

Abstract
Mutations in the leucine-rich repeat kinase 2 gene (LRRK2) cause late-onset Parkinson's disease (PD) with a clinical appearance indistinguishable from idiopathic PD. Initial studies suggest that LRRK2 mutations are the most common yet identified determinant of PD susceptibility, transmitted in an autosomal-dominant mode of inheritance. Herein, we characterize the LRRK2 gene and transcript in human brain and subclone the predominant ORF. Exogenously expressed LRRK2 protein migrates at ≈280 kDa and is present largely in the cytoplasm but also associates with the mitochondrial outer membrane. Familial-linked mutations G2019S or R1441C do not have an obvious effect on protein steady-state levels, turnover, or localization. However, in vitro kinase assays using full-length recombinant LRRK2 reveal an increase in activity caused by familial-linked mutations in both autophosphorylation and the phosphorylation of a generic substrate. These results suggest a gain-of-function mechanism for LRRK2-linked disease with a central role for kinase activity in the development of PD.
Keywords: dardarin, parkinsonism, PARK8
Recent studies exploring the etiology of Parkinson's disease (PD) suggest an important role for the leucine-rich repeat kinase 2 gene (LRRK2, PARK8, dardarin, OMIM 609007). The PARK8 locus encompassing LRRK2 was originally identified by use of parametric two-point analysis in a large Japanese family with autosomal-dominant PD (1). Using a positional cloning approach, two groups concurrently identified mutations within the LRRK2 gene (2, 3). The frequency of a novel mutation (G2019S) was subsequently reported in 5-6% of autosomal-dominant PD patients (4, 5) and even in ≈1% of PD patients with sporadic late-onset disease (6). Additional studies confirmed the presence of the G2019S mutation in PD cohorts (7, 8). The unprecedented prevalence of LRRK2 mutations in PD cases suggests a critical role for LRRK2 in disease susceptibility.
Most PARK8-linked families described to date exhibit a clinical and in vivo neurochemical phenotype indistinguishable from that of sporadic PD (9). In contrast, pathological heterogeneity in affected individuals examined so far ranges from pure nigral degeneration without Lewy bodies to nigral degeneration associated with Lewy bodies typical of PD, widespread Lewy bodies consistent with diffuse Lewy body disease, or neurofibrillary τ-positive tangles (2, 10). Clarification of the function of LRRK2 is therefore relevant toward deciphering the biochemical pathway(s) that is important for the development of late-onset PD and for understanding the underlying pathology.
The LRRK2 gene contains at least 51 exons predicted to encode a 2,527-aa protein. Northern analysis indicated a possible 9-kbp transcript ubiquitously expressed in multiple tissues (3). In this report, we describe the isolation and characterization of the LRRK2 transcript as present in human substantia nigra tissue and the functional analysis of two familial-linked LRRK2 mutations localized to the GTPase domain (R1441C) or the mixed-lineage kinase (MLK)-like domain (G2019S). We demonstrate that LRRK2 possesses MLK-like activity and that familial-associated mutations appear to enhance protein kinase activity consistent with a possible gain-of-function model for LRRK2-linked disease.
Materials and Methods
5′ RACE and Plasmid Generation. FirstChoice adapter-mediated RACE kit (Ambion, Austin, TX) was used with human brain cDNA as previously described in ref. 11. PCR was performed with oligonucleotides (Invitrogen) specific for the 5′ adapter and LRRK2 gene (exon 1 outer, 5′-TGG ACA TTG TTC AGC CTG ACT ATC AAC-3′; exon 3 outer, 5′-CAC CAA GGA CTT CCC AAT CAT TTC-3′; exon 5 outer, 5′-AGA CTC TCT CAA ACA GCA CAT GTA AAG C-3′). A secondary PCR was performed by using nested primers [exon 1 inner (forward), 5′-GAG TTT CCT CGT CCT CTT CGC AC-3′; exon 3 inner, 5′-GGG TCC CAT TAA GCT TTG CAT TG-3′; exon 5 inner, 5′-TGG CTG GAA ATG AGT GCA TGG-3′]. Products were analyzed by agarose gel electrophoresis. PCR products were subcloned into pCR2.1 by using the TOPO-TA kit (Invitrogen), and insert DNA was sequenced.
LRRK2 exonic structure and sequence were determined by using cDNA derived from fresh-frozen human substantia nigra tissue from a pathologically confirmed normal brain (courtesy of Deborah Mash, University of Miami, Miami), first by isolating total RNA by using the RNAqueous kit (Ambion) and then through reverse transcription by using the SuperScript III RT-PCR kit and oligo(dT) priming (Invitrogen). For alternative splicing and sequence analysis, oligonucleotide primers were designed in the forward and reverse direction in every other LRRK2 exon, and PCR was carried out by using human substantia nigra cDNA as template in a standard 60-50°C touch-down program over 35 cycles. Products were analyzed by agarose gel electrophoresis, and selected products were dye-terminator sequenced on a PRISM 3100 Genetic Analyzer (Applied Biosystems).
To clone full-length LRRK2 cDNA into a mammalian expression vector, two sets of primers were designed with internal BamHI and XhoI restriction sites to amplify and insert 5′ and 3′ overlapping halves of LRRK2 into pcDNA3.1 (F1, 5′-TAG GAT CCT GCC GGT TCC CTG AGC-3′; R1, 5′-TGC TCG AGA ATC ACA GGG GAA GAA GAA G-3′; F2, 5′-TAG GAT CCC CAT TTT ATG ACG CAG C-3′; R2, 5′-CTC TCG AGC TCA ACA GAT GTT CGT CTC-3′). PCR was carried out by using Expand High Fidelity Taq (Roche Diagnostics) and human substantia nigra cDNA, prepared as above. The two halves of the LRRK2 ORF were then joined together by virtue of an endogenous BbvCI site in LRRK2, and sequence of the full-length LRRK2 insert was verified and compared with the Entrez database entry for human LRRK2.
The familial-linked mutations G2019S (6055G → A) and R1441C (4321C → T) were introduced into LRRK2 cDNA by PCR-mediated site-directed mutagenesis by using the QuikChange kit (Stratagene) followed by sequencing of the entire insert to confirm their correct incorporation.
Immunoblotting. The following peptides were synthesized and used to generate antibodies to human LRRK2: JH5514, CHEVQNLEKHIEVR (amino acids 2,500-2,515), JH5517, KNENQENDDEGEEDC (amino acids 334-347), and JH5518, EFHHKLNMVKKC (amino acids 527-537). Cysteine residues (in italics) were added to facilitate conjugation to keyhole limpet hemocyanin. Immunization of rabbits and antibody purification using affinity chromatography on peptide-immobilized Sulfolink coupling gel matrix columns (Pierce, Rockford, IL) was performed as previously described (12). Cellular protein extracts were prepared, and protein was separated by 6% SDS/PAGE and blotted as described (13). For internal controls and fractionation experiments, membranes were stripped and reprobed with antibodies against β-actin (1:2,000, Sigma-Aldrich), PGK (1:1,000, Abgent, San Diego), porin (1:2,500, Calbiochem), c-myc (1:2,000, clone 9E10, Roche Applied Science, Indianapolis), and HA (1:1,000, clone 12CA5, Roche Applied Science).
Cell Culture and Protein Purification. HEK-293T and SH-SY5Y cells were cultured in Opti-MEM media (Invitrogen) supplemented with 10% FBS, penicillin (100 units/ml), and streptomycin (100 μg/ml). Transfection was accomplished by using FuGENE 6.0 reagent (Roche Applied Science) according to the manufacturer's instructions. To prepare recombinant LRRK2 protein for kinase assays, immunoprecipitation was carried out as previously described (12). Briefly, HEK-293T cells transiently transfected with each myc-tagged LRRK2 plasmid were harvested in immunoprecipitation (IP) buffer, and resulting lysates were rotated at 4°C for 1 h followed by centrifugation at 17,500 × g for 15 min. The supernatant fractions were combined with Protein G Sepharose 4 Fast Flow (Amersham Pharmacia Biotech) precomplexed with mouse monoclonal anti-c-myc antibody (clone 9E10) followed by rotating overnight at 4°C. The protein G Sepharose complex was pelleted and stringently washed five times with IP buffer supplemented with 500 mM NaCl and once with PBS. Where necessary, immunoprecipitated proteins were eluted in 2× SDS sample buffer containing 5% 2-mercaptoethanol by heating at 75°C for 10 min. Silver staining was performed by using the SilverSNAP stain kit (Pierce).
For ubiquitination assays, cells were transiently transfected with myc-tagged LRRK2 with or without hemagglutinin-tagged ubiquitin. Cells were treated with or without the proteasome inhibitor MG-132 (5 μM) for 24 h before harvesting in RIPA buffer (150 mM NaCl/10 mM Tris·HCl, pH 7.2/1% Triton X-100/1% sodium deoxycholate/0.1% SDS) and anti-c-myc immunoprecipitation as detailed above. Immunprecipitates were resolved by 7.5% SDS/PAGE and analyzed by Western blot with anti-HA (Roche Applied Science) or anti-c-myc (Roche Applied Science) antibodies directly conjugated to horseradish peroxidase.
Subcellular Fractionation and Immunofluorescence. For cell fractionation, HEK-293T and SH-SY5Y cells were transfected as above with WT or mutant LRRK2. At 48 h posttransfection, cells were harvested by scraping into hypotonic buffer as previously described (14). Cells were incubated on ice for at least 25 min, and complete lysis of the plasma membrane was visualized by using a 2× trypan blue solution containing DAPI. Lysates were centrifuged at 800 × g for 5 min to generate crude supernatant and pellet (P1, nuclear) fractions. The supernatant fraction was further centrifuged at 15,000 × g for 10 min to generate a crude pellet fraction (P10) containing mitochondria and a supernatant fraction (S10) containing cytosol. The nuclear (P1) and mitochondrial (P10) fractions were solubilized in RIPA buffer. Protein content was determined by using the BCA protein assay kit (Pierce). For experiments indicating the use of trypsin, 0.05% wt/vol trypsin (Invitrogen) was added to lysates and incubated at 37°C for 10 min before fractionation.
HEK-293T cells were plated onto poly-l-lysine-coated glass coverslips (BD Biosciences, Franklin Lakes, NJ) and transfected with WT or mutant LRRK2 constructs as above. The coverslips were washed and fixed as previously described (14). Coverslips were incubated for 24 h at 4°C in a solution containing 0.1% Triton X-100 and anti-myc-FITC-conjugated antibody (1:1,000; Cell Signaling Technology, Beverly, MA) in PBS. Coverslips were washed with PBS, incubated with DAPI (300 nM in PBS), and washed a final three times with PBS. Coverslips were mounted on glass slides, and cells were visualized on a Ziess 510 META multiphoton confocal microscope.
In Vitro Kinase Assays. Myc-tagged LRRK2 (WT or mutant) or control protein (myc-tagged DJ-1) bound to protein G Sepharose was resuspended in kinase assay buffer (20 mM Hepes, pH 7.4/15 mM MgCl2/5 mM EGTA/20 mM β-glycerol phosphate) on ice together with 25 μM biotinylated myelin basic protein (MBP; Upstate Biotechnology, Lake Placid, NY). The kinase reaction was initiated by adding together 0.5 μCi of [γ-32P]ATP (PerkinElmer) and 15 μM ATP and incubating the reaction at 30°C for 15 min. The reaction was then placed on ice and terminated by the addition of 2.5 M guanidine-HCl (Calbiochem). The universal kinase assay kit (Calbiochem) was used to quantify incorporated radioisotope in biotinylated MBP by adding avidin solution (Calbiochem) to the reaction and incubating at 25°C for 10 min. The reaction was centrifuged at 800 × g for 2 min, and supernatant was placed onto a column containing an avidin-binding membrane and washed according to protocol. Incorporated radioisotope was measured by liquid scintillation, and background and nonspecific activity was substracted from samples containing WT or mutant LRRK2.
LRRK2 autophosphorylation activity was determined by using the kinase-reaction conditions above except for the lack of MBP in the reaction. After 15 min at 30°C, the reactions were placed on ice and terminated by the addition of Laemmli sample buffer with 5% 2-mercaptoethanol. Reactions were heated at 75°C for 10 min and placed on ice. Reactions were then centrifuged at 800 × g for 2 min, and supernatant was resolved by 6% SDS/PAGE and transferred to a poly(vinylidene difluoride) membrane. Membranes were subsequently exposed to BioMax film (Kodak) for 24 h, and then immunoblotting with anti-c-myc antibody (Roche Applied Science) was performed to determine the amount of LRRK2 present in each reaction. Autoradiography and Western blotting bands were quantified by using imagequant 5.0.
Results
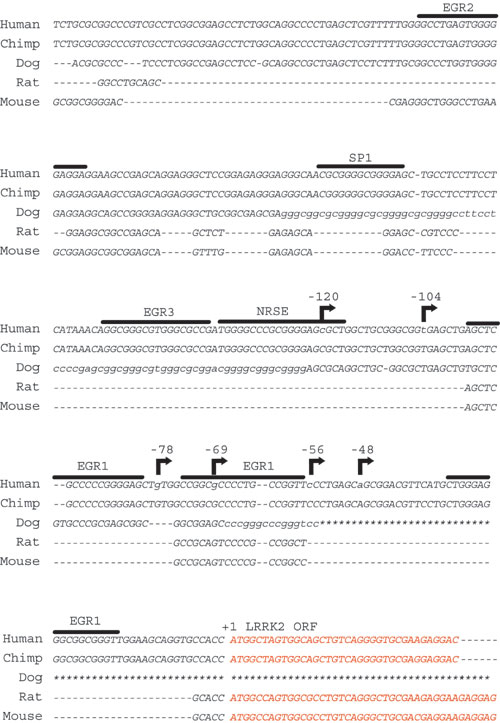
Characterization of the LRRK2 Transcript. At the onset of this study LRRK2 was a largely predicted gene with an unknown potential for alternative ORFs, promoters, or exon splicing. To determine the correct and predominant LRRK2 transcript as it exists in human tissue, we performed adapter-mediated 5′ RACE using cDNA derived from human brain. Reverse oligonucleotides positioned in the predicted first, third, or fifth exon of LRRK2 revealed multiple transcription initiation sites (Fig. 1A and Fig. 7, which is published as supporting information on the PNAS web site). In the brain, at least six transcription start sites are present between 48 and 120 bp upstream of the first predicted Kozak sequence. A single and distinct transcription initiation site downstream of the first predicted exon (within intron 1) excludes the complete LRRK2 ORF (Fig. 1B). However, the transcript does not correlate to an obvious ORF that would produce protein of appreciable size. Therefore, the biological significance of this transcript remains unknown. No evidence of alternate LRRK2 exons or protein coding sequence was identified. We verified the presence of endogenous LRRK2 transcript in human tissue and HEK-293T cells by using RT-PCR with oligonucleotide primers situated on the 3′ end of LRRK2 mRNA (Fig. 1C).
Fig. 1.

Characterization of expression of LRRK2. (A) 5′-RACE nested PCR using human total brain cDNA, as resolved on an ethidium bromide agarose gel. The exon position of the corresponding reverse oligonucleotide in the predicted LRRK2 transcript is given. (B) Diagram depicting the 5′ end of the LRRK2 gene. The ATG represents the presence of the first consensus Kozak sequence downstream of the initiation region and the start of the predominant LRRK2 ORF. (C) RT-PCR using oligonucleotide primers spanning the 3′ end of the predicted LRRK2 transcript. Sources of cDNA derived from human tissue or cell lines are indicated. (D) Protein domain structure of LRRK2 where the ankryin (Ank-like) repeat region, the leucine-rich repeat (LRR) domain, the GTPase domain, the MLK-like domain, and the WD40 domain positions are indicated. Lysate derived from HEK-293T cells transiently expressing myc-tagged LRRK2 (+) or empty (-) vector were analyzed by Western blotting by using LRRK2-specific antibodies with recognition sites near the N terminus (JH5517 and JH5518) or the C terminus (JH5514 and anti-myc).
Next, the exon structure and potential for alternative splicing within LRRK2 was determined in mRNA derived from human substantia nigra tissue. Using oligonucleotides positioned in the identified 5′ and 3′ untranslated regions of LRRK2, full-length LRRK2 transcript (≈7.5 kb) was sequentially amplified and subcloned into a plasmid library (pCR2.1). Twenty clones containing LRRK2 were isolated and examined for alternative splicing by using PCR and sequence analysis. In all 20 clones from this tissue source, 51 exons were present. A cryptic splice site found between exons 50 and 51 (within intron 50) in 3 of 20 clones results in the exclusion of 6 aa near the WD40 domain. Thus, no prevalent alternative splicing in LRRK2 occurs in this tissue source although a cryptic splice site likely produces an alternate LRRK2 protein isoform with a partially altered amino acid sequence near the C terminus.
Expression of LRRK2 in Cell Lines. The complete LRRK2 ORF was subcloned into the mammalian expression plasmid pcDNA3.1 in frame with a C-terminal myc-His tag, and the resultant amino acid sequence was compared with the current Entrez database entry for LRRK2 (accession no. AAV63975). Deviations in LRRK2 amino acid sequence used in this study include a Thr for Met at position 2397, corresponding to a common nonsynonomous polymorphism with a heterozygousity near 0.5 (dbSNP accession no. rs3761863). In addition, a Leu is present at position 212 in contrast to Ser; however, the sequence encoding a Ser was not detected in human DNA despite sequencing over 1,000 European cases and controls for the underlying polymorphism (S.B., unpublished observations).
Plasmid containing full-length LRRK2 was transiently transfected into HEK-293T cells and analyzed by Western blotting using antibodies directed against either the C-terminal myc tag or three peptide-derived LRRK2 antibodies raised against two distinct regions of the N terminus (JH5517 and JH5518) or the C terminus (JH5514). A single protein corresponding to LRRK2 (≈280 kDa) is detected in transfected cells in addition to several crossreactive proteins apparent in both transfected and nontransfected cells (Fig. 1D). Despite the presence of endogenous LRRK2 transcript in HEK-293T cells (Fig. 1C), no endogenous LRRK2 protein of the appropriate size is apparent by Western blot using the antibodies described here. Similarly, endogenous LRRK2 protein appears expressed at very low levels in adult human brain or whole mouse brain (data not shown).
Subcellular Localization of Overexpressed WT and Mutant LRRK2. Mutations in several genes related to neurodegeneration have previously been described to alter cellular localization and hence function of the encoded protein. To determine the cellular localization of overexpressed WT LRRK2 as well as LRRK2 harboring the G2019S and R1441C mutations, transfected HEK-293T cells were imaged by using confocal microscopy (Fig. 2A). Subcellular fractionation performed on cells overexpressing WT or mutant LRRK2 were separated into fractions enriched in cytoplasmic, nuclear, and mitochondrial proteins with successful enrichment assessed by relevant marker proteins (Fig. 2B). Transient expression of either WT or mutant LRRK2 is similar in cells compared with marker proteins. LRRK2 is largely excluded from the nucleus and present in a diffuse organization in the cytosol (Fig. 2 A and B). A significant amount (≈10%) of total LRRK2 is associated with the P10 mitochondrial-enriched fraction. The subcellular distribution of LRRK2 is similar in transfected SH-SY5Y cells (data not shown).
Fig. 2.

Subcellular localization of WT and mutant LRRK2. (A) Representative merged confocal images of transiently transfected HEK-293T cells. DAPI stain is indicated as blue, and green represents myc-FITC antibody staining for LRRK2. (Red scale bars, 10 μm.) (B and C) Subcellular fractionation of HEK-293T cells transiently transfected with myc-tagged WT or mutant LRRK2 and analyzed by Western blotting. Enrichment of each fraction was assessed by stripping the membrane and reprobing with the indicated marker antibody. Two micrograms of protein per lane was loaded, and LRRK2 expression was evaluated by using anti-myc antibody.
To determine whether LRRK2 is associated with the inside or outside of mitochondria, cell lysate enriched in cytosolic and mitochondrial proteins was partially digested with trypsin before fractionation. In this scenario, DJ-1, a known mitochondrial protein that is present in both the matrix and intermembrane space (14), is protected from digestion, whereas LRRK2 is completely digested (Fig. 2C). In addition, the mitochondrial outer-transmembrane protein porin is largely unaffected by brief trypsin exposure as opposed to LRRK2, which implies that LRRK2 is associated with the outer membrane of mitochondria but is unlikely to enter mitochondria. The G2019S and R1441C mutations have no robust effect on steady-state levels or cellular distribution.
Metabolism of WT and Mutant LRRK2. Familial-PD-linked mutations in the α-synuclein gene, DJ-1 gene, and PINK1 gene may adversely affect protein turnover contributing to the pathogenesis of PD (15). To determine whether mutations in LRRK2 may affect protein turnover, transiently transfected SH-SY5Y cells were treated with the proteasome inhibitor MG-132 or the lysosomal degradation inhibitor ammonium chloride (Fig. 3A). Whereas inhibition of the lysosomal degradation pathway has no effect on LRRK2 turnover, treatment with proteasome inhibitors results in the accumulation of LRRK2 (≈2-fold over 24 h) and the appearance of higher-molecular-weight LRRK2, which may correspond to polyubiquitinated protein. To determine whether LRRK2 is ubiquitinated under normal cellular conditions in SH-SY5Y cells, myc-tagged WT LRRK2 was cotransfected with or without HA-tagged ubiquitin followed by treatment with or without MG-132. Analysis of immunprecipitates confirmed the covalent attachment of ubiquitin to LRRK2 upon proteasome inhibition (Fig. 3B) consistent with polyubiquitination. As expected, ubiquitination of LRRK2 was less pronounced in the absence of MG-132, which is consistent with the idea that the proteasomal turnover of LRRK2 is mediated, at least in part, by polyubiquitination. Experiments using cycloheximide-chase assays to assess protein turnover demonstrate a long but equivalent half-life (>12 h) for WT and mutant LRRK2 in cell lines (data not shown). Thus, familial-linked mutations have no appreciable effect on the ubiquitin-mediated proteasomal turnover of LRRK2. Experiments were repeated using HEK-293T cells with similar results (data not shown).
Fig. 3.

Metabolism of WT and mutant LRRK2. (A) SH-SY5Y cells overexpressing WT or mutant LRRK2 were treated with either 5 μM MG-132 or 50 mM ammonium chloride for 24 h, and lysates were evaluated by Western blotting. LRRK2 was probed with anti-myc antibody, and the membrane was stripped and reprobed for β-actin. (B) SH-SY5Y cells were cotransfected with myc-LRRK2 with or without HA-ubiquitin, treated with or without 5 μM MG-132 for 24 h, and subjected to immunoprecipitation with anti-c-myc antibody. Immunprecipitates were probed with anti-HA or anti-c-myc antibodies as indicated.
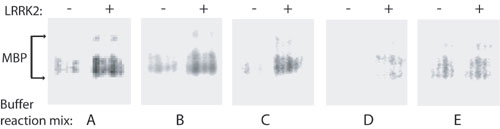
Kinase Activity of WT and Mutant LRRK2. LRRK2 encodes a kinase domain with highest similarity to the MLK motif found in proteins that commonly have both Ser/Thr and Tyr kinase activity. PD-linked mutations in LRRK2 have previously been suggested to affect intrinsic kinase activity, thereby leading to disease (16). To assess the kinase activity of WT and mutant LRRK2, we derived microgram quantities of recombinant LRRK2 at high purity by immunoprecipitation as assessed by silver stain (Fig. 4A). We next developed a kinase assay to measure the ability of WT or mutant LRRK2 to phosphorylate biotinylated MBP. Kinase buffer conditions were first optimized to increase the ratio of LRRK2-mediated phosphorylated MBP versus both background and nonspecific phosphorylation (Fig. 8, which is published as supporting information on the PNAS web site). In preliminary experiments, kinase reaction conditions previously reported for MLK activity also induced the highest level of LRRK2 kinase activity (Fig. 8, buffer C). Interestingly, buffer compositions previously used for other tyrosine kinases or Ser/Thr kinases performed poorly in activity assays, and assays using the tyrosine kinase-specific substrate poly(Glu)tyrosine revealed low but significant levels of LRRK2-mediated phosphorylation (data not shown), consistent with the assignment of LRRK2 as an MLK.
Fig. 4.

In vitro LRRK2 kinase assay. (A) Immunoprecipitated LRRK2 preparation derived from transfected HEK-293T cells as analyzed by SDS/PAGE and subsequent silver stain. (B) Assessment of WT or mutant LRRK2 protein levels by Western blot using anti-c-myc antibody. (C) In vitro kinase activity of recombinant WT or mutant LRRK2 against biotinylated MBP. Incorporated 32P in MBP was assessed by liquid scintillation, and results are representative of at least three independent experiments. Error bars represent mean ± SEM. n.s., not significant. *, P < 0.05 assessed by two-tailed unpaired Student's t test.
The amount of WT or mutant LRRK2 protein per kinase reaction was directly assessed by Western blotting, thereby normalizing the amount of phosphorylated MBP to the amount of LRRK2 in each experiment (Fig. 4B). Background and nonspecific phosphorylation was subtracted from all reactions containing LRRK2 by using immunoprecipitated myc-tagged DJ-1 protein as a negative control (data not shown). The G2019S mutant LRRK2 had significantly greater phosphorylation activity than WT protein (P < 0.05). In addition, we observed a trend toward increased kinase activity of R1441C mutant LRRK2 compared with WT; however, despite repeated experiments, the trend was not significant (P < 0.1, Fig. 4B). Recombinant LRRK2 was unable, under the conditions described here, to appreciably phosphorylate other recombinant proteins with importance in PD including parkin, MAPT, and α-synuclein in preliminary in vitro kinase assays.
Autophosphorylation Activity of WT and Mutant LRRK2. Using the same buffer and reaction conditions as in kinase activity assays with MBP, we measured the ability of recombinant LRRK2 protein to phosphorylate itself in the absence of any potential cofactors or activators. The amount of phosphorylated LRRK2 was normalized to the amount of input LRRK2 protein as determined by Western blotting (Fig. 5). Similar to experiments using MBP as a substrate, the G2019S mutant LRRK2 protein demonstrates increased autophosphorylation activity compared with WT protein (Fig. 5C). In these experiments, the R1441C mutation (located outside of the MLK domain) has significantly increased autophosphorylation activity versus WT protein. No other phosphorylation signals were apparent in these experiments, suggesting the absence of contaminating protein kinases, cofactors, or substrates in our immunoprecipitated LRRK2 preparations.
Fig. 5.

Autophosphorylation of LRRK2. (A) Autoradiogram of incorporated 32P in mutant or WT LRRK2 recombinant protein resolved by SDS/PAGE. (B) LRRK2 protein levels were determined by Western blot analysis by using anti-c-myc antibody. (C) Normalization of incorporated 32P compared with LRRK2 protein content. Data represent at least three independent experiments. a.u., arbitrary units. Error bars are mean ± SEM. *, P < 0.01; **, P < 0.001 assessed by two-tailed unpaired Student's t test.
Discussion
Recent advances in the molecular genetics of PD have unambiguously identified numerous genes that, when mutated, give rise to disease that mimics the clinical and pathologic characteristics of sporadic PD (17). The identification of variants within the LRRK2 gene in a small percentage of idiopathic late-onset PD cases suggests that LRRK2 may have a unique contribution to the development of PD. As yet, only a fraction of the entire LRRK2 gene has been assessed in PD cases; thus, it remains possible that the contribution of genetic variation in LRRK2 in PD susceptibility is underestimated. The functional characterization of LRRK2 and placement in biochemical pathways thought to have importance in PD may provide a critical link toward developing targeted therapeutics.
In this study, we defined the LRRK2 gene and transcript in human brain and characterized basic aspects of its biology. Despite the large size of LRRK2 protein, in silico analysis failed to indicate the presence of hydrophobic transmembrane domains or obvious mitochondrial or nuclear targeting sequences. The location of overexpressed LRRK2 protein was then determined on both an immunocytochemical and biochemical level. LRRK2 resides diffusely throughout the cytosol but also associates with the mitochondrial outer membrane, similar to the distribution of the E3 ubiquitin ligase parkin, also involved in the development of PD (18). More detailed analyses are now required to fully characterize the interaction of LRRK2 with mitochondria.
The familial mutations R1441C and G2019S, in the GTPase and MLK domains of LRRK2, respectively, did not have an effect on protein steady-state levels, localization, or turnover. Experiments demonstrate a long half-life for LRRK2 in cell lines; however, no LRRK2 accumulation was observed upon inhibition of the lysosomal degradation pathway. Rather, LRRK2 appears to be a substrate of the ubiquitin-proteasome system. The factors targeting LRRK2 for ubiquitination are as yet unknown, and preliminary experiments suggest that the E3 ligase protein parkin does not target LRRK2 for degradation (data not shown).
It is unclear at this time what aspects of LRRK2 function are relevant toward PD. Mutations in the serine/threonine kinase PINK1 were recently identified in autosomal-recessive PD cases (19). Therefore, we hypothesized that kinase activity of LRRK2 may be a critical feature of downstream function and developed a kinase assay using the generic kinase substrate MBP to determine whether the mutations G2019S and R1441C have an effect on more subtle enzymatic characteristics. The LRRK2 kinase domain shares greatest homology to proteins that are members of the MLK family, proteins known to have both serine/threonine and tyrosine kinase activity and have in vitro phosphorylation activity toward MBP. Consistent with this notion are our observations that reaction conditions that favor MLK activity promote greatest LRRK2 kinase activity.
In regard to both phosphorylation of a substrate and autophosphorylation activity, the familial LRRK2 mutations G2019S and R1441C enhance kinase activity. On a hypothetical structural basis, an increase in kinase activity for the G2019S alteration may be due to the perturbation of a stretch of amino acids termed the “activation segment” (Fig. 6). For many kinases, activation requires phosphorylation of the activation segment, and alteration due to mutations within this region may have the overall effect of increasing kinase activity, a condition associated with cancer and other diseases (20). If the critical feature of LRRK2 function related to PD susceptibility is kinase activity, then mutations that increase kinase activity fit well within a paradigm of the gain-of-function mechanism observed in families that inherit LRRK2 mutations in an autosomal-dominant mode, although the possibility of gene dosage or dominant negative effects cannot yet be ruled out.
Fig. 6.

Hypothetic effect of LRRK2 G2019S mutation on kinase activity. (A) Protein alignment of the activation segment of known protein kinase. Red amino acids represent alterations in the bRaf protein that overactivate corresponding kinase activity and predispose to cancer. Green amino acids represent PD-linked mutations in LRRK2. Amino acids in bold represent the conserved protein sequence that defines the activation segment. (B) An * indicates the position of the G2019S mutation within the activation segment. The diagram is adapted from ref. 20.
Known mutations in LRRK2 associated with PD are restricted neither to the activation segment of the kinase domain nor to the kinase domain as a whole. More than a dozen identified mutations suggest a broad distribution of mutation in the various functional domains of LRRK2 in PD cases. In this study, we describe an increase (albeit less dramatic) in kinase activity due to the R1441C mutation, an alteration within the GTPase domain of LRRK2. Because many kinases such as RAF and phosphatidylinositol 3-kinase family members associate with a corresponding activated GTPase protein to regulate kinase activity, the GTPase domain within LRRK2 may serve a self-regulatory role in defining the specificity and activity of its own kinase domain. The R1441C alteration may therefore serve to disrupt the normal regulatory interaction of the GTPase domain with the kinase domain, with the end result being a corresponding increase in kinase activity. Other LRRK2 mutations outside the kinase and GTPase domain might conceivably affect protein conformation or interaction with cofactors that regulate the specificity and kinase activity of LRRK2, perhaps with the final outcome being modulation of kinase activity. Our findings suggest that development of agents that abrogate LRRK2 kinase activity may offer therapeutic potential for the treatment of PD.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health, National Institute of Neurological Disorders and Stroke Grant NS 38377 and by grants from the Lee Martin Trust, the Sylvia Nachlas Trust, the National Parkinson Foundation, and the American Parkinson's Disease Association. T.M.D. is the Leonard and Madlyn Abramson Professor in Neurodegenerative Diseases.
Author contributions: A.B.W., D.J.M., and T.M.D. designed research; A.B.W., D.J.M., and A.B. performed research; A.B.W. contributed new reagents/analytic tools; A.B.W., D.J.M., S.B., C.A.R., W.W.S., V.L.D., and T.M.D. analyzed data; and A.B.W., D.J.M., and T.M.D. wrote the paper.
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: HA, hemagglutinin; LRRK2, leucine-rich repeat kinase 2; MBP, myelin basic protein; MLK, mixed-lineage kinase; PD, Parkinson's disease.
See Commentary on page 16535.
References
- 1.Funayama, M., Hasegawa, K., Kowa, H., Saito, M., Tsuji, S. & Obata, F. (2002) Ann. Neurol. 51, 296-301. [DOI] [PubMed] [Google Scholar]
- 2.Zimprich, A., Biskup, S., Leitner, P., Lichtner, P., Farrer, M., Lincoln, S., Kachergus, J., Hulihan, M., Uitti, R. J., Calne, D. B., et al. (2004) Neuron 44, 601-607. [DOI] [PubMed] [Google Scholar]
- 3.Paisan-Ruiz, C., Jain, S., Evans, E. W., Gilks, W. P., Simon, J., van der Brug, M., Lopez de Munain, A., Aparicio, S., Gil, A. M., Khan, N., et al. (2004) Neuron 44, 595-600. [DOI] [PubMed] [Google Scholar]
- 4.Nichols, W. C., Pankratz, N., Hernandez, D., Paisan-Ruiz, C., Jain, S., Halter, C. A., Michaels, V. E., Reed, T., Rudolph, A., Shults, C. W., et al. (2005) Lancet 365, 410-412. [DOI] [PubMed] [Google Scholar]
- 5.Di Fonzo, A., Rohe, C. F., Ferreira, J., Chien, H. F., Vacca, L., Stocchi, F., Guedes, L., Fabrizio, E., Manfredi, M., Vanacore, N., et al. (2005) Lancet 365, 412-415. [DOI] [PubMed] [Google Scholar]
- 6.Gilks, W. P., Abou-Sleiman, P. M., Gandhi, S., Jain, S., Singleton, A., Lees, A. J., Shaw, K., Bhatia, K. P., Bonifati, V., Quinn, N. P., et al. (2005) Lancet 365, 415-416. [DOI] [PubMed] [Google Scholar]
- 7.Zabetian, C. P., Samii, A., Mosley, A. D., Roberts, J. W., Leis, B. C., Yearout, D., Raskind, W. H. & Griffith, A. (2005) Neurology 65, 741-744. [DOI] [PubMed] [Google Scholar]
- 8.Farrer, M., Stone, J., Mata, I. F., Lincoln, S., Kachergus, J., Hulihan, M., Strain, K. J. & Maraganore, D. M. (2005) Neurology 65, 738-740. [DOI] [PubMed] [Google Scholar]
- 9.Adams, J. R., van Netten, H., Schulzer, M., Mak, E., McKenzie, J., Strongosky, A., Sossi, V., Ruth, T. J., Lee, C. S., Farrer, et al. (August 4, 2005) Brain, 10.1093/brain/awh607. [DOI]
- 10.Wszolek, Z. K., Pfeiffer, R. F., Tsuboi, Y., Uitti, R. J., McComb, R. D., Stoessl, A. J., Strongosky, A. J., Zimprich, A., Muller-Myhsok, B., Farrer, M. J., et al. (2004) Neurology 62, 1619-1622. [DOI] [PubMed] [Google Scholar]
- 11.West, A. B., Lockhart, P. J., O'Farell, C. & Farrer, M. J. (2003) J. Mol. Biol. 326, 11-19. [DOI] [PubMed] [Google Scholar]
- 12.Moore, D. J., Zhang, L., Dawson, T. M. & Dawson, V. L. (2003) J. Neurochem. 87, 1558-1567. [DOI] [PubMed] [Google Scholar]
- 13.Moore, D. J., Zhang, L., Troncoso, J., Lee, M. K., Hattori, N., Mizuno, Y., Dawson, T. M. & Dawson, V. L. (2005) Hum. Mol. Genet. 14, 71-84. [DOI] [PubMed] [Google Scholar]
- 14.Zhang, L., Shimoji, M., Thomas, B., Moore, D. J., Yu, S. W., Marupudi, N. I., Torp, R., Torgner, I. A., Ottersen, O. P., Dawson, T. M. & Dawson, V. L. (2005) Hum. Mol. Genet. 14, 2063-2073. [DOI] [PubMed] [Google Scholar]
- 15.Moore, D. J., West, A. B., Dawson, V. L. & Dawson, T. M. (2004) Annu. Rev. Neurosci. 28, 57-87. [DOI] [PubMed] [Google Scholar]
- 16.Kachergus, J., Mata, I. F., Hulihan, M., Taylor, J. P., Lincoln, S., Aasly, J., Gibson, J. M., Ross, O. A., Lynch, T., Wiley, J., et al. (2005) Am. J. Hum. Genet. 76, 672-680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dawson, T. M. & Dawson, V. L. (2003) Science 302, 819-822. [DOI] [PubMed] [Google Scholar]
- 18.Darios, F., Corti, O., Lucking, C. B., Hampe, C., Muriel, M. P., Abbas, N., Gu, W. J., Hirsch, E. C., Rooney, T., Ruberg, M. & Brice, A. (2003) Hum. Mol. Genet. 12, 517-526. [DOI] [PubMed] [Google Scholar]
- 19.Valente, E. M., Abou-Sleiman, P. M., Caputo, V., Muqit, M. M., Harvey, K., Gispert, S., Ali, Z., Del Turco, D., Bentivoglio, A. R., Healy, D. G., et al. (2004) Science 304, 1158-1160. [DOI] [PubMed] [Google Scholar]
- 20.Nolen, B., Taylor, S. & Ghosh, G. (2004) Mol. Cell 15, 661-675. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}