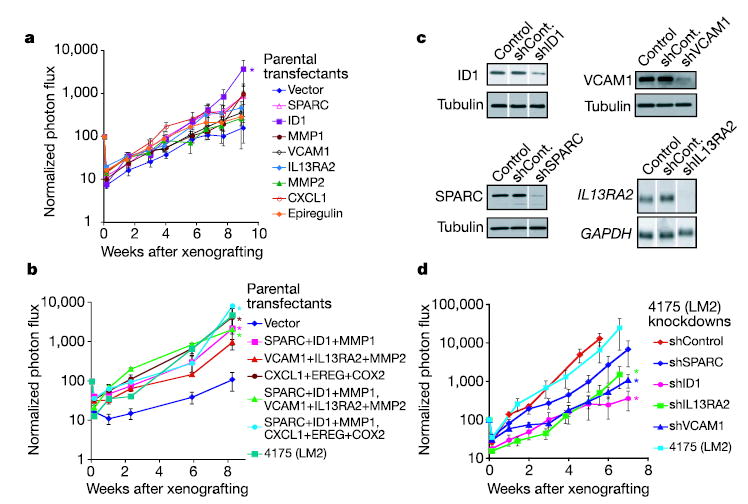
a, b, Retrovirus-mediated expression of selected genes from the lung metastasis signature in weakly metastatic parental MDA-MB-231 cells. Genes were tested individually (a) or in groups of three or six genes (b). c, Stable short hairpin (sh) RNAi constructs were introduced retrovirally into 4175 lung-metastatic cells, and their effectiveness at knocking down the expression of their intended target was validated at the protein level (ID1, VCAM1, SPARC) or at the mRNA level (IL13RA2). Controls were uninfected 4175 (LM2) cells, and shCont refers to 4175 cells transduced with a nonfunctional shRNAi. d, 4175 knockdown cell lines were xenografted through the tail vein to assess lung metastatic activity. One shRNAvector against ID1 that was ineffective at decreasing expression of this gene served as a negative control. Results are means ± s.e.m. for each cohort. Asterisks, P < 0.05 with a one-sided rank test.
Lung metastasis signature in primary tumours
A biologically meaningful and clinically relevant gene profile that mediates lung metastasis might be expressed uniquely by a subgroup of patients that suffered relapse to the lung and it should be associated with the clinical outcome. To test this, a cohort of 82 breast cancer patients treated at our institution was used in a univariate Cox proportional hazards model to relate the expression level of each lung metastasis signature gene with clinical outcome. Twelve of the 54 genes are significantly associated with lung-metastasis-free survival, including MMP1, CXCL1 and PTGS2 (Supplementary Table 5). A cross-validated multivariate analysis using a linear combination of each of the 54 genes weighted by the univariate results26 distinguished between patients with a high risk and those with a low risk for developing lung metastasis (10-year lung-metastasis-free survival of 56% versus 89%, P = 0.0018; see Supplementary Fig. S5) but not bone metastasis (70% versus 79%, P = 0.31). When a similar multivariate analysis was performed by weighting each gene by a t-statistic derived from a comparison of its expression between the LM2 cell lines with that of the parental MDA-MD-231 cells, the 54 genes again distinguished patients at high risk for developing lung metastasis (62% versus 88%, P = 0.01; see Supplementary Fig. S5) but not bone metastasis (75% versus 79%, P = 0.49). These results indicate that a clinically relevant subgroup of patients might express certain combinations of lung metastasis signature genes.
To determine directly the extent to which breast cancers express the lung metastasis signature in a manner resembling the LM2 cell lines, the 54 genes were used to cluster the Memorial Sloan-Kettering Cancer Center (MSKCC) data set hierarchically. Manual inspection of branches in the dendogram revealed a group of primary tumours that concordantly expressed many elements of this signature (Fig. 4a, dashed red box). In particular, a subgroup of primary tumours expressed to various degrees most of the nine genes that were functionally validated. Many patients who developed lung metastasis were among this group. Tumours in this group predominantly expressed markers of clinically aggressive disease, including negative oestrogen receptor (ER)/progesterone receptor status, a Rosetta-type poor-prognosis signature8, and a basal cell subtype of breast cancer27. There was no association of our signature with a high expression of HER2. A molecularly similar subgroup of breast cancer was identified when the clustering analysis was repeated on a previously published Rosetta microarray data set of breast cancer patients9 (Supplementary Fig. S6), indicating that the findings might not be unique to our cohort of patients.
Figure 4. Lung metastasis signature in human primary breast tumours.
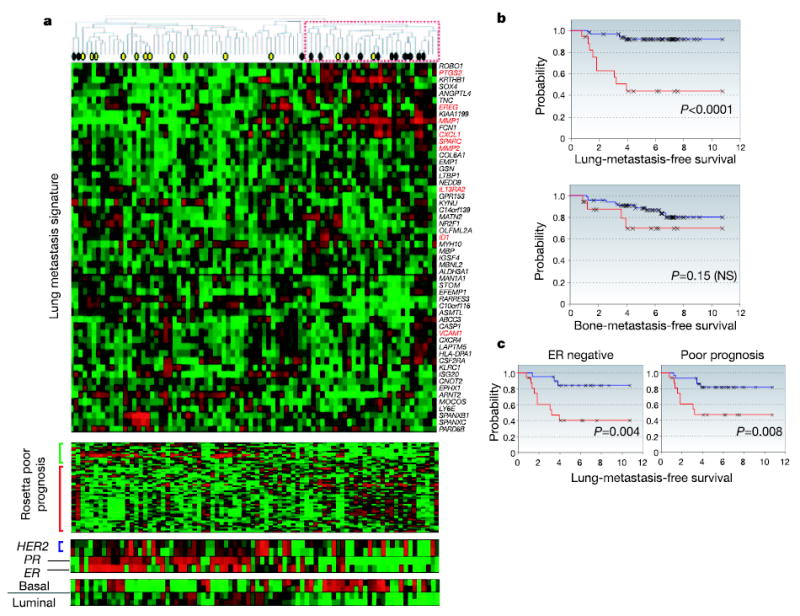
a, Hierarchical clustering of primary breast carcinomas from a cohort of 82 breast cancer patients was performed with the 54 lung metastasis signature genes. A dendrogram of the tumours is shown at the top, with tumours from patients who developed lung metastasis (black circles) or metastasis at non-pulmonary sites (yellow circles) denoted. A subcluster with a reproducibility index of 0.71 (dashed red box) groups tumours that tended to express the lung metastasis signature in a manner resembling the LM2 cell lines. The genes were also clustered; gene names are on the right. Functionally validated genes are in red. The Rosetta poor-prognosis signature is displayed with the genes underexpressed (green bar) and overexpressed (red bar) in poor-prognosis tumours indicated at the left. The expression of HER2, progesterone receptor (PR), oestrogen receptor (ER) and basal and luminal keratins is shown. Expression of the lung metastasis signature was confirmed in the independent Rosetta breast cancer cohort (Supplementary Fig. S6). b, Lung-metastasis-free survival and bone-metastasis-free survival for MSKCC patients who either expressed (red line) or did not express (blue line) the lung metastasis signature based on a classifier trained with the Rosetta cohort (Supplementary Fig. S7 and Supplementary Methods). The P value for each survival curve is shown. c, Lung-metastasis-free survival restricted to patients with ER-negative tumours or Rosetta-type poor-prognosis tumours.
Although the results of the hierarchical clustering are indicative, this approach can lead to arbitrary class assignments and is generally not ideal for class prediction28. We therefore took advantage of the repeated observation of our signature in two independent data sets. For training purposes the Rosetta data set was used to define a group of patients expressing the lung metastasis signature most resembling the LM2 cell lines (Supplementary Fig. S7). All 48 of the 54 lung metastasis genes that were shared between the MSKCC and Rosetta data set microarray platforms were subsequently used to generate a classifier to distinguish these tumours from the remaining tumours in the cohort (Supplementary Table 6). This classifier was then applied to the MSKCC cohort to identify tumours expressing the lung metastasis signature in a manner resembling the LM2 cell lines. These patients had a markedly worse lung-metastasis-free survival (P < 0.001; Fig. 4b) but not bone-metastasis-free survival (P = 0.15; Fig. 4b). These results were independent of ER status and classification as a Rosetta-type poor-prognosis tumour (Fig. 4c). Six of the nine genes that we tested in functional validation studies (MMP1, CXCL1, PTGS2, ID1, VCAM1 and EREG) were among the 18 most univariately significant (P < 0.05) genes that distinguished the patients used to train the classifier (Table 1 and Supplementary Fig. S7, cluster 3), and classification using only these 18 genes gave similar results (data not shown). The three remaining genes (SPARC, IL13RA2 and MMP2) are members of the lung metastasis virulence subset and were expressed only in the most highly metastatic cell lines in our model system (Fig. 2c).
Table 1.
Partial list of lung metastasis signature genes used to classify primary breast cancers expressing the lung metastasis signature
| UG cluster | Gene symbol | Description | P |
|---|---|---|---|
| Hs.118400 | FSCN1 | Fascin homologue 1, actin-bundling protein (Strongylocentrotus purpuratus) | <0.000001 |
| Hs.83169 | MMP1 | Matrix metalloproteinase 1 (interstitial collagenase) | <0.000001 |
| Hs.9613 | ANGPTL4 | Angiopoietin-like 4 | <0.000001 |
| Hs.74120 | C10orf116 | Chromosome 10 open reading frame 116 | 0.000006 |
| Hs.789 | CXCL1 | Chemokine (C-X-C motif) ligand 1 (melanoma growth-stimulating activity, alpha) | 0.00002 |
| Hs.196384 | PTGS2 | Prostaglandin-endoperoxide synthase 2 (prostaglandin G/H synthase and cyclooxygenase) | 0.000355 |
| Hs.185568 | KRTHB1 | Keratin, hair, basic, 1 | 0.000444 |
| Hs.109225 | VCAM1 | Vascular cell adhesion molecule 1 | 0.000506 |
| Hs.17466 | RARRES3 | Retinoic acid receptor responder (tazarotene induced) 3 | 0.000627 |
| Hs.368256 | LTBP1 | Latent transforming growth factor beta binding protein 1 | 0.001263 |
| Hs.444471 | KYNU | Kynureninase (L-kynurenine hydrolase) | 0.004365 |
| Hs.421986 | CXCR4 | Chemokine (C-X-C motif) receptor 4 | 0.005179 |
| Hs.77667 | LY6E | Lymphocyte antigen 6 complex, locus E | 0.006426 |
| Hs.410900 | ID1 | Inhibitor of DNA binding 1, dominant-negative helix–loop–helix protein | 0.007153 |
| Hs.255149 | MAN1A1 | Mannosidase, alpha, class 1A, member 1 | 0.010871 |
| Hs.388589 | NEDD9 | Neural precursor cell expressed, developmentally downregulated 9 | 0.032361 |
| Hs.115263 | EREG | Epiregulin | 0.03713 |
| Hs.98998 | TNC | Tenascin C (hexabrachion) | 0.046859 |
There are 48 unique genes shared between MSKCC and Rosetta microarray platforms. Patients from the Rosetta training set were used to define a class label for patients who either expressed or did not express the lung metastasis signature. Shown is the P value of a t-test comparing the difference in gene expression between these two classes (Supplementary Fig. S7, cluster 3). Only 18 genes with P < 0.05 are shown.
Breast tumorigenicity versus lung metastagenicity
It is unknown how and when metastasis genes are activated29. One explanation for the expression of a lung metastasis signature in a subgroup of primary breast cancers is that these genes may confer a growth advantage on the primary tumour while allowing growth at distant sites7. To test this hypothesis, MDA-MB-231 cells were injected orthotopically into the mammary fat pad of immunodeficient mice. We found that the 1834 (LM0) and 4175 (LM2) cell populations were progressively more aggressive at growing in the mammary fat pad than in the parental cell line. This was correlated with expression of lung metastagenicity genes (Figs 2c and 5a) and was not due to a general enhancement of growth because the 4175, 1834 and parental populations had comparable abilities to metastasize to bone (refer to Fig. 1b). Furthermore, the 4175 and 1834 populations were also more metastatic to the lungs from the orthotopic site after primary tumour resection, recapitulating the phenotypes observed with the tail vein metastasis assay (Fig. 5b). In contrast, the virulently bone-metastatic population 1833 (ref. 4) was only marginally more aggressive in the mammary fat pad than the parental cells and did not metastasize to lung after primary tumour resection (Fig. 5a, b).
Figure 5. Breast tumorigenicity and lung metastagenicity partially overlap.
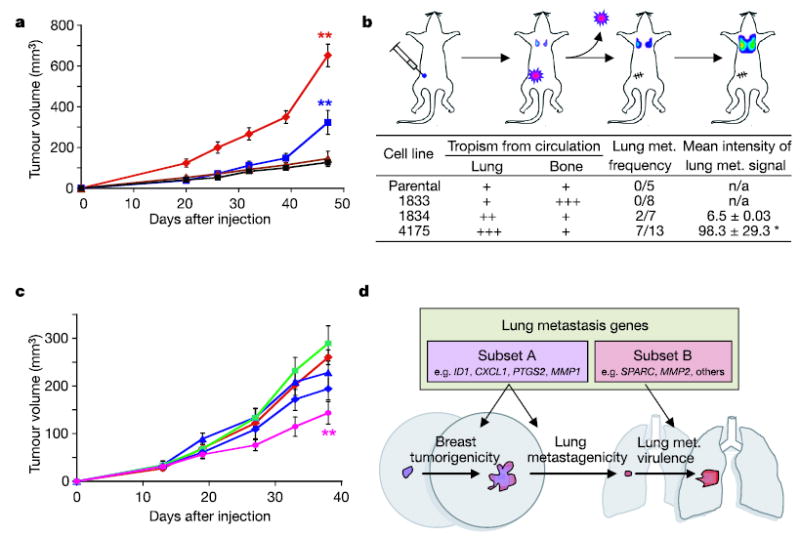
a, Representative MDA-MB-231 cell populations were injected into the mammary fat pad of immunodeficient mice and monitored for tumour growth. Red diamonds, 4175 cells (n = 9, where n is the number of mice in each cohort); blue squares, 1834 cells (n = 10); brown triangles, 1833 cells (n = 5); black squares, parental cells (n = 5). Each curve shows tumour volumes in cubic millimetres (means ± s.e.m.). b, As shown in the diagram, mice were inoculated with the indicated MDA-MB-231 cells into the mammary fat pad and tumours were removed after reaching a volume of 300 mm3. Lung metastasis was monitored with BLI, and normalized photon flux was measured 2 weeks after removal of the primary tumour. Asterisk, a mouse in the 4175 cohort with an unusually high normalized photon flux of 36,400 was excluded. c, Growth in mammary fat pad of highly lung- metastatic 4175 (LM2) cells after stable shRNA knockdown of the following gene products: red diamonds, shControl; blue triangles, shVCAM; green squares, shIL13RA2; blue diamonds, shSPARC; pink circles, shID1. shControl refers to a cell line transduced with a short hairpin construct that did not result in effective knockdown of its target gene. Two asterisks, P < 0.01 by a one-sided rank test. Each curve shows tumour volumes in cubic millimetres (means ± s.e.m.). d, A model of two classes of genes contained within the lung metastasis signature. The first class (subset A) confers both breast tumorigenicity and basal lung metastagenicity. Examples may include ID1, CXCL1, PTGS2 and MMP1. The second class (subset B) confers functions specific to the lung microenvironment, facilitating lung metastatic virulence. Examples may include SPARC and MMP2.
To identify which of the genes in the lung metastasis signature might be conferring growth at the primary tumour site, we quantified mammary-fat-pad tumour growth of 4175 cell populations with stable knockdown of various lung metastasis genes that were previously assayed for effects on metastatic behaviour (refer to Fig. 3c, d). Whereas knockdown of IL13Rα2, SPARC and VCAM1 decreased lung metastatic ability but not orthotopic tumour growth, knockdown of ID1 resulted in a statistically significant reduction in both (Figs 3d and 5c). These data indicate that some lung metastasis genes might facilitate both breast tumorigenicity and lung metastagenicity, whereas others confer growth advantages exclusively in the lung microenvironment.
Discussion
We have identified a set of genes that mediates breast cancer metastasis to lung and is clinically correlated with the development of lung metastasis when expressed in primary breast cancers. Many of the genes in this signature have not previously been linked to metastasis. Together with bone, the lung is one of the most frequent targets of breast cancer metastasis in humans. We provide evidence that these two sites impose different requirements for the establishment of metastases by circulating cancer cells. In addition to providing clinical validation, potential prognostic tools and possible targets for cancer treatment, the present findings shed new light on the biology of breast cancer metastasis.
Many of the genes in the lung metastasis signature are frequently expressed in all MDA-MB-231 subpopulations that metastasize to the lungs, regardless of whether these cells were randomly picked from the parental cell line or selected in vivo. Most of these genes, which we denote as promoting lung metastagenicity, encode extra-cellular products including growth and survival factors (for example the HER/ErbB receptor ligand epiregulin), chemokines (CXCL1), cell adhesion receptors (for example ROBO1) and extracellular proteases (MMP1). They also include intracellular enzymes (for example COX2) and transcriptional regulators (for example ID1), as well as several other downregulated genes. Their expression pattern is tightly correlated with lung metastatic activity. When tested by overexpression in poorly metastatic cells or by RNAi-mediated knockdown in highly metastatic cells, several genes in this group function as mediators of lung metastasis but not bone metastasis. Furthermore, in the cohort of human breast cancer primary tumours examined, those expressing the lung metastasis signature had a significantly poorer lung-metastasis-free survival but not bone-metastasis-free survival. This signature therefore seems to include a set of clinically relevant genes that mediate a metastagenicity function30,31 with selectivity for the lung.
Beside our data, other recent findings reveal the existence of metastasis gene signatures expressed by primary tumours. It is unclear at what point these metastasis gene signatures are acquired during the process of tumorigenesis because the selection pressure for this acquisition is unknown. One possibility is that elements of metastasis gene signatures might have a function in primary tumour growth. Consistent with this idea is the observation that the in vivo selected cell lines expressing the lung metastagenicity signature are more tumorigenic when implanted in the mammary glands of mice. Despite promoting growth in the mammary gland and in the lung, these genes are not general mediators of neoplastic growth. Many lung metastasis signature genes therefore seem to enhance growth both within the breast and the lung (Fig. 5d). These overlapping functions might explain how cells expressing genes involved in metastasis can be selected for in the primary tumour, providing insight into the interpretation of primary tumour microarray data.
Another subset of the lung metastasis genes is overexpressed only in rare, virulently metastatic cells selected in vivo. Several of these genes mediate lung metastasis in our functional assays. Many in this class encode extracellular proteins (for example SPARC and MMP2). With some exceptions (for example the receptors IL13RA2 and VCAM1) this group of genes is sporadically expressed in human primary breast tumours. We propose that these genes act mainly as virulence genes30,31 that may allow tumours to aggressively invade, colonize and grow in the lungs without markedly contributing to primary tumour growth (Fig. 5d). Thus, their expression may be rare in primary tumours but strongly selected for once such cells reach the lung. Supporting this model, a recent study analysing MMP2 expression in matched primary breast cancers and pleural effusions found that MMP2 levels are specifically enriched at the metastatic site32.
Breast cancer is a heterogeneous disease with diverse metastatic behaviour. As a consequence, patients differ widely in prognosis and survival. Attempts to classify this disease molecularly have yielded several useful markers of poor prognosis. However, to our knowledge none of these markers have yet been shown to act as functional mediators that account for the diversity of breast cancer metastases. In contrast, our lung metastasis signature seems to identify poor-prognosis patients who are at high risk of developing lung metastasis, which is consistent with the functional testing done experimentally. Further studies with additional patient cohorts, and a delineation of the role of these genes in specific steps of the metastatic process, should lead to a better understanding of the biology of metastasis and its susceptibilities to treatment.
METHODS
Cell lines
The parental MDA-MB-231 cell line was obtained from the American Type Tissue Collection. Its derivative cell lines and SCPs were described previously4. Cells were grown in high-glucose DMEM medium with 10% fetal bovine serum. For bioluminescent tracking, cell lines were retrovirally infected with a triple-fusion protein reporter construct encoding herpes simplex virus thymidine kinase 1, green fluorescent protein (GFP) and firefly luciferase13,33. GFP-positive cells were enriched by fluorescence-activated cell sorting.
Animal studies
All animal work was done in accordance with a protocol approved by the Institutional Animal Care and Use Committee. Balb/c nude mice (NCI) 4–6 weeks old were used for all xenografting studies. For lung metastasis formation, 2 × 105 viable cells were washed and harvested in PBS and subsequently injected into the lateral tail vein in a volume of 0.1 ml. Endpoint assays were conducted at 15 weeks after injection unless significant morbidity required that the mouse be euthanized earlier. For bone metastasis, 105 cells in PBS were injected into the left ventricle of anaesthetized mice (100 mg kg−1 ketamine, 10 mg kg−1 xylazine)4. Mice were imaged for luciferase activity immediately after injection to exclude any that were not successfully xenografted.
For mammary-fat-pad tumour assays, cells were harvested by trypsinization, washed twice in PBS and counted. Cells were then resuspended (107 cells ml−1) in a 50:50 solution of PBS and Matrigel. Mice were anaesthetized, a small incision was made to reveal the mammary gland and 106 cells were injected directly into the mammary fat pad. The incision was closed with wound clips and primary tumour outgrowth was monitored weekly by taking measurements of the tumour length (L) and width (W). Tumour volume was calculated as πLW2/6. For metastasis assays, tumours were surgically resected when they reached a volume greater than 300 mm3. After resection, the mice were monitored by bioluminescent imaging for the development of metastases.
Bioluminescent imaging and analysis
Mice were anaesthetized and injected retro-orbitally with 1.5 mg of d-luciferin (15 mg ml−1 in PBS). Imaging was completed between 2 and 5 min after injection with a Xenogen IVIS system coupled to Living Image acquisition and analysis software (Xenogen). For BLI plots, photon flux was calculated for each mouse by using a rectangular region of interest encompassing the thorax of the mouse in a prone position. This value was scaled to a comparable background value (from a luciferin-injected mouse with no tumour cells), and then normalized to the value obtained immediately after xenografting (day 0), so that all mice had an arbitrary starting BLI signal of 100.
RNA isolation, labelling and microarray hybridization
Methods for RNA extraction, labelling and hybridization for DNA microarray analysis of the cell lines have been described previously4. For the primary breast tumour data, tissues from primary breast cancers were obtained from therapeutic procedures performed as part of routine clinical management. Samples were snap-frozen in liquid nitrogen and stored at −80 °C. Each sample was examined histologically in cryostat sections stained with hematoxylin and eosin. Regions were dissected manually from the frozen block to provide a consistent tumour cell content of greater than 70% in tissues used for analysis. All studies were conducted under protocols approved by the MSKCC Institutional Review Board. RNA was extracted from frozen tissues by homogenization in TRIzol reagent (Gibco/BRL) and evaluated for integrity. Complementary DNA was synthesized from total RNA by using a dT primer tagged with a T7 promoter. The RNA target was synthesized by transcription in vitro and labelled with biotinylated nucleotides (Enzo Biochem). The labelled target was assessed by hybridization to Test3 arrays (Affymetrix). All gene expression analysis was performed with an HG-U133A GeneChip (Affymetrix). Gene expression was quantified with MAS 5.0 or GCOS (Affymetrix).
Statistical analysis
The Kaplan–Meier method was used to estimate survival curves, and the log-rank test was used to test for differences between curves using WinSTAT (R. Fitch Software). The site of distant metastasis for the patients in the MSKCC data set was determined from patient records. Patients with lung metastasis developed metastasis to the lung only or to the lung within months of metastasis to other sites. A detailed description of analytical methods used in the paper is provided in Supplementary Methods.
Additional procedures
Descriptions of additional experimental procedures used are given in Supplementary Methods.
Acknowledgments
We thank R. Benezra, Y. Kang, C. Hudis, L. Norton, N. Rosen and C. VanPoznak for insights and discussions, and K. Manova and the staff of the Molecular Cytology Core Facility for assistance with immunohistochemistry. A.J.M is a recipient of the Leonard B. Holman Research Pathway fellowship. G.P.G. is supported by an NIH Medical Scientist Training Program grant, a fellowship from the Katherine Beineke Foundation and a Department of Defense Breast Cancer Research Program pre-doctoral traineeship award. J.M. is an Investigator of the Howard Hughes Medical Institute. This research is supported by the W.M. Keck Foundation and an NIH grant to J.M., and a US Army Medical Research grant to W.G.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Information All microarray data have been submitted to the Gene Expression Omnibus (GEO) under accession number GSE2603. Reprints and permissions information is available at npg.nature.com/reprintsandpermissions. The authors declare no competing financial interests.
References
- 1.Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nature Rev Cancer. 2002;2:563–572. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 2.Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nature Rev Cancer. 2003;3:453–458. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 3.Yokota J. Tumor progression and metastasis. Carcinogenesis. 2000;21:497–503. doi: 10.1093/carcin/21.3.497. [DOI] [PubMed] [Google Scholar]
- 4.Kang Y, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3:537–549. doi: 10.1016/s1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- 5.Clark EA, Golub TR, Lander ES, Hynes RO. Genomic analysis of metastasis reveals an essential role for RhoC. Nature. 2000;406:532–535. doi: 10.1038/35020106. [DOI] [PubMed] [Google Scholar]
- 6.Yang J, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumour metastasis. Cell. 2004;117:927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 7.Bernards R, Weinberg RA. A progression puzzle. Nature. 2002;418:823. doi: 10.1038/418823a. [DOI] [PubMed] [Google Scholar]
- 8.van de Vijver MJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- 9.van ’t Veer LJ, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 10.Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nature Genet. 2003;33:49–54. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- 11.Solomayer EF, Diel IJ, Meyberg GC, Gollan C, Bastert G. Metastatic breast cancer: clinical course, prognosis and therapy related to the first site of metastasis. Breast Cancer Res Treat. 2000;59:271–278. doi: 10.1023/a:1006308619659. [DOI] [PubMed] [Google Scholar]
- 12.Cailleau R, Olive M, Cruciger QV. Long-term human breast carcinoma cell lines of metastatic origin: preliminary characterization. In Vitro. 1978;14:911–915. doi: 10.1007/BF02616120. [DOI] [PubMed] [Google Scholar]
- 13.Minn AJ, et al. Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J Clin Invest. 2005;115:44–55. doi: 10.1172/JCI22320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shelly M, et al. Epiregulin is a potent pan-ErbB ligand that preferentially activates heterodimeric receptor complexes. J Biol Chem. 1998;273:10496–10505. doi: 10.1074/jbc.273.17.10496. [DOI] [PubMed] [Google Scholar]
- 15.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nature Rev Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 16.Balkwill F. Cancer and the chemokine network. Nature Rev Cancer. 2004;4:540–550. doi: 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- 17.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nature Rev Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 18.Duffy MJ, Maguire TM, Hill A, McDermott E, O’Higgins N. Metalloproteinases: role in breast carcinogenesis, invasion and metastasis. Breast Cancer Res. 2000;2:252–257. doi: 10.1186/bcr65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Framson PE, Sage EH. SPARC and tumour growth: where the seed meets the soil? J Cell Biochem. 2004;92:679–690. doi: 10.1002/jcb.20091. [DOI] [PubMed] [Google Scholar]
- 20.Wood N, et al. Enhanced interleukin (IL)-13 responses in mice lacking IL-13 receptor alpha 2. J Exp Med. 2003;197:703–709. doi: 10.1084/jem.20020906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amatschek S, et al. Tissue-wide expression profiling using cDNA subtraction and microarrays to identify tumour-specific genes. Cancer Res. 2004;64:844–856. doi: 10.1158/0008-5472.can-03-2361. [DOI] [PubMed] [Google Scholar]
- 22.O’Hanlon DM, et al. Soluble adhesion molecules (E-selectin, ICAM-1 and VCAM-1) in breast carcinoma. Eur J Cancer. 2002;38:2252–2257. doi: 10.1016/s0959-8049(02)00218-6. [DOI] [PubMed] [Google Scholar]
- 23.Desprez PY, Sumida T, Coppe JP. Helix–loop–helix proteins in mammary gland development and breast cancer. J Mammary Gland Biol Neoplasia. 2003;8:225–239. doi: 10.1023/a:1025957025773. [DOI] [PubMed] [Google Scholar]
- 24.Ruzinova MB, Benezra R. Id proteins in development, cell cycle and cancer. Trends Cell Biol. 2003;13:410–418. doi: 10.1016/s0962-8924(03)00147-8. [DOI] [PubMed] [Google Scholar]
- 25.Arun B, Goss P. The role of COX-2 inhibition in breast cancer treatment and prevention. Semin Oncol. 2004;31:22–29. doi: 10.1053/j.seminoncol.2004.03.042. [DOI] [PubMed] [Google Scholar]
- 26.Beer DG, et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nature Med. 2002;8:816–824. doi: 10.1038/nm733. [DOI] [PubMed] [Google Scholar]
- 27.Perou CM, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 28.Simon R. Diagnostic and prognostic prediction using gene expression profiles in high-dimensional microarray data. Br J Cancer. 2003;89:1599–1604. doi: 10.1038/sj.bjc.6601326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hynes RO. Metastatic potential: generic predisposition of the primary tumour or rare, metastatic variants-or both? Cell. 2003;113:821–823. doi: 10.1016/s0092-8674(03)00468-9. [DOI] [PubMed] [Google Scholar]
- 30.Heimann R, Hellman S. Clinical progression of breast cancer malignant behaviour: what to expect and when to expect it. J Clin Oncol. 2000;18:591–599. doi: 10.1200/JCO.2000.18.3.591. [DOI] [PubMed] [Google Scholar]
- 31.Schairer C, Mink PJ, Carroll L, Devesa SS. Probabilities of death from breast cancer and other causes among female breast cancer patients. J Natl Cancer Inst. 2004;96:1311–1321. doi: 10.1093/jnci/djh253. [DOI] [PubMed] [Google Scholar]
- 32.Davidson B, et al. Altered expression of metastasis-associated and regulatory molecules in effusions from breast cancer patients: a novel model for tumour progression. Clin Cancer Res. 2004;10:7335–7346. doi: 10.1158/1078-0432.CCR-04-0183. [DOI] [PubMed] [Google Scholar]
- 33.Ponomarev V, et al. A novel triple-modality reporter gene for whole-body fluorescent, bioluminescent, and nuclear noninvasive imaging. Eur J Nucl Med Mol Imaging. 2004;31:740–751. doi: 10.1007/s00259-003-1441-5. [DOI] [PubMed] [Google Scholar]