

Abstract
The exon/intron architecture of genes determines whether components of the spliceosome recognize splice sites across the intron or across the exon. Using in vitro splicing assays, we demonstrate that splice-site recognition across introns ceases when intron size is between 200 and 250 nucleotides. Beyond this threshold, splice sites are recognized across the exon. Splice-site recognition across the intron is significantly more efficient than splice-site recognition across the exon, resulting in enhanced inclusion of exons with weak splice sites. Thus, intron size can profoundly influence the likelihood that an exon is constitutively or alternatively spliced. An EST-based alternative-splicing database was used to determine whether the exon/intron architecture influences the probability of alternative splicing in the Drosophila and human genomes. Drosophila exons flanked by long introns display an up to 90-fold-higher probability of being alternatively spliced compared with exons flanked by two short introns, demonstrating that the exon/intron architecture in Drosophila is a major determinant in governing the frequency of alternative splicing. Exon skipping is also more likely to occur when exons are flanked by long introns in the human genome. Interestingly, experimental and computational analyses show that the length of the upstream intron is more influential in inducing alternative splicing than is the length of the downstream intron. We conclude that the size and location of the flanking introns control the mechanism of splice-site recognition and influence the frequency and the type of alternative splicing that a pre-mRNA transcript undergoes.
Keywords: alternative splicing, bioinformatics, EST database, intron length
Pre-mRNA splicing is an essential process that accounts for many aspects of regulated gene expression. Of the ≈25,000 genes encoded by the human genome (1), >60% are believed to produce transcripts that are alternatively spliced. Thus, alternative splicing of pre-mRNAs can lead to the production of multiple protein isoforms from a single pre-mRNA, exponentially enriching the proteomic diversity of higher eukaryotic organisms (2, 3). Because regulation of this process can determine when and where a particular protein isoform is produced, changes in alternative-splicing patterns modulate many cellular activities.
The spliceosome assembles onto the pre-mRNA in a coordinated manner by binding to sequences located at the 5′ and 3′ ends of introns. Spliceosome assembly is initiated by the stable associations of the U1 small nuclear ribonucleoprotein particle with the 5′ splice site, branch-point-binding protein/SF1 with the branch point, and U2 snRNP auxiliary factor with the pyrimidine tract (4). ATP hydrolysis then leads to the stable association of U2 snRNP at the branch-point and functional splice-site pairing (5).
Intron size has been correlated with rates of evolution (6) and the regulation of genome size (7, 8). The exon/intron architecture has also been shown to influence splice-site recognition (9–11). For example, increasing the size of mammalian exons results in exon skipping. However, the same enlarged exons were included when the flanking introns were small (11). Thus, splice-site recognition is more efficient when introns or exons are small. Because, in the human genome, the majority of exons are short and introns are long (12), it is expected that the vast majority of splice sites in the human genome are recognized across the exon. Lower eukaryotes have a genomic architecture that is typified by small introns and flanking exons with variable length, suggesting that splice-site recognition occurs across the intron (10, 13, 14). Consistent with this model, expansion of small introns in yeast or Drosophila causes loss of splicing, cryptic splicing, or intron retention (9, 15). Taken together, these observations suggest that splice sites are recognized across an optimal nucleotide length.
It is unknown whether splice-site recognition across the intron or across the exon results in similar efficiencies of spliceosomal assembly and/or splice-site pairing. Here, we demonstrate that splice-site recognition across the intron ceases when the intron reaches a length between 200 and 250 nt. Because splice-site recognition is more efficient across the intron, alternative splicing is less likely for exons flanked by short introns. This influence is supported experimentally and by computational analyses of Drosophila and human alternative-splicing databases. We conclude that the size and location of the flanking introns control the mechanism of splice-site recognition and influence the frequency and the type of alternative pre-mRNA splicing.
Methods
RNA and Splicing Reactions. A detailed description of the construction of the two-exon (A–D) and the three-exon (L/L–S/S) pre-mRNA substrates is summarized in Supporting Text, which is published as supporting information on the PNAS web site. All plasmids were linearized with XhoI, in vitro transcribed with SP6 RNA polymerase (Promega), uniformly labeled with 32P, and gel purified on 7 M urea polyacrylamide gel. In vitro splicing reactions were performed in 30% HeLa nuclear extract as described in ref. 16. Bands were visualized and quantitated by using PhosphorImager analysis and quantity one software (Bio-Rad). Percent spliced is defined as spliced products/(unspliced RNA + spliced products). To derive kinetic rate constants, time points were fit to a first-order rate description for product appearance. Transfection experiments with Lipofectamine (Life Technologies) were performed in HeLa cells grown in MEM supplemented with 2 mM glutamine and 10% FBS according to manufacturer protocols. Each splicing experiment was repeated at least three times.
Computational Analysis. The computational analysis was based on the Alternative Splicing Database (ASD) (17). ASD is a computer-generated data set of transcript-confirmed splice patterns, alternative-splice events, and the associated annotations. ASD is downloadable and provides statistics and coverage similar to other databases analyzing alternative splicing (18, 19). We used a large EST database to determine the frequency of alternative splicing, because it included alternative-splicing information for not only exon-skipping events but also alternative 3′ and 5′ splice-site usage and other complex alternative-splicing events. For the human genome, ASD records ≈26,000 alternative 5′ or 3′ splice-site events and ≈12,000 exon-skipping events for 137,197 confirmed exons (17). For Drosophila, ASD records 1,332 exon-skipping events and 1,563 alternative 5′ or 3′ splice-site events for 40,758 confirmed exons. For each species analyzed, the reference transcript-structure file and the alternative-splicing-events files were downloaded from ASD. Perl scripts were used to parse the transcript-structure and alternative-splicing files to produce an output file that lists all exons, the length of their upstream and downstream flanking introns, and their mode of alternative splicing. We then estimated the conditional probability of an exon's being alternatively spliced, given various length categorizations of its flanking introns. Because this correlation was performed independent of splice-site strength or the presence of splicing enhancers or silencers, the incidence of alternative splicing in each category was normalized to the total number of exons in each data set. For exon skipping, an exon was considered to be alternatively spliced if it was absent in a given construct, but both its upstream and downstream exons were present. For alternative 5′ and 3′ splicing events, an exon was considered to be alternatively spliced if its length was increased or reduced, and both its upstream and downstream exons were present. Initial and terminal exons were removed from the analysis.
Results
Switch of Splice-Site Recognition from Cross-Intron Interactions to Cross-Exon Interactions. Previous studies have suggested that genes with small introns tend to be recognized across the intron, and genes with large introns are recognized across the exon (11). To determine the distance at which recognition of splice sites switches from cross-intron interactions to cross-exon interactions occurs, we took advantage of an in vitro kinetic splicing assay that was originally used to demonstrate that exonic splicing enhancers (ESEs) activate both splice sites of an exon simultaneously (16). We designed a series of pre-mRNAs with intron lengths ranging from 120 to 425 nt. Within each set, the pre-mRNAs differ only in the presence or absence of a well characterized 13-nt ESE derived from the Drosophila doublesex and Drosophila fruitless pre-mRNAs (Fig. 1A). Each pre-mRNA harbors the same weak 5′ and 3′ splice sites that require the activities of ESEs for recognition in their natural context (20, 21). Because splicing factors present in HeLa cell nuclear extracts activate the ESEs used (21), the presence of functional or mutant enhancer elements within each test substrate determine its splicing efficiency. If the splice sites are recognized across the exon, it is expected that the activation of the splice sites on each exon constitutes a different step during spliceosomal assembly, because the ESE located on each exon will only aid in the recognition of its weak splice site. Thus, the activities of the separate ESEs are expected to display synergistic kinetics, because the activation of each ESE accelerates an independent step during spliceosomal assembly. However, if the splice sites are recognized across the intron, the ESE located on each exon will aid in the recognition of both weak splice sites, because the recruited spliceosomal components define the entire intron within one step. In this scenario, the activities of the separate ESEs are expected to display additive kinetics, because the activation of each ESE accelerates the same rate-limiting step during spliceosomal assembly.
Fig. 1.

Switch from splice-site recognition across the intron to splice-site recognition across the exon. (A) Schematic illustration of the pre-mRNAs used. Boxes represent exons. Each pre-mRNA is identical, except for the presence or absence of an ESE sequence and the intron size. ESEs are depicted as black boxes. Intron size was varied from 120 to 425 nt. (B) Representative autoradiogram depicting the efficiency of splicing for substrates A, B, C, and D, with an intron length of 120 nt, after a 90-min incubation. The diagrams (Right) denote pre-mRNA, spliced product, and the lariat intermediate. (C) Bar graph displaying the fold activation of observed rate constants for each set of four constructs at various intron lengths (Table 1). The switch from splice-site recognition across the intron to splice-site recognition across the exon is indicated by the change from gray to white bars.
In vitro splicing assays were performed with each of the four pre-mRNA sets over a 3-h time course to determine the apparent rates of splicing (see Fig. 5, which is published as supporting information on the PNAS web site). As illustrated for a single time point, pre-mRNAs with an intron size of 120 nt display additive kinetics (Fig. 1B). Using Drosophila nuclear extract (Kc), we were also able to demonstrate additive kinetics for substrates containing the 120-nt intron; however, we were unable to detect sufficient splicing for our substrates containing longer introns (data not shown). These results are consistent with previous in vitro studies demonstrating that splicing of pre-mRNAs with long introns is supported in HeLa nuclear extract but not in Kc extract (15, 22, 23). The results from all rate experiments are summarized in Fig. 1C and Table 1. The kinetics of pre-mRNAs containing an intron 200 nt or less in length were observed to be additive. This behavior indicates that the spliceosomal components required for the recognition of both splice sites are recruited to the intron simultaneously. However, constructs with introns >200 nt demonstrated synergistic kinetics. We conclude that the change from splice-site recognition across the intron to splice-site recognition across the exon occurs when the intronic length is between 200 and 250 nt.
Table 1. Rate constants and fold activation for test substrates at various intron lengths.
| Substrate
|
||||
|---|---|---|---|---|
| Intron length | A | B | C | D |
| 120-nt | 12 ± 7 | 42 ± 15 | 44 ± 23 | 65 ± 13 |
| Fold activation | 1 | 3.5 | 3.7 | 5.4 |
| Normalized | 18.5 | 65 | 68 | 100 |
| 200-nt | 1.4 ± 0.9 | 45 ± 3 | 62 ± 11 | 94 ± 19 |
| Fold activation | 1 | 32 | 44 | 67 |
| Normalized | 1.5 | 48 | 65 | 100 |
| 250-nt | 0.9 ± 0.4 | 6.4 ± 4 | 12 ± 2 | 45 ± 6 |
| Fold activation | 1 | 7 | 13 | 50 |
| Normalized | 2 | 14 | 26 | 100 |
| 340-nt | 0.5 ± 0.1 | 14 ± 3 | 12 ± 6 | 60 ± 17 |
| Fold activation | 1 | 28 | 24 | 120 |
| Normalized | 0.8 | 23 | 20 | 100 |
| 425-nt | 0.3 ± 0.1 | 5 ± 1 | 1.9 ± 0.2 | 50 ± 8 |
| Fold activation | 1 | 17 | 6.3 | 165 |
| Normalized | 0.6 | 10 | 4 | 100 |
All rate constants were determined from time points taken over a 3-h splicing reaction and are expressed in units of (10-2·hr-1). Fold activations were normalized to construct D. Experimental error for each rate determination was within 20%. Rates determined from different experiments varied <50%.
Mechanisms of Splice-Site Recognition and Alternative Splicing. The kinetic analysis summarized in Fig. 1 demonstrates that the upstream 5′ splice site and the downstream 3′ splice site are recognized simultaneously across introns <200 nt. Significantly, in the absence of ESEs, splice-site recognition across the intron is a much more efficient process than splice-site recognition across the exon (Fig. 2). Thus, splice-site recognition across the intron may be able to rescue the inclusion of internal exons harboring weak splice sites. To test this hypothesis, we designed a series of pre-mRNA substrates containing three exons for in vitro splicing analysis in which the internal exon contains splice sites that are insufficiently recognized in the absence of ESEs. The four substrates generated differed only in their ability to be recognized across each intron by changing the length of the intron from <200 to >250 nt (Fig. 3A), thus permitting or discouraging splice-site recognition across the intron. As expected, the internal exon is predominantly excluded when flanked by two long introns (Fig. 3B, substrate L/L, lane 2). However, we observed significant inclusion of the internal exon if one of the flanking introns is short enough to support splice-site recognition across the intron (Fig. 3B, substrates S/L, S/S, and L/S, lanes 4, 6, and 8). In fact, two short introns increase exon inclusion ≈30 times greater than two long introns (Fig. 3B, substrate S/S, lane 6).
Fig. 2.

Splice-site recognition across the intron is more efficient than splice-site recognition across the exon. (A) Representative autoradiogram depicting the efficiency of splicing for substrates containing weak 5′ and 3′ splice sites that differ only in the length of their intron, as indicated above each panel. The diagrams (Right) represent pre-mRNA and spliced products. (B) Quantitation of the data in A.
Fig. 3.

Intron definition promotes exon inclusion in vitro and in cell culture. (A) Schematic of three-exon pre-mRNA substrates with variable intron lengths. The intron sizes for each pre-mRNA are indicated above each intron. The internal exon contains weak splice sites and an ESE specified by a black box. (B) Representative autoradiograph displaying the efficiencies of internal-exon inclusion. Pre-mRNA and spliced mRNA products are indicated (Right). Odd-numbered lanes display input pre-mRNA. The internal exon inclusion/exclusion ratio for each substrate is shown in a logarithmic bar graph (Lower). (C) Representative RT-PCR analysis of transfection experiments displaying the efficiencies of internal-exon inclusion. (Right) Pre-mRNA and spliced mRNA products are indicated. (Lower) Quantitation of the data are shown as in B.
To further analyze the effect of intron size on alternative splicing, we examined splicing of the same pre-mRNA substrates in cell culture. Qualitatively, results similar to those seen in Fig. 3B were observed in transfection experiments (Fig. 3C). When the weak internal exon is flanked by long introns, it is predominantly excluded from the splicing pattern (Fig. 3C, lane 1). However, the presence of small neighboring introns rescued internal exon inclusion (Fig. 3C, lanes 2–4). We conclude that splice-site recognition across the intron can promote inclusion of exons flanked by weakly defined splice sites in vitro and in vivo.
Exon/Intron Architecture and Alternative Splicing in the Drosophila and Human Genomes. To estimate the fractions of splice sites that may be recognized through cross-intron interactions, we recorded the flanking-intron lengths for every internal exon within the human and Drosophila genomes. Genome information was obtained from the ASD, which contains information about the exon/intron structure and EST-verified alternative-splicing events of several thousand genes (17). Within the human genome, many exons are flanked by at least one short intron, creating two separate populations, separated roughly by the intron length that we propose to represent the transition of splice-site recognition from across the intron to across the exon (Fig. 4A). As expected from previous intron-length analyses (7, 24), a very different distribution is seen in the Drosophila genome, where ≈85% of exons are flanked by at least one short intron (Fig. 4B). An overlay of the Drosophila and human genomes demonstrates that the minimum intron length in the human genome is at the same location that demarcates the maximum intron length of the major Drosophila exon population (see Fig. 6, which is published as supporting information on the PNAS web site). This difference in genome constraint may reflect specific compositional variations between the Drosophila and human spliceosomes.
Fig. 4.

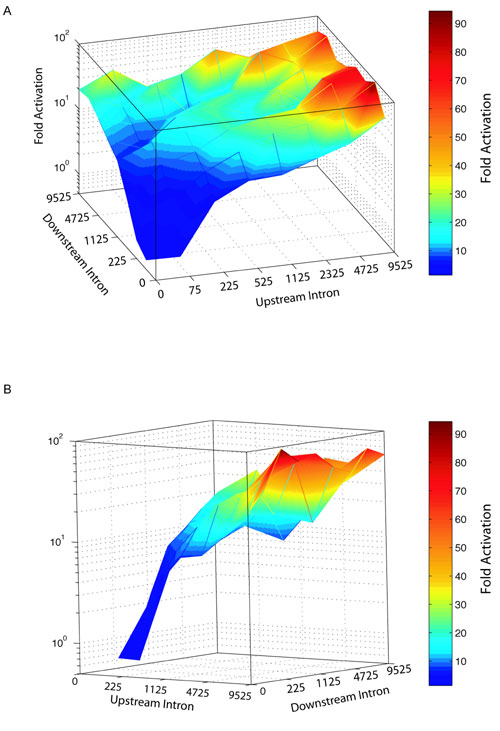
Computational analysis of intron size and alternative splicing in the Drosophila and human genomes. (A and B) Every internal exon within the human or the Drosophila genomes is displayed as a function of the nucleotide length of its upstream (x axis) and downstream (y axis) introns. Each point within the scatter plots represents a unique exon. The x and y axes are shown in log scale. (A) Exon profile of the human genome. The majority of introns are long; however, ≈25% of exons are flanked by at least one short intron. The vertical and horizontal lines demarcate the experimentally determined 200- to 250-nt transition from cross-intron to cross-exon recognition. (B) Exon profile of the Drosophila genome. The majority (>85%) of exons are flanked by at least one short intron. (C–F) Color diagrams displaying the probability of an exon's undergoing alternative splicing as a function of the length of its flanking introns. The upstream and downstream intron length increases exponentially along the x and y axes. The color scale (Right) represents the fold increase in the probability of an exon's being alternatively spliced relative to the probability calculated for exons that are flanked by introns <225 nt. (C) Probability diagram for exon skipping within the Drosophila genome. (D) Drosophila alternative 5′ or 3′ splice-site usage. (E) Human exon skipping. (F) Human alternative 5′ or 3′ splice-site usage.
Because splice-site recognition across the intron rescued exon inclusion (Fig. 3), we investigated whether intron length influences alternative splicing within the Drosophila and human genomes. To do so, the flanking-intron information of each exon was correlated with exon-skipping and alternative-splice-site-activation events reported in the ASD to compute the probability that an exon is involved in alternative splicing, without taking into consideration the contributions made by splice-site signal strength and splicing enhancers or silencers. Thus, the correlation simply tests whether the influence of the exon/intron architecture on alternative splicing is significant enough to be detectable amid all other splicing determinants. Our computational analysis of the Drosophila genome supports a significant role for intron length in defining the likelihood of alternative splicing. A striking influence of the exon/intron architecture is observed for simple exon-skipping events (Fig. 4C). Exons flanked by very long introns are up to 90-fold more likely to be skipped than exons that are flanked by two short introns. Significantly, the most drastic increase in the probability of alternative splicing (>10-fold) was observed when the length of flanking introns increased from 225 to 525 nt (Fig. 4C; for a 3D representation, see Fig. 7, which is published as supporting information on the PNAS web site). In agreement with our experimental results (Fig. 3), we observe a greater probability that an exon is alternatively spliced when the upstream intron is long. This polarity could be the consequence of coupling pre-mRNA splicing to transcription by RNA polymerase II (25). Even in the category of alternative 5′ or 3′ splice-site activation, alternative splicing is up to 10-fold more likely for exons that are flanked by long introns (Fig. 4D). We conclude that, in Drosophila, exon skipping is a rare event for exons flanked by short introns and that the length of the upstream intron is of greater importance than the length of the downstream intron in determining whether an exon will be involved in exon skipping.
Within the human genome, a similar correlation between the exon/intron architecture and the probability of exon skipping is observed; however, the ≈5-fold maximal variance calculated is significantly lower than that observed for Drosophila (Fig. 4E). As for Drosophila, the length of the upstream intron is more important in determining the frequency of alternative splicing. In the case of alternative 5′ or 3′ splice-site usage, the opposite distribution of alternative splicing is seen in the human genome (Fig. 4F). The activation of alternative splice sites is less likely if the flanking introns are long. We conclude that exon/intron architecture influences the frequency and type of alternative splicing that an exon may undergo in the Drosophila and human genomes.
Discussion
The experiments presented here support the existence of two different mechanisms for splice-site recognition, splice-site recognition across the intron, and splice-site recognition across the exon. Splice-site recognition across the intron ceases when the intron size reaches the threshold length of >200 nt. Importantly, splice-site recognition across the intron is more efficient and increases the inclusion of exons with weak splice sites. These results demonstrate that the distance between splice sites affects efficient spliceosomal assembly. Presumably, the pairing of cross-exon-defined splice sites requires the interaction between two sets of pre-spliceosomes across an intron of variable length. In contrast, splice-site recognition across the intron already identifies the splice sites that will be paired. It is also possible that the kinetics of splice-site pairing are slowed because longer introns associate with an increased number of hnRNP proteins. HnRNP proteins coat nascent pre-mRNAs and are thought to interfere with the splicing reaction (26). Therefore, larger introns may reduce splicing by decreasing the relative concentration of splicing components through competition with hnRNPs.
Additive kinetics of splice-site activation demonstrate that splice-site recognition across the intron is achieved through the recruitment of a multicomponent complex that contains components of the splicing machinery required for 5′ and 3′ splice-site definition. Interestingly, the activation of a single ESE resulted in a significant increase in splicing activity (Table 1), suggesting that ESEs influence splice-site activation of adjacent exons. As anticipated from ESE distance/activity correlations (27), this effect depended on intron length. Given the unique combination of splice sites and cis-acting elements, it is possible that the precise transition from splice-site recognition across the intron to splice-site recognition across the exon may vary for different substrates. The presence of strong splice sites and enhancers or silencers could modulate the cross-intron recognition by increasing or decreasing the strength of interaction between spliceosomal components and the pre-mRNA.
The observation that increasing exon length decreases exon inclusion (28) suggests that similar distance limitations exist for splice-site recognition across the exon. Approximately 80% of human exons are <200 bp in length, the average being 170 bp. Importantly, exon length is tightly distributed when compared with intron length (12). These results demonstrate that maintaining exon size in the human genome is more important to the architecture and evolution of a gene than is maintaining intron size. In contrast to the human genome, exon size varies much more than intron size in yeast (7). The maximum intron length of 182 nt lies well within the size limitations of splice-site recognition across the intron. Taken together, these considerations support the notion that the majority of splice sites in higher eukaryotes are recognized across the exon, whereas lower eukaryotes employ splice-site recognition across the intron (10).
It is well established that several types of exon and intron elements influence splice-site choice. The most prominent include the exon/intron junction signals and splicing enhancers and silencers (4). Our results show that the exon/intron architecture is an additional parameter that affects the efficiency of splice-site recognition and alternative pre-mRNA splicing. When compared in otherwise isogenic test substrates, splice-site recognition across the intron could rescue the inclusion of a weak internal exon by >10-fold (Fig. 3). Even though our computational analysis ignored the contributions made by variable splice sites, enhancers, and silencers, a striking increase in the probability of alternative splicing was observed for Drosophila exons, whose splice sites are recognized across the exon. Thus, the exon/intron architecture in Drosophila is a major determinant in governing the probability of alternative splicing. Within the human genome, we observed a qualitatively similar trend for exon-skipping events but with a reduced magnitude (Fig. 4). One major difference between the Drosophila and human gene architecture is intron length. Human genes are dominated by long introns (87% of introns are >250 nt), whereas short introns are much more common in Drosophila (66% are <250 nt). One possible explanation for the small intron size in Drosophila could be the pressure to maintain a constrained genome size in these fast-replicating organisms (29).
Alternative splicing is extensive in both species, supporting the argument that both species benefit from expanded proteomes generated from alternative splicing. However, our genome analysis suggests that there are significant differences in the weight of the mechanisms by which alternative splicing can be induced. In Drosophila, intron length is a major determinant in promoting alternative splicing patterns. In the human, additional mechanisms of controlling alternative splicing may have gained more influence on intron expansion to maintain balanced levels of alternative splicing.
Supplementary Material
Acknowledgments
We thank Jonathan Gruber for assistance in the computational analysis; Brent Graveley (University of Connecticut Health Center, Farmington) for Drosophila Kc extract; and Marian Waterman, Kristen Lynch, and members of the laboratory for helpful comments on the manuscript. This work was supported by National Institutes of Health (NIH) Training Grant GM 07311 (to B.J.L.) and NIH Grant GM 62287 (to K.J.H.).
Author contributions: K.L.F.-W., P.F.B., and K.J.H. designed research; K.L.F.-W. and Y.D. performed research; Y.D., B.J.L., and S.-p.H. contributed new reagents/analytic tools; K.L.F.-W., Y.D., and K.J.H. analyzed data; and K.L.F.-W. and K.J.H. wrote the paper.
Conflict of interest statement: No conflicts declared.
Abbreviations: ESEs, exonic splicing enhancers; ASD, Alternative Splicing Database.
References
- 1.International Human Genome Sequencing Consortium (2004) Nature 431, 931–945. [DOI] [PubMed] [Google Scholar]
- 2.Maniatis, T. & Tasic, B. (2002) Nature 418, 236–243. [DOI] [PubMed] [Google Scholar]
- 3.Johnson, J. M., Castle, J., Garrett-Engele, P., Kan, Z., Loerch, P. M., Armour, C. D., Santos, R., Schadt, E. E., Stoughton, R. & Shoemaker, D. D. (2003) Science 302, 2141–2144. [DOI] [PubMed] [Google Scholar]
- 4.Black, D. L. (2003) Annu. Rev. Biochem. 72, 291–336. [DOI] [PubMed] [Google Scholar]
- 5.Lim, S. R. & Hertel, K. J. (2004) Mol. Cell 15, 477–483. [DOI] [PubMed] [Google Scholar]
- 6.Marais, G., Nouvellet, P., Keightley, P. D. & Charlesworth, B. (2005) Genetics 170, 481–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deutsch, M. & Long, M. (1999) Nucleic Acids Res. 27, 3219–3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moriyama, E. N., Petrov, D. A. & Hartl, D. L. (1998) Mol. Biol. Evol. 15, 770–773. [DOI] [PubMed] [Google Scholar]
- 9.Talerico, M. & Berget, S. M. (1994) Mol. Cell. Biol. 14, 3434–3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berget, S. M. (1995) J. Biol. Chem. 270, 2411–2414. [DOI] [PubMed] [Google Scholar]
- 11.Sterner, D. A., Carlo, T. & Berget, S. M. (1996) Proc. Natl. Acad. Sci. USA 93, 15081–15085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sakharkar, M. K., Chow, V. T. & Kangueane, P. (2004) In Silico Biol. 4, 0032. [PubMed] [Google Scholar]
- 13.Guthrie, C. (1991) Science 253, 157–163. [DOI] [PubMed] [Google Scholar]
- 14.Ruby, S. W. & Abelson, J. (1991) Trends Genet. 7, 79–85. [DOI] [PubMed] [Google Scholar]
- 15.Guo, M., Lo, P. C. & Mount, S. M. (1993) Mol. Cell. Biol. 13, 1104–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lam, B. J. & Hertel, K. J. (2002) RNA 8, 1233–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thanaraj, T. A., Stamm, S., Clark, F., Riethoven, J. J., Le Texier, V. & Muilu, J. (2004) Nucleic Acids Res. 32, D64–D69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee, C., Atanelov, L., Modrek, B. & Xing, Y. (2003) Nucleic Acids Res. 31, 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leipzig, J., Pevzner, P. & Heber, S. (2004) Nucleic Acids Res. 32, 3977–3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tian, M. & Maniatis, T. (1992) Science 256, 237–240. [DOI] [PubMed] [Google Scholar]
- 21.Lam, B. J., Bakshi, A., Ekinci, F. Y., Webb, J., Graveley, B. R. & Hertel, K. J. (2003) J. Biol. Chem. 278, 22740–22747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mount, S. M., Burks, C., Hertz, G., Stormo, G. D., White, O. & Fields, C. (1992) Nucleic Acids Res. 20, 4255–4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo, M. & Mount, S. M. (1995) J. Mol. Biol. 253, 426–437. [DOI] [PubMed] [Google Scholar]
- 24.Lim, L. P. & Burge, C. B. (2001) Proc. Natl. Acad. Sci. USA 98, 11193–11198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kornblihtt, A. R., de la Mata, M., Fededa, J. P., Munoz, M. J. & Nogues, G. (2004) RNA 10, 1489–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reed, R. & Magni, K. (2001) Nat. Cell Biol. 3, E201–E204. [DOI] [PubMed] [Google Scholar]
- 27.Graveley, B. R., Hertel, K. J. & Maniatis, T. (1998) EMBO J. 17, 6747–6756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sterner, D. A. & Berget, S. M. (1993) Mol. Cell. Biol. 13, 2677–2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bingham, P. M., Chou, T. B., Mims, I. & Zachar, Z. (1988) Trends Genet. 4, 134–138. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}