

Abstract
A detailed understanding of the molecular events underlying the conversion and self-association of normally soluble proteins into amyloid fibrils is fundamental to the identification of therapeutic strategies to prevent or cure amyloid-related disorders. Recent investigations indicate that amyloid fibril formation is not just a general property of the polypeptide backbone depending on external factors, but that it is strongly modulated by amino acid side chains. Here, we propose and address the validation of the premise that the amyloidogenicity of a protein is indeed localized in short protein stretches (amyloid stretch hypothesis). We demonstrate that the conversion of a soluble nonamyloidogenic protein into an amyloidogenic prone molecule can be triggered by a nondestabilizing six-residue amyloidogenic insertion in a particular structural environment. Interestingly enough, although the inserted amyloid sequences clearly cause the process, the protease-resistant core of the fiber also includes short adjacent sequences from the otherwise soluble globular domain. Thus, short amyloid stretches accessible for intermolecular interactions trigger the self-assembly reaction and pull the rest of the protein into the fibrillar aggregate. The reliable identification of such amyloidogenic stretches in proteins opens the possibility of using them as targets for the inhibition of the amyloid fibril formation process.
Keywords: amyloid core, amyloid fibril formation, amyloid pattern, β-sheet
Over the past several years, much effort in the protein folding field has been directed toward the understanding of the factors controlling the deposition of normally soluble proteins into insoluble aggregates known as amyloid fibers. The importance of this process relies on the fact that amyloid fibril formation is associated with a range of prevalent human disorders, such as Alzheimer's disease, spongiform encephalopathies, and light chain amyloidosis (1). Under some circumstances, nonpathogenic (2) and designed peptides and proteins (3, 4) can also self-assemble into fibrils that closely resemble those formed by natural amyloid proteins. The apparent ubiquity of the amyloid feature in proteins and the common basic cross-β-structure displayed by all amyloid fibrils (5) suggests that amyloid fibril formation is a general property of the polypeptide backbone (6), which to a large extent is modulated by amino acid side chains (7).
Recent investigations indicate that the amyloidogenic capability of a protein seems to be concentrated in particular protein regions and, more specifically, in small sequence fragments therein (8–12). Small protein fragments have been identified as critical for the amyloidogenic nature of some proteins and proved to recapitulate in vitro the self-assembly (11) and cytotoxic capability of the full-length protein (13). The comparison of two homologous proteins that differ in amyloidogenic properties has served to identify short divergent sequence fragments, that once swapped into the nonamyloidogenic protein can trigger amyloid formation (8, 9). The relevance of particular stretches in protein amyloidogenesis is also in agreement with some recent models of the amyloid filament architecture (the amyloid spine model), which propose that particular protein segments, the so-called amyloid domains, stack to form a β–sheet core that is surrounded by the rest of the protein (14, 15).
The relationship between sequence and amyloid feature in short amyloid stretches has been explored in detail by using a peptide model system designed de novo by our group (4). From the full positional scanning mutagenesis of STVIIE, we have extracted a sequence pattern (amyloid pattern) that is able to identify amyloidogenic stretches shown experimentally to be fundamental for the amyloidogenicity of some natural amyloid proteins (7). This study has revealed that, even in such a short peptide, one can find positions that are tolerant (edges) or very restrictive (core) to mutation with respect to the final amyloid propensities.
The results mentioned above suggest that even a short amino acid stretch bearing a highly amyloidogenic motif might provide most of the driving force needed to trigger the self-assembly process of a protein (amyloid stretch hypothesis). Here, we report on our strategy to demonstrate this premise unambiguously: the conversion of a nonamyloidogenic protein into an amyloid-prone molecule by inserting just a six-residue amyloidogenic stretch. Previously, it has been shown that insertion of long amyloid domains (30–80 aa) from naturally occurring amyloid proteins can trigger amyloid formation of some nonamyloidogenic proteins both in vitro (16, 17) and in vivo (18). Although shorter amyloidogenic fragments (10- and 7-mer) from the N-terminus region of the yeast prion Sup35 have been used with the same aim, these fragments have been inserted either into an already amyloidogenic protein (19) or no compelling evidence has been provided in favor of the amyloid stretch hypothesis (20).
In the present work, we have chosen the α-spectrin Src homology 3 (SH3) domain (α–SH3) as a target nonamyloidogenic protein. This protein has been shown not to be amyloidogenic under any conditions tested in our laboratory (21). For amyloidogenic sequences we have selected a de novo-designed peptide, STVIIE (4), highly amyloidogenic in vitro. To validate the amyloid pattern within the context of a protein, we have designed point mutants of this sequence at different position categories (core and edges). We have also introduced an amyloidogenic six-residue fragment of the Aβ amyloid peptide identified with the amyloid pattern, 16KLVFFA21 (7). Residues 16–20 have been shown experimentally to be fundamental to the amyloidogenicity of the full-length Aβ (10). Sequences have been inserted at different positions of the protein to assess the influence of the structural environment of the sequence on its amyloidogenic capabilities.
Modified versions of the α–SH3 carrying these short amyloidogenic sequences (amyloid SH3 variants) are as stable as the WT protein, but they fibrillate under conditions where the original domain still remains soluble. Thus, the amyloidogenic behavior shown by these proteins is not caused by an extra destabilization of the protein, but rather by the amyloidogenic properties and location of the inserted sequence. Finally, we have demonstrated that the amyloid domain of the fibrils consists partly of the amyloidogenic insertion plus a short additional fragment of the protein. All of these results unambiguously demonstrate that amyloid fibril formation from the otherwise soluble α-SH3 is triggered by the inserted amyloid exogenous sequences. On these bases, we propose a mechanism for amyloid formation.
Materials and Methods
Cloning and Expression of Amyloidogenic α-SH3 Variants. The amyloidogenic insertions (hexapeptide plus a two glycine linker) were introduced at the N or C terminus of the SH3 domain of chicken brain spectrin (residues 1–62) by PCR. Protein cloning, expression, and purification procedures can be found in Supporting Text, which is published as supporting information on the PNAS web site. After purification, salts were removed, and the protein was concentrated and, for storage purposes, lyophilized and kept at –80°C.
Amyloid Fibril Formation Studies. Sample preparation from lyophilized protein powder is described in detail in Supporting Text. All samples were prepared in buffer with 20 mM Gly/HCl, pH 2.6. Fibrillation assays were set up at two concentration ranges (100 and 250–300 μM). Protein stability measurements were performed at 50 μM in buffered solution with Gly/HCl, pH 2.6. Phosphate salts were present in the range from 1 to 10 mM depending on the protein sample. For two of the protein variants we checked that removal of the salt decreased the melting temperature (Tm) only ≈1 K, which was within the error of the fitting.
Details on far- and near-UV CD spectra acquisition and analysis are reported in Supporting Text. For temperature denaturation experiments, data points were acquired by monitoring ellipticity changes at 235 nm through the temperature range from 277 to 375 K at a heating rate of 40°C per h in a 0.2-cm sealed quartz cuvette. Reversibility of the transition was checked by complete recovery of the CD signal after cooling down the sample to 298 K.
Thioflavin binding assays are described in Supporting Text, and EM was carried out as described (4).
Thermal Denaturation Curve Fitting and Data Analysis. Denaturation curves were fitted to Eq. 1 (22) for a two-state thermal denaturation by using profit 6.0 (QuantumSoft, Uetikon am See, Switzerland).
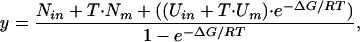 |
[1] |
where Nin is the ellipticity of the folded state, Nm is the dependence of Nin with temperature, Uin is the ellipticity of the unfolded state and Um is the dependence of Uin with temperature.
ΔG of the unfolding reaction was calculated by using Eq. 2 (22)
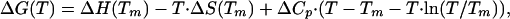 |
[2] |
where ΔH(Tm) and Tm were obtained from the fitting of the experimental denaturation curves to Eq. 1. We assumed for ΔCp the experimental value calculated from calorimetric data for the WT protein, ΔCp = 3.3 kJ·mol–1·K–1 (23).
The percentage of denatured (%D) protein was calculated by using Eq. 3
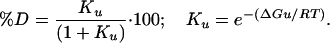 |
[3] |
Errors were estimated by using the general error propagation Eq. 4 with the assumption that 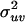 was 0,
was 0,
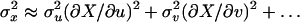 |
[4] |
Extensive Proteolysis. Mature fibrils of 1-SH3 were obtained by ultracentrifugation of ≈300-μM solutions incubated for 8 months. Fibrils were separated from the supernatant by ultracentrifugation to avoid contamination of the results by monomer digestion (Supporting Text and see Fig. 7a). The separation of fibrils from monomeric and soluble oligomeric species was carried out by ultracentrifugation at 300,000 × g for 3 h at 10°C. The resulting pellet was resuspended in buffer to a final concentration of 0.5–1 mg/ml. The concentration of supernatant and resuspended fibrils was determined by UV spectroscopy in buffer containing 6 M urea. Because the sample was incubated for a long period under acidic conditions, before digestion, we checked by MS that the fibrils obtained under these conditions consisted of the full-length protein. Fibrils were incubated for 48 h with pepsin (Sigma P6887) at room temperature. The ratio of monomeric protein/enzyme in the proteolysis reaction was 10:1. The reaction was quenched with 1 M NaOH. The insoluble material was collected by centrifugation as explained above. The pellet was dissolved in buffer containing 6 M urea for MS determination or in buffer with Gly/HCl, pH 2.6 for EM.
Aminopeptidase proteolysis must be carried out at pH 7.4 and 37°C. Thus, before the experiment, we checked that these conditions did not affect the integrity of preformed fibrils. Mature fibrils obtained as explained above were incubated with ≈0.15 units of aminopeptidase (Roche Diagnostics 1010276800) per μM of protein for 48 h. Aliquots were taken after 24 and 48 h of incubation and diluted with 10 M urea for MS determination.
Results and Discussion
Design of Amyloidogenic α-SH3 Variants: Strategy, Amyloid Stretches, and Insertion Sites. The amyloid behavior of a given protein can be switched by mutating at selected regions or increasing the homology to an already known amyloid sequence (24). Predictive tools can be also used to design mutations that at particular sequence regions may augment the amyloidogenicity of the protein (7, 12, 25). However, this mutation-based approach raises the question as to whether sequence modifications have altered essential structural elements of the WT protein, and whether or not amyloidogenicity relates to stability changes rather than to an increase of sequence propensity to form amyloids. Thus, our strategy to demonstrate the amyloid stretch hypothesis unambiguously has been the insertion of short nondestabilizing amyloidogenic sequences at different regions of a soluble nonamyloidogenic protein. Proteins designed this way keep the original sequence of the WT protein unchanged and, in principle, any amyloid propensity shown derives from the exogenous sequences.
The α-SH3 has been extensively studied as a model for protein folding (23, 26). In contrast to the structurally and evolutionary related phosphatidylinositol 3–SH3 domain, α-SH3 does not form amyloid fibrils under any of the conditions tested in our laboratory (21). All of these characteristics make it a very suitable target for proving the amyloid stretch hypothesis and understanding changes derived from the insertions.
As insertions we chose a set of sequence-related hexapeptides designed de novo along with an unrelated amyloidogenic stretch of the Aβ amyloid peptide. The de novo-designed sequence STVIIE (1) was chosen because of its strong amyloidogenic properties. Moreover, the insertion of 1 is very convenient for the purposes of the present work, because the self-assembly of 1 and its single-point mutants into amyloid-like fibrils have been characterized in detail. To demonstrate the applicability of the amyloid sequence pattern extracted from 1 in the context of full-length proteins, we designed a series of mutations at different position categories of 1 (edges vs. core). According to the pattern, Glu-6–Thr is a mutation in a tolerant position of 1 and should keep the aggregating properties provided by the core VII. In fact, the peptide generated, STVIIT (2), forms fibrils (7). Thus, 2 should also trigger amyloid formation of the protein. The mutation Ile-5–Lys at the most restrictive position of 1 has been shown to prevent peptide amyloid formation. Therefore, proteins carrying the insertion STVIKT (3) should not form amyloid fibrils and provide a negative control.
To demonstrate that sequence stretches identified with the amyloid pattern do not only trigger amyloid formation from the natural amyloid polypeptides to which they belong, but also from an unrelated soluble globular domain, the amyloidogenic fragment of the Aβ amyloid peptide, 16KLVFFA21 (Aβ), was also selected.
To assess the influence of the structural environment of the amyloid stretch, we inserted the same amyloidogenic sequences at different regions of the protein. These locations should allow the inserted sequences to be accessible for intermolecular interactions, while only minimally perturbing the structure of the α-SH3. According to these criteria three protein sites were selected: the N and C terminus of the protein and the distal loop (Fig. 1). The C terminus is structured into a β-strand, whereas the N terminus is disordered (27). This structural difference between the two termini provides an interesting framework for comparison of the effect of the insertions. In addition, the evaluation of the effect of amyloid-prone stretches at the N and C terminus of a protein is of biological relevance as several natural amyloid proteins present their amyloid domains at the protein termini (28, 29). The insertion of a de novo-designed β-hairpin at the distal loop of the α-SH3 (Bergerac SH3) has been reported not to disrupt the original folding of the domain despite being located in the middle of the sequence of the protein (30). Thus, this site is also suitable for the intended exercise.
Fig. 1.

Ribbon representation of the α-SH3. The sites selected for insertion of amyloidogenic sequences are indicated by arrows.
Sequences 1–3 and Aβ wer inserted at the N and C terminus of the α-SH3. A linker consisting of two Glys was placed between the amyloidogenic insertion and the globular domain to allow for molecular flexibility. Because of the location of the cloning sites, amyloid sequences inserted at the N terminus were also preceded by one Gly, except 1, which was preceded by two (GG-STVIIE-GG-SH3). Proteins generated this way were labeled by the number of the appended peptide followed by SH, if the insertion was performed at the N terminus (i.e., 1-SH), and in the other way around, if the insertion was at the C terminus (i.e., SH-1).
Based on the antecedents mentioned above, at the distal loop we designed an amyloidogenic β-hairpin as insertion. Structural (4) and molecular dynamics studies of peptide 1 (31) suggest an antiparallel arrangement of the polymeric β-sheets from which amyloid fibrils propagate. Thus, we designed a β-hairpin of sequence STVIIT-NG-STVIIT. NG should adopt a type I′ β-turn (32) that would favor the antiparallel arrangement of the amyloidogenic strands that consist of the sequence of peptide 2. We generated the corresponding protein by replacing Asn-47 and Asp-48 of the WT protein by the amyloidogenic β-hairpin. The protein generated has been termed AmyBergerac.
Conformation of α-SH3 Proteins with Amyloidogenic Insertions. Before the evaluation of the amyloidogenic properties of the generated proteins, we analyzed their conformation in solution by CD. The far- and near-UV CD spectra of the α-SH3 are very characteristic and strongly influenced by the presence of two consecutive Trp residues (residues 41 and 42) (26). At pH 2.6, the far-UV CD spectrum displays an inflexion point at 221 nm that is characteristic of the folded protein and that disappears upon unfolding (33).
We characterized the conformation of the soluble form of these proteins under the same conditions as the peptide self-assembly assays were conducted (pH 2.6 and room temperature) (7, 10). As shown in Fig. 2A, WT and proteins with insertions display a typical α-SH3 far-UV signature. According to the ellipticity measured at 221 nm, the proteins can be classified into three groups. The first group consists of WT, 1-SH3, Aβ-SH3, and SH-Aβ. Proteins 3-SH, SH-1, SH-2, and SH-3 constitute a second group of proteins that exhibit a small decrease of the ellipticity at 221 nm, which might indicate a slight loss of structure. A larger decrease of ellipticity is found for the group consisting of 2-SH and AmyBergerac. There exists no obvious reason for 2-SH to have a different conformation to the other proteins with sequence-related insertions. In the case of AmyBergerac, however, the change of the far-UV signature might result either from a destabilization of the protein or the addition of a structured element. According to the design, the 14-residue peptide inserted could form an amyloid β-hairpin. Unfortunately, the folding of this peptide into an antiparallel β-hairpin in solution cannot be confirmed and its amyloidogenic properties cannot be demonstrated, because it could not be purified after chemical synthesis because of its highly aggregating capability.
Fig. 2.

Far-UV (Left) and near-UV (Right) CD spectra recorded for the α-SH3 with insertions at the N terminus and distal loop (A) and the C terminus (B). Spectra have been recorded at t = 0, pH 2.6, and c ≈ 300 μM, except for 2-SH and AmyBergerac (t = 0, pH 2.6, and c ≈ 100 μM). The spectrum of the WT protein is displayed as a reference. Arrows in the far-UV CD plots indicate the inflexion point at 221 nm. The arrow in the near-UV CD plot (A Right) indicates the new peak at 280 nm exhibited by 1-SH.
α-SH3 variants with amyloidogenic insertions display the same near-UV signature as the WT protein (Fig. 2B). Only 1-SH shows a new peak ≈280 nm. This new peak suggests some changes in the environment of the Trp side chain. However, neither the shape nor the mean residue ellipticity of the spectrum changed upon dilution (data not shown). This result indicates that the aggregation state of the protein is the same within the analyzed concentration range (25–300 μM) (data not shown). This observation is not yet unambiguous evidence of the monomeric state of the protein, because it is also consistent with the existence of highly stable soluble oligomers that do not disassemble upon dilution. Nevertheless, such stable oligomers seem to be unlikely as the addition of a high concentration of a denaturant additive (2 M urea) to a solution of 1-SH (250 μM) did not alter the spectra either (data not shown). Moreover, no kind of aggregates were observed by EM at t = 0 for the concentrations checked.
Stability of α-SH3 Proteins with Amyloidogenic Insertions. A destabilization of the domain caused by the exogenous sequence would confound the amyloid stretch hypothesis. Thus, to prove that the amyloid insertions do not affect the stability of the globular domain, we estimated the stability of the α-SH3 variants under conditions where the proteins are monomeric.
At low salt concentrations, the WT protein exhibits a two-state reversible folding transition that has been kinetically and thermodynamically characterized (23). We applied this model to fitting the data from the α-SH3 variants that show a reversible thermally induced unfolding transition as monitored by CD (Table 1). From the estimated Tm, we calculated the ΔG of the unfolding reaction and the percentage of folded protein in solution (see Materials and Methods). It has to be mentioned that at pH 2.6 the α-SH3 is destabilized with respect to pH 7.0 (23). According to our data, 9% of the WT protein already populates the denatured state at pH 2.6. Thus, the effect of the insertions in the stability of the domain was assessed by comparing relative changes in the population of the folded state in respect to the WT domain. The N-terminus fusion proteins populate the folded state to the same extent as the original α-SH3. Interestingly enough, while the insertion of KLVFFA at the C terminus (SH-Aβ) did not seem to affect the stability of the domain, the insertions of STVIIE and its single-point mutants at this site determined a similar increase, of ≈10%, in the population of unfolded protein with respect to the WT.
Table 1. Stability of the α-SH3 with amyloidogenic insertions as estimated by thermal denaturation monitored by CD.
| Protein* | Tm,† K | ΔGu,† Kcal/mol | Denatured protein,† % | Folded protein,† % |
|---|---|---|---|---|
| WT | 321.5 ± 0.3 | 1.39 ± 0.05 | 8.7 ± 0.7 | 91.3 ± 0.7 |
| 1-SH | 319.0 ± 0.3 | 1.46 ± 0.07 | 7.9 ± 0.8 | 92.1 ± 0.8 |
| 2-SH | —‡ | —‡ | —‡ | —‡ |
| 3-SH | 316.9 ± 0.2 | 1.32 ± 0.05 | 9.7 ± 0.7 | 90.2 ± 0.7 |
| Aβ-SH | 319.7 ± 0.2 | 1.61 ± 0.05 | 7.0 ± 0.6 | 93.0 ± 0.6 |
| SH-1 | 313.0 ± 0.6 | 0.77 ± 0.04 | 21.7 ± 1.2 | 78.3 ± 1.2 |
| SH-2 | 313.4 ± 0.4 | 0.96 ± 0.05 | 16.7 ± 1.2 | 83.3 ± 1.2 |
| SH-3 | 321.5 ± 0.3 | 0.94 ± 0.04 | 17.4 ± 0.9 | 82.6 ± 0.9 |
| SH-Aβ | 322.2 ± 0.2 | 1.54 ± 0.05 | 6.1 ± 0.5 | 93.9 ± 0.5 |
| AmyBergerac | —‡ | —‡ | —‡ | —‡ |
Protein variants that form amyloid fibrils are in italics.
Thermal denaturation has been performed on freshly prepared protein solutions (pH 2.6, c = 50 μM).
Stability parameters and errors have been calculated as explained in Materials and Methods.
Nonreversible transition.
The stability of two of the designed proteins, 2-SH and AmyBergerac, has not been estimated as they do not present a reversible two-state transition. Visible aggregates, that are amyloid filaments in the case of AmyBergerac, were observed at the end of the thermal denaturation experiment. In the case of 2-SH one can hypothesize that the inserted sequence is more amyloidogenic than 1 and Aβ and once the protein is sufficiently unfolded, the equilibrium is shifted toward the amyloid pathway faster. For AmyBergerac, however, the insertion in the distal loop might have largely destabilized the globular domain. Thus, no conclusion can be obtained in favor of the amyloid hypothesis by using this α-SH3 variant. Characterization of the fibrils formed by AmyBergerac can be seen in Fig. 4, which is published as supporting information on the PNAS web site.
Conformational Transition and Amyloid Fibril Formation of α-SH3 Variants with Amyloidogenic Insertions. We tested the amyloidogenic behavior of all SH3 variants at pH 2.6 and two protein concentrations (≈100 and ≈300 μM). Aliquots from these solutions were taken and checked by CD for a transition to a polymeric β-sheet and by EM for amyloid fibril formation. To compare with the amyloidogenic behavior shown by the peptides, checks were performed at t = 0, 1 week, 1 month, and 3 months. Within this time range 1-mM solutions of the amyloidogenic peptides already displayed a β-sheet spectrum and formed amyloid fibrils (4).
EM analysis showed the presence of amyloid fibrils in aliquots taken from solutions of 1-SH, 2-SH, and Aβ-SH (t = 3 months, Fig. 3). 1-SH fibrils from solutions incubated for 3 months were shown to bind thioflavin T (Fig. 5, which is published as supporting information on the PNAS web site). No fibrils were detected for 3-SH, C-terminus variants, and the WT protein under the conditions tested. After 3 months of incubation time, only a very small amount of aggregated material was detected for these proteins (Fig. 3). A large amount of fibrillar material was observed only for 1-SH and Aβ-SH. Indeed, these proteins were the only two α-SH3 variants that formed fibrils after 1 week (data not shown). At t = 1 week nuclei were detected only for 2-SH samples (data not shown).
Fig. 3.

Electron micrographs of the α-SH3 variants that form amyloid fibrils (pH 2.6, c ≈ 300 μM, t = 3 months, and room temperature). 2-SH has been assayed only at the lowest concentration (c ≈ 100 μM) because of solubility problems.
Under the conditions tested and after an incubation time of 3 months, none of the protein solutions exhibited a far-UV CD transition from the typical SH3 signature to a polymeric β-sheet, nor were any large changes observed in the near-UV spectra (Fig. 6, which is published as supporting information on the PNAS web site). Only 1-SH and Aβ-SH displayed the beginning of a WT → β transition and a certain loss of amplitude of the near UV-spectra (Fig. 6). This result correlates well with the fact that they were the only mutants that showed abundant amyloid formation within this incubation time.
Usually, a CD β-sheet transition is a concomitant of amyloid fibril formation (34). The fact that only small CD changes were observed in solutions where amyloid fibrils were found could be caused by two reasons: (i) only a small percentage of the protein population becomes amyloidogenic, whereas most protein remains soluble and properly folded; or (ii) most protein is incorporated into the fibrils in a close-to-native conformation. To distinguish between these two possibilities, we separated the insoluble fraction from the supernatant for a sample of Aβ-SH incubated for 8 months. After this long incubation period the far-UV spectra of the total protein solution was still native-like (Fig. 7, which is published as supporting information on the PNAS web site) and <15% of the protein was in the pellet. The soluble fraction hardly presented amyloid fibrils, and its CD spectrum overlapped with the spectrum of the total protein solution recorded before ultracentrifugation. However, the resuspended pellet contained abundant fibrillar material that displayed a typical β-sheet spectrum (Fig. 7), results that confirm the β-sheet structure of the fibrils detected by EM.
Further evidence on the β-sheet structure of the fibrils was obtained from aged samples of 1-SH and salt-containing solutions. For samples of 1-SH incubated at 300 μM, although fibrils were detected after 1 week, a full β-sheet transition was observed only for samples incubated for a year and a half. This result indicates that longer incubation times than those checked systematically in this work (up to 3 months) are needed for the observation of a full β-transition of all protein population in this case (Fig. 8, which is published as supporting information on the PNAS web site). A typical β-sheet signature was also displayed by 300-μM solutions of 1-SH containing phosphate salts in the range from 1 to 10 mM after 1 week, which highlights how much salts can shorten the conformational transition preceding amyloid fibril formation (Fig. 9, which is published as supporting information on the PNAS web site).
Core vs. Edge Mutations at the Amyloid Stretch: Effect on the Amyloidogenicity of the Protein. The amyloidogenicity showed by the proteins with the insertion at the N terminus compares to the amyloidogenic behavior exhibited by the peptides (7). Mutation Ile-5–Lys at the most restrictive position of the core completely abolishes amyloid fibril formation (3-SH3). Only very little amorphous material was found on EM grids after 3 months (Fig. 3). Despite being slightly destabilizing, the insertion with this positively charged mutation blocks the formation of unwanted misfolded or amyloid prone states (gatekeeper residue) (24) and prevents self-assembly into amyloid fibrils from the percentage of unfolded protein in solution (amyloid breaker residue) (7). Introduction of positive charges has been reported as a way to avoid β-sheet aggregation in natural proteins (35) and β-sheet libraries (36). Mutation Glu-6–Thr at position 6 of the more tolerant edges (2-SH3) keeps the amyloidogenic feature of 1 and leads to the formation of fibrils of similar twisted morphology to those formed by 1-SH.
These results suggest that the amyloidogenic properties of proteins containing six-residue stretches that match the amyloid pattern can be modified by designing mutations according to the amino acid tolerance at each position provided by the pattern.
Effect of the Structural Environment of the Amyloid Stretch on the Amyloidogenicity of the Protein. As described above, only N-terminus variants formed amyloid fibrils under the conditions tested in this work. Interestingly enough, under these conditions α-SH3 proteins with insertions at the C terminus were less stable than proteins with the insertions at the N terminus (Table 1), which rules out stability effects (extra destabilization) as being responsible for the differential amyloidogenic ability shown by the N- and C-terminus variants. This result rather illustrates how differences in the structural environment of the amyloidogenic sequence can influence amyloid formation. The N terminus of α-SH3 is disordered, whereas the C terminus is structured into a β-strand (27). Therefore, the amyloid tail should be more exposed for mutants with the insertion at the N terminus, and thus more accessible for intermolecular processes such as amyloid fibril formation.
The results obtained with our model proteins are in agreement with the behavior shown by natural proteins whose aggregating behavior is triggered by an amyloidogenic stretch that is not accessible under native-like conditions. As an example, β2-microglobulin (β2m) forms fibrils at neutral pH only under destabilizing conditions that allow for the exposure of the β-strand shown experimentally to be important for amyloid formation (11) and identified as amyloidogenic by the amyloid pattern (7). Therefore, as in the case of β2m, the C-terminus proteins contain a sequence encoding for amyloid formation, but trapped into a stable conformation.
Isolation of the Protease-Resistant Core of 1-SH Fibrils. According to the amyloid stretch hypothesis, we would expect that the amyloid stretch inserted in the α-SH3 constitutes or takes part of the protease-resistant fiber core. The fiber core typically contains the region that confers the amyloidogenic properties to the protein, that is, the so-called amyloid domain (9). To this end, we performed extensive proteolysis (pepsin, t = 48 h; see Materials and Methods) of mature fibrils of 1-SH collected from an aged solution incubated for 8 months. The proteolysis reaction produced sedimentable material consisting of very thin filaments (Fig. 10a, which is published as supporting information on the PNAS web site). MS analysis of this fraction yielded a clear unique peak corresponding to a molecular weight of 1,245 (Fig. 10a). Sequencing of this peak has identified a 12-aa peptide that belongs to the N-terminal region of the protein (VIIE-GG-DETGKE) (Fig. 10b). The theoretical mass of this fragment matches perfectly with the experimental value. Thus, the fiber core resistant to proteolysis consists of at least the last four amino acids of the inserted sequence (VIIE).
Because GGST is not an expected cleavage site for pepsin, we investigated why GGST has been cleaved off from the fibril core. To assess the degree of protection toward unspecific cleavage of the N terminus of the protein within the fibril, we digested mature fibrils of 1-SH with aminopeptidase for 48 h. Immediately after addition of the enzyme, degradation commenced, and two peaks, corresponding to the degradation of the two initial glycines and the cleavage of the GGST fragment, respectively, were found. These two peaks increased gradually in time (see time points t = 24 and 48 h in Fig. 11, which is published as supporting information on the PNAS web site).
These results suggest that the inserted sequence is still accessible for degradation until the highly amyloidogenic VII core, which is protected despite the very aggressive conditions of proteolysis. In fact, it has been suggested by molecular dynamics that VII forms a hydrophobic cluster that provides most stabilizing side-chain interactions to the formation of an ordered β-sheet (31). VII has been shown to drive copolymerization of many different sequences that share only these three residues (7). Its highly amyloidogenic capability, together with the fact that the N terminus of the protein is already unstructured in the native fold, might explain why some neighboring residues have been incorporated in the amyloid domain as well.
Therapeutical and Biological Implications of the Amyloid Stretch Hypothesis
Amyloid Stretches as Targets for Amyloid Inhibition. As the self-association process relies on the establishment of stabilizing contacts between self-complementary stretches of a protein, different strategies can be devised to prevent, cap, or disrupt the amyloid spine. Drugs can be designed to stabilize a protein so that the dangerous amyloidogenic stretches are not accessible for molecular interactions (37). As the information contained by the amyloid pattern on short amyloid stretches is also valid within the context of a protein (7), by combining the amyloid pattern with protein design algorithms (38) mutations can be designed that prevent amyloid formation without changing the stability of the protein. A similar approach has been recently used to reduce the propensity of human calcitonin to form amyloid fibrils without affecting its physiological activity (39). Amyloid protein sequences also can be scanned for stretches that match the amyloid pattern, and drugs can be designed to specifically interact with these fragments. Stretches identified this way can also be synthesized and used to screen for molecules that can effectively block the formation of the amyloid spine by the hexapeptide and thereby amyloid formation of the full-length protein (unpublished work).
Amyloid Formation Signals in Proteins. One of the dogmas in biology is that all of the information required for a protein to fold is encoded in its primary structure. In past years, however, it has been demonstrated that short consensus sequences within this primary structure also determine important events in the life of a protein, such as localization, posttranslational modifications, or degradation (40). The demonstration of the amyloid stretch hypothesis adds a function to this list: amyloid formation. As the energetic barrier for amyloid fibril formation is very high (15), the amyloid reaction pathway is activated only under exceptional cellular conditions.
Conclusions
By using a minimalist approach, we have provided strong support in favor of the amyloid stretch hypothesis. We have shown that just a short amyloidogenic stretch is required to trigger amyloid fibril formation from a soluble globular domain. Our results also highlight that the structural environment of the amyloidogenic stretch plays a fundamental role in whether or not an amyloid-prone sequence can productively trigger the amyloid self-assembly process. Under native-like conditions, the lag phase of assembly will depend enormously on the accessibility of the amyloid sequence. Under denaturing conditions or for exposed amyloid sequences, the nucleation phase will represent the time required by these sequences to search out for productive interactions to form an extended β-sheet. Once the self-assembly process has been triggered by the amyloidogenic stretch, some other residues also may be incorporated into the amyloid spine, but the major part of the protein sequence will not.
Supplementary Material
Acknowledgments
We thank I. Angrand for assistance with protein purification; N. Kuemmerer for assistance with the EM measurements; the European Molecular Biology Laboratory Proteomic Core Facility for help with MS determination; Dr. I. E. Sánchez for critical review of the manuscript and assistance with protein stability analysis; and Dr. M. T. Pastor for useful discussions. This work was supported by a European Union grant (Research Training Network Project 2001-00364, Protein Folding, Misfolding, and Disease) (to A.E.-C.) and a European Union grant (APOPIS, Abnormal Proteins in the Pathogenesis of Neurodegenerative Disorders) (to M.L.d.l.P.).
Author contributions: L.S. and M.L.d.l.P. designed research; A.E.-C. performed research; A.E.-C. and M.L.d.l.P. analyzed data; and A.E.-C., L.S., and M.L.d.l.P. wrote the paper.
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: SH3, Src homology 3; α-SH3, α-spectrin SH3 domain; Tm, melting temperature.
References
- 1.Ross, C. A. & Poirier, M. A. (2004) Nat. Med. 10, Suppl., S10–S17. [DOI] [PubMed] [Google Scholar]
- 2.Fandrich, M., Fletcher, M. A. & Dobson, C. M. (2001) Nature 410, 165–166. [DOI] [PubMed] [Google Scholar]
- 3.Fezoui, Y., Hartley, D. M., Walsh, D. M., Selkoe, D. J., Osterhout, J. J. & Teplow, D. B. (2000) Nat. Struct. Biol 7, 1095–1099. [DOI] [PubMed] [Google Scholar]
- 4.López de la Paz, M., Goldie, K., Zurdo, J., Lacroix, E., Dobson, C. M., Hoenger, A. & Serrano, L. (2002) Proc. Natl. Acad. Sci. USA 99, 16052–16057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sunde, M., Serpell, L. C., Bartlam, M., Fraser, P. E., Pepys, M. B. & Blake, C. C. (1997) J. Mol. Biol. 273, 729–739. [DOI] [PubMed] [Google Scholar]
- 6.Chiti, F., Taddei, N., Bucciantini, M., White, P., Ramponi, G. & Dobson, C. M. (2000) EMBO J. 19, 1441–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.López de la Paz, M. & Serrano, L. (2004) Proc. Natl. Acad. Sci. USA 101, 87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ventura, S., Zurdo, J., Narayanan, S., Parreno, M., Mangues, R., Reif, B., Chiti, F., Giannoni, E., Dobson, C. M., Aviles, F. X. & Serrano, L. (2004) Proc. Natl. Acad. Sci. USA 101, 7258–7263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ivanova, M. I., Sawaya, M. R., Gingery, M., Attinger, A. & Eisenberg, D. (2004) Proc. Natl. Acad. Sci. USA 101, 10584–10589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tjernberg, L. O., Naslund, J., Lindqvist, F., Johansson, J., Karlstrom, A. R., Thyberg, J., Terenius, L. & Nordstedt, C. (1996) J. Biol. Chem. 271, 8545–8548. [DOI] [PubMed] [Google Scholar]
- 11.Jones, S., Manning, J., Kad, N. M. & Radford, S. E. (2003) J. Mol. Biol. 325, 249–257. [DOI] [PubMed] [Google Scholar]
- 12.Pawar, A. P., Dubay, K. F., Zurdo, J., Chiti, F., Vendruscolo, M. & Dobson, C. M. (2005) J. Mol. Biol. 350, 379–392. [DOI] [PubMed] [Google Scholar]
- 13.Tenidis, K., Waldner, M., Bernhagen, J., Fischle, W., Bergmann, M., Weber, M., Merkle, M. L., Voelter, W., Brunner, H. & Kapurniotu, A. (2000) J. Mol. Biol. 295, 1055–1071. [DOI] [PubMed] [Google Scholar]
- 14.Sambashivan, S., Liu, Y., Sawaya, M. R., Gingery, M. & Eisenberg, D. (2005) Nature 437, 266–269. [DOI] [PubMed] [Google Scholar]
- 15.Nelson, R., Sawaya, M. R., Balbirnie, M., Madsen, A. O., Riekel, C., Grothe, R. & Eisenberg, D. (2005) Nature 435, 773–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baxa, U., Taylor, K. L., Wall, J. S., Simon, M. N., Cheng, N., Wickner, R. B. & Steven, A. C. (2003) J. Biol. Chem. 278, 43717–43727. [DOI] [PubMed] [Google Scholar]
- 17.Tanaka, M., Machida, Y., Nishikawa, Y., Akagi, T., Morishima, I., Hashikawa, T., Fujisawa, T. & Nukina, N. (2002) Biochemistry 41, 10277–10286. [DOI] [PubMed] [Google Scholar]
- 18.Wigley, W. C., Stidham, R. D., Smith, N. M., Hunt, J. F. & Thomas, P. J. (2001) Nat. Biotechnol. 19, 131–136. [DOI] [PubMed] [Google Scholar]
- 19.He, Y., Tang, H., Yi, Z., Zhou, H. & Luo, Y. (2005) FEBS Lett. 579, 1503–1508. [DOI] [PubMed] [Google Scholar]
- 20.Chae, Y. K., Cho, K. S. & Chun, W. (2002) Protein Pept. Lett. 9, 315–321. [DOI] [PubMed] [Google Scholar]
- 21.Ventura, S., Lacroix, E. & Serrano, L. (2002) J. Mol. Biol. 322, 1147–1158. [DOI] [PubMed] [Google Scholar]
- 22.Becktel, W. J. & Schellman, J. A. (1987) Biopolymers 26, 1859–1877. [DOI] [PubMed] [Google Scholar]
- 23.Viguera, A. R., Martinez, J. C., Filimonov, V. V., Mateo, P. L. & Serrano, L. (1994) Biochemistry 33, 2142–2150. [DOI] [PubMed] [Google Scholar]
- 24.Otzen, D. E., Kristensen, O. & Oliveberg, M. (2000) Proc. Natl. Acad. Sci. USA 97, 9907–9912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fernández-Escamilla, A. M., Rousseau, F., Schymkowitz, J. & Serrano, L. (2004) Nat. Biotechnol. 22, 1302–1306. [DOI] [PubMed] [Google Scholar]
- 26.Viguera, A. R., Jimenez, M. A., Rico, M. & Serrano, L. (1996) J. Mol. Biol. 255, 507–521. [DOI] [PubMed] [Google Scholar]
- 27.Musacchio, A., Noble, M., Pauptit, R., Wierenga, R. & Saraste, M. (1992) Nature 359, 851–855. [DOI] [PubMed] [Google Scholar]
- 28.Peretz, D., Williamson, R. A., Matsunaga, Y., Serban, H., Pinilla, C., Bastidas, R. B., Rozenshteyn, R., James, T. L., Houghten, R. A., Cohen, F. E., et al. (1997) J. Mol. Biol. 273, 614–622. [DOI] [PubMed] [Google Scholar]
- 29.Balbirnie, M., Grothe, R. & Eisenberg, D. S. (2001) Proc. Natl. Acad. Sci. USA 98, 2375–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Viguera, A. R. & Serrano, L. (2001) J. Mol. Biol. 311, 357–371. [DOI] [PubMed] [Google Scholar]
- 31.López de la Paz, M., de Mori, G. M., Serrano, L. & Colombo, G. (2005) J. Mol. Biol. 349, 583–596. [DOI] [PubMed] [Google Scholar]
- 32.López de la Paz, M., Lacroix, E., Ramirez-Alvarado, M. & Serrano, L. (2001) J. Mol. Biol. 312, 229–246. [DOI] [PubMed] [Google Scholar]
- 33.Spagnolo, L., Ventura, S. & Serrano, L. (2003) Protein Sci. 12, 1473–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pastor, M. T., Esteras-Chopo, A. & López de la Paz, M. (2005) Curr. Opin. Struct. Biol. 15, 57–63. [DOI] [PubMed] [Google Scholar]
- 35.Richardson, J. S. & Richardson, D. C. (2002) Proc. Natl. Acad. Sci. USA 99, 2754–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang, W. & Hecht, M. H. (2002) Proc. Natl. Acad. Sci. USA 99, 2760–2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Petrassi, H. M., Johnson, S. M., Purkey, H. E., Chiang, K. P., Walkup, T., Jiang, X., Powers, E. T. & Kelly, J. W. (2005) J. Am. Chem. Soc. 127, 6662–6671. [DOI] [PubMed] [Google Scholar]
- 38.Schymkowitz, J., Borg, J., Stricher, F., Nys, R., Rousseau, F. & Serrano, L. (2005) Nucleic Acids Res. 33, W382–W388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fowler, S. B., Poon, S., Muff, R., Chiti, F., Dobson, C. M. & Zurdo, J. (2005) Proc. Natl. Acad. Sci. USA 102, 10105–10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aitken, A. (1999) Mol. Biotechnol. 12, 241–253. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.