

Abstract
This is a multinanosecond molecular dynamics study of a bio–nano complex formed by a carbon nanoparticle, a buckyball C60, and a biological molecule, an antibody, with high binding affinity and specificity. In the simulation, the ball is completely desolvated by the binding site of the antibody by means of a nearly perfect shape complementarity and extensive side-chain interactions, with the exception that about 17% of the surface is persistently exposed to solvent and could be used for functional derivatization. The interactions are predominantly hydrophobic, but significant polar interactions occur as well. There exists a rich body of various π-stacking interactions. Aromatic side chains are involved in both double and triple stackings with the ball. Some ionic side chains, such as the guanidinium group of arginine, also form π-stackings with the ball. The results suggest that π-stackings are very efficient and common modes of biological recognition of π-electron-rich carbon nanoparticles. Most importantly, the results demonstrate that, in general, an ordinary protein binding site, such as that of an antibody, can readily bind to a carbon nanoparticle with high affinity and specificity through recognition modes that are common in protein–ligand recognition.
Keywords: molecular dynamics simulation‖molecular recognition‖bio–nano conjugate‖π-stackings
In 1985, a third allotropic form of carbon was discovered (1). The molecule was named Buckministerfullerene, commonly known as the buckyball, because of its geodesic structure (2). Six years later, the fullerene family was expanded with the discovery of nanotubes (3). Because of the unique structural properties associated with these molecules (4), there is great interest in using them in real-world applications (5–9), including integrating nanoparticles into biological systems, a fast-emerging field known as bio–nanotechnology. Examples of potential applications in bio–nanotechnology are transporting devices for drug delivery (10, 11), carriers of radioactive agents for biomedical imaging (12, 13), and templates for designing pharmaceutical agents, such as HIV type 1 protease inhibitors (8, 9), antioxidant (14–16), chemotactic agents (17), and neuroprotectants (18).
However, to introduce artificial nanomaterials into living cells, one must deal with issues such as water solubility, biocompatibility, and biodegradability. This requires a comprehensive understanding of the interactions of nanomaterials with biological molecules such as proteins, nucleic acids, membrane lipids, and even water molecules. As in the studies of protein interactions (19, 20), computer simulation techniques are very useful to investigate the interfacial properties of bio–nano systems, especially the dynamic, thermodynamic, and mechanical properties, at different spatial and temporal resolutions (21, 22). One particularly interesting subject is the study of the interactions of nanoparticles within the binding sites of proteins, and optimizing the interactions for improved bio–nano recognition.
Recent biochemical and structural studies reveal the existence of certain natural proteins that can recognize specific nanoparticles (23, 24). One such example is an antibody that was selected from the mouse immune repertoire to specifically recognize derivatized C60 fullerenes (23, 24) and had a binding affinity of ≈25 nM (23). The crystal structure of the Fab fragment of this antibody has been determined (23) (Fig. 1). Although the fullerene–antibody complex structure is not available, it was speculated that the fullerene-binding site is formed at the interface of the antibody light and heavy chains lined with a cluster of shape-complementary hydrophobic amino acid residues (23). The covalent modifications of the functionalized buckyball used in the experiments for solubility purpose occupy only a small fraction of the ball surface (see figure 1. in ref. 24); therefore, the unoccupied surface area would be large enough to interact with the antibody.
Figure 1.
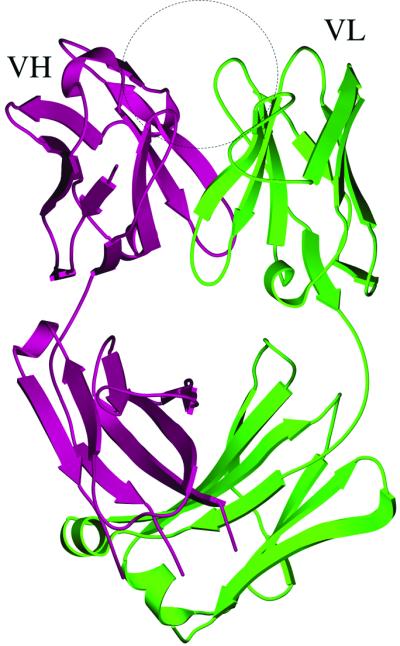
Ribbon diagram of the crystal structure of the Fab fragment of the fullerene-specific antibody (ref. 23; PDB ID code, 1EMT). The two polypeptide chains, variable region heavy chain (VH) and light chain (VL), are marked. The suggested binding site of the buckyball substrate is indicated by the circle. The figure is made by using software MOLSCRIPT (39) and rendered by RASTER3D (40).
To understand the detailed interactions of the buckyball in the binding site of the antibody, we study the buckyball–antibody complex by using molecular dynamics simulation (19). The purpose of our computational modeling/simulation is to identify the energetically favorable binding modes between the antibody and the buckyball. These results will be useful in developing new biologically compatible fullerene molecules.
Methods
We performed molecular dynamics simulations of a buckyball–antibody complex. The initial coordinates of the antibody were available from the Protein Data Bank (PDB ID code 1EMT) (23). The coordinates of the buckyball (C60) were provided by Richard E. Smalley at Rice University (Houston). Although original biochemical experiments were done on a derivatized buckyball for solubility reasons (24), we omitted the derivatizations in our simulation and focused on the ball–protein interactions. In this particular case, it seems reasonable to assume, as a first approximation, that the hydrophilic derivatizations on the ball do not play a critical role in the predominantly hydrophobic interactions between the ball and the antibody. Because all of the derivatizations were attached to the balls by two neighboring carbon atoms, we argue that the electronic structures of the derivatized balls, at the least the aromaticity, may not be disturbed dramatically at the opposite face, where the ball contacts the antibody.
Because the original x-ray structure of the antibody does not contain the buckyball substrate, we docked the ball into the suggested binding site (23). We then performed 200 steps of minimization, using the steepest descent method and 300 steps of minimization using the Adapted Basis Newton Raphson method. To reduce the necessary simulation time, the stochastic boundary molecular dynamics (SBMD) method was used (see ref. 25 for details). This is a highly efficient method for simulating the localized interactions in the active site of a protein as exemplified in a recent study of enzyme catalytic mechanism (26). The charmm program (27) was used for the simulation. Polar-hydrogen potential function (PARAM19) (28) was used for the protein and a modified TIP3P water model (29) was used for the solvent. Atomic partial charges for the buckyball were set to neutral (30) and the van der Waals parameters of its atoms were the same as an aromatic carbon atom (31) carried in the charmm force field (28). The system was separated into a reaction zone and a reservoir region, and the reaction zone was further divided into a reaction region and a buffer region (25). The reference point for partitioning the system in SBMD was chosen to be near the center of the buckyball. The reaction region around the active site was a sphere of radius r of 14 Å, the buffer region of 14 Å < r < 16 Å, and the reservoir region of r > 16 Å; all atoms in the reservoir region were deleted. The simulation system, shown in Fig. 2, consisted of 106 protein residues, a buckyball C60, and 166 water molecules. Atoms inside the reaction region were propagated by molecular dynamics, whereas atoms in the buffer region were propagated by the Langevin dynamics. Atoms inside the buffer region were retained by harmonic restoring forces with constants derived from the temperature factors in the crystal structure. Water molecules were confined to the active-site region by a deformable boundary potential (32). The friction constant in the Langevin dynamics was 250 ps−1 for protein atoms and 62 ps−1 for water molecules. During the simulation, all bonds with hydrogen atoms were fixed by using the shake algorithm (33). A 1-fs time step was used for integrating the equations of motion during the molecular dynamics simulation, whereas initial random velocities were sampled from the Boltzmann distribution (34). The system was equilibrated for 50 ps at 300 K, and was followed by a 5-ns production run.
Figure 2.

The molecular dynamics simulation system with the stochastic boundary condition. It contains 106 protein residues (ribbon), the buckyball (space-filling model in yellow), and 166 water molecules (ball-and-stick).
As an approximation, the simulated buckyball was treated as a nonpolarizable hydrophobic entity. To a first approximation, this treatment is reasonable based on the experimental observation that an unmodified buckyball is insoluble in water. The overwhelmingly large number of hydrophobic interactions in the binding site also justifies such a treatment.
We also simulated the systems containing the whole antibody molecule submerged in a large periodic water box with and without the presence of the buckyball in the binding site for a shorter period (200 ps). The results were compared with those from the SBMD simulation.
Results
We first observed during the 5-ns simulation that a single buckyball C60 molecule can be readily accommodated in the suggested binding site of the antibody. The ball inside the binding site undergoes a small relative translational motion, but a significant rotational motion. Further analysis of the angular momentum reveals no favored axis of rotation. The ball is nearly rigid, therefore the deformational motion of the ball is negligible. About 17% of the surface of the ball is exposed to solvent throughout the simulation, with the antibody covering the remaining surface. Fig. 3 shows the exposed surface area as a function of time in a 5-ns simulation window. The persistently solvent-exposed ball surface could be used for additional functional derivatization (24).
Figure 3.

Solvent-exposed surface area as a function of time in a 5-ns simulation window. The average value is about 17%.
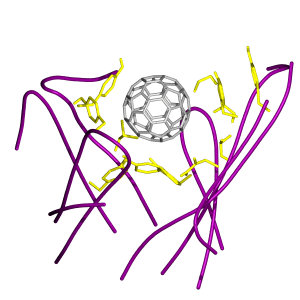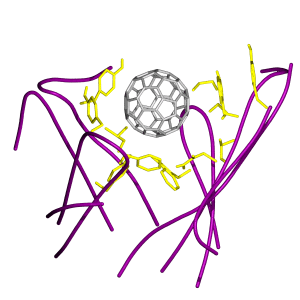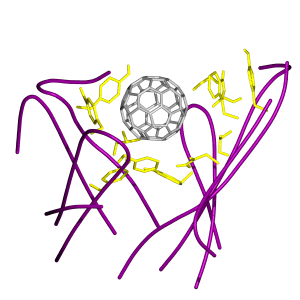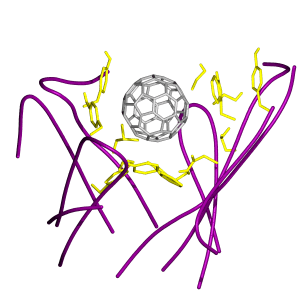
Although some of the ball–antibody interactions had been suggested from an earlier docking study (23), our results from molecular dynamics simulation are more thorough and reliable. Fig. 4a is a snapshot of the ball in the binding site, surrounded predominantly by hydrophobic amino acid side chains. Some of the important side chains of the antibody are explicitly shown, and the rest of the protein matrix is represented by a dotted surface. Of particular interest is the presence of rich π-interactions between the ball and the aromatic side chains of the antibody. Phe-96 (VL), Tyr-49 (VL), and Tyr-91 (VL) residues all form π-stacking arrangements with the ball. A three-tiered π-stacking interaction is observed between the ball, Tyr-52 (VH), and Trp-33 (VH) (Fig. 4b).
Figure 4.

(a) Stereo pair snapshot of the buckyball inside the binding site of the antibody. Some key protein side chains are explicitly drawn and the rest of the protein matrix is given in a dotted surface representation. The view is from the top in Fig. 1. (b) Stereo pair of a triple π-stacking. A piece of the buckyball and the side chains of Trp-33 (VH) and Tyr-52 (VH) are shown. (c) Stereo pair of stacking interactions made by Trp-47 (VH) and Phe-96 (VL). The Hɛ atom of Trp-47 (VH) points directly toward the ball, which can induce a weak hydrogen bond with the π-electron of the ball. Phe-96 (VL) is in stacking with the ball as well. (d) Stereo pair of a different configuration of the interactions in c. The side chain motions bring the guanidinium group of Arg-50 (VH) to a triple π-stacking with Trp-47 (VH) and the ball. The side chain of Phe-96 (VL) moves aside, but remains in stacking with the ball.
Another interesting interaction arises from the motion of the side chains of Trp-47 (VH) and Arg-50 (VH). Two distinct interaction modes made by these side chains were observed. Fig. 4c shows that Phe-96 (VL) is π-stacking with the ball while the Hɛ atom of Trp-47 (VH) points toward the ball, which induces a weak hydrogen bond with the rich π-electrons of the ball. In Fig. 4d, however, a rotation of the side chain of Trp-47 (VH) results in a triple π-stacking between the ball, the guanidinium group of Arg-50 (VH), and the side chain of Trp-47 (VH). In this case, the side chain of Phe-96 (VL), though moved aside, remains in π-stacking with the ball.
Similar π-stacking interactions have been observed in different antibody–antigen complexes (35) and other proteins (36). The interaction modes of aromatic side chains with the buckyball are also remarkably similar to those observed in the x-ray structure of a buckyball cocrystallized with benzene molecules (37). Moreover, in a recent experimental study (38), π-stacking was found to be very effective in noncovalently immobilizing functional groups on nanotubes. These results indicate that π-stacking is indeed a very efficient and common mode for biological recognition of π-electron-rich carbon nanoparticles.
In addition to the π-stacking interactions, the complementary shape of the antibody pocket also plays an important role in recognition. The interface between the ball and the antibody-binding pocket is nearly seamless and completely desolvated. Several hydrogen-bonded side chains and van der Waals contacts contribute to the formation of the complementary pocket. Trp-103 (VH) lies at the ball–antibody interface, but is not oriented in a manner expected for π-stacking; it is hydrogen bonded to another interfacial residue, Tyr-36 (VL). This hydrogen-bonding network extends to residues Asn-34 (VL) and Gln-89 (VL), both of which make contacts with the ball. Other residues that are in van der Waals contacts with the ball are Leu-46 (VL), Ala-97 (VH), and Ala-101 (VH).
For comparison, we also ran simulations of the systems containing the whole antibody molecule submerged in a large periodic water box with and without the buckyball in the binding site for a shorter period (200 ps). In the absence of the buckyball, we observed a big vacuum bubble around the hydrophobic binding site of the antibody. When the buckyball is present inside the binding site, the observed interactions are qualitatively similar to those from the SBMD simulation. It is worth pointing out that no electronic polarizability effect of the ball was included in our simulations. The observed rich π-stacking interactions arise primarily from the shape complementarity between the stacking aromatic side chains and the buckyball, which is a prerequisite for the stacking interactions.
Concluding Discussion
We have conducted a molecular dynamics study of a bio–nano complex formed by a carbon nanoparticle, a buckyball C60, and a biological molecule, an antibody with a high binding affinity and specificity. The results agree well with known biochemical (24) and structural (23) data of the system. The simulation shows that the high binding affinity and specificity are achieved through complementary shape and extensive side-chain interactions, including a set of rich π-stacking interactions. This finding also suggests that π-stacking is a very efficient and common mode for biological recognition of π-electron-rich carbon nanoparticles. It is notable that, in addition to tight binding, there is about 17% of the surface of the ball persistently exposed to the solvent. This amount of exposure may leave enough room for further manipulation of biocompatible buckyballs. Finally, the simulation results demonstrate that, in general, an ordinary protein binding site, such as that of an antibody, can readily bind to a carbon nanoparticle with high affinity and specificity through recognition modes that are common in protein–ligand recognition. A dynamic animation of the motion of the ball inside the binding site of the antibody can be found as Movie 1, which is published as supporting information on the PNAS web site, www.pnas.org.
Supplementary Material
Acknowledgments
We thank Prof. Richard E. Smalley and Dr. Kevin Ausman for their encouragement on the project and for providing us the atomic coordinates of the buckyball C60. We also thank Prof. Robert Hauge for helpful discussions. This work is supported by grants from National Science Foundation, American Heart Association, and The Robert A. Welch Foundation (to J.M.).
Abbreviations
- SBMD
stochastic boundary molecular dynamics
- VL
variable region light chain
- VH
variable region heavy chain
Footnotes
This paper results from the Arthur M. Sackler Colloquium of the National Academy of Sciences, “Nanoscience: Underlying Physical Concepts and Phenomena,” held May 18–20, 2001, at the National Academy of Sciences in Washington, DC.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Kroto H W, Heath J R, O'Brien S C, Curl R F, Smalley R E. Nature (London) 1985;318:162–163. [Google Scholar]
- 2.Yakobson B I, Smalley R E. Am Sci. 1997;85:324–337. [Google Scholar]
- 3.Iijima S. Nature (London) 1991;354:56–58. [Google Scholar]
- 4.Dresselhaus M S, Dresselhaus G, Eklund P C. Science of Fullerenes and Carbon Nanotubes. San Diego: Academic; 1996. [Google Scholar]
- 5.Collins P G, Arnold M S, Avouris P. Science. 2001;292:706–709. doi: 10.1126/science.1058782. [DOI] [PubMed] [Google Scholar]
- 6.Balavoine F, Schultz C, Richard C, Mallouh V, Ebbesen T W, Mioskowski C. Angew Chem Int Ed. 1999;38:1912–1915. doi: 10.1002/(SICI)1521-3773(19990712)38:13/14<1912::AID-ANIE1912>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 7. Da Ros, T. & Prato, M. (1999) J. Chem. Soc. Chem. Commun., 663–669.
- 8.Friedman S H, DeCamp D L, Sijbesma R P, Srdanov G, Wudl F, Kenyon G L. J Am Chem Soc. 1993;115:6506–6509. [Google Scholar]
- 9.Sijbesma R, Srdanov G, Wudl F, Castoro J A, Wilkins C, Friedman S H, DeCamp D L, Kenyon G L. J Am Chem Soc. 1993;115:6510–6512. [Google Scholar]
- 10.Wilson L J, Cagle D W, Thrash T P, Kennel S J, Mirzadeh S, Alford J M, Ehrhardt G J. Coord Chem Rev. 1999;190:199–207. [Google Scholar]
- 11.Wilson L J. Interface. 1999;8:24–28. [Google Scholar]
- 12.Cagle D W, Kennel S J, Mirzadeh S, Alford J M, Wilson L J. Proc Natl Acad Sci USA. 1999;96:5182–5187. doi: 10.1073/pnas.96.9.5182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wharton T, Alford J M, Husebo L O, Wilson L J. Recent Advances in the Chemistry and Physics of Fullerenes and Related Materials. Vol. 9. Pennington, NJ: Electrochem. Soc.; 2000. pp. 258–266. [Google Scholar]
- 14.Straface E, Natalini B, Monti D, Franceschi C, Schettini G, Bisaglia M, Fumelli C, Pincelli C, Pellicciari R, Malorni W. FEBS Lett. 1999;454:335–340. doi: 10.1016/s0014-5793(99)00812-1. [DOI] [PubMed] [Google Scholar]
- 15.Lin A M, Chyi B Y, Wang S D, Yu H H, Kanakamma P P, Luh T Y, Chou C K, Ho L T. J Neurochem. 1999;72:1634–1640. doi: 10.1046/j.1471-4159.1999.721634.x. [DOI] [PubMed] [Google Scholar]
- 16.Chueh S C, Lai M K, Lee M S, Chiang L Y, Ho T I, Chen S C. Transplant Proc. 1999;31:1976–1977. doi: 10.1016/s0041-1345(99)00234-1. [DOI] [PubMed] [Google Scholar]
- 17.Toniolo C, Bianco A, Maggini M, Scorrano G, Prato M, Marastoni M, Tomatis R, Spisani S, Palu G, Blair E D. J Med Chem. 1994;37:4558–4562. doi: 10.1021/jm00052a015. [DOI] [PubMed] [Google Scholar]
- 18.Dugan L L, Turetsky D M, Du C, Lobner D, Wheeler M, Almli C R, Shen C K, Luh T Y, Choi D W, Lin T S. Proc Natl Acad Sci USA. 1997;94:9434–9439. doi: 10.1073/pnas.94.17.9434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brooks C L, III, Karplus M, Pettitt B M. Adv Chem Phys. 1988;71:1–249. [Google Scholar]
- 20.McCammon J A, Harvey S. Dynamics of Proteins and Nucleic Acids. Cambridge, U.K.: Cambridge Univ. Press; 1987. [Google Scholar]
- 21.Xu C, Scuseria G E. Phys Rev Lett. 1994;72:669–672. doi: 10.1103/PhysRevLett.72.669. [DOI] [PubMed] [Google Scholar]
- 22.Yakobson B I, Brabec C J, Bernholc J. J Comp Aided Mater Des. 1996;3:173–182. [Google Scholar]
- 23.Braden B C, Goldbaum F A, Chen B X, Kirschner A N, Wilson S R, Erlanger B F. Proc Natl Acad Sci USA. 2000;97:12193–12197. doi: 10.1073/pnas.210396197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen B X, Wilson S R, Das M, Coughlin D J, Erlanger B F. Proc Natl Acad Sci USA. 1998;95:10809–10813. doi: 10.1073/pnas.95.18.10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brooks C L, III, Karplus M. J Mol Biol. 1989;208:159–181. doi: 10.1016/0022-2836(89)90093-4. [DOI] [PubMed] [Google Scholar]
- 26.Ma J, Zheng X, Schnappauf G, Braus G, Karplus M, Lipscomb W N. Proc Natl Acad Sci USA. 1998;95:14640–14645. doi: 10.1073/pnas.95.25.14640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brooks B R, Bruccoleri R E, Olafson B D, States D J, Swaminathan S, Karplus M. J Comput Chem. 1983;4:187–217. [Google Scholar]
- 28.Neria E, Fischer S, Karplus M. J Chem Phys. 1996;105:1902–1921. [Google Scholar]
- 29.Jorgensen W L. J Am Chem Soc. 1981;103:335–340. [Google Scholar]
- 30.Andreoni W. Annu Rev Phys Chem. 1998;49:405–439. doi: 10.1146/annurev.physchem.49.1.405. [DOI] [PubMed] [Google Scholar]
- 31.Klein D J, Schmalz T G, Hite G E, Seitz W A. J Am Chem Soc. 1986;108:1301–1302. [Google Scholar]
- 32.Brooks C L, III, Karplus M. J Chem Phys. 1983;79:6312–6325. [Google Scholar]
- 33.Ryckaert J P, Ciccotti G, Berendsen H J C. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 34.Allen M P, Tildesley D J. Computer Simulation of Liquids. Oxford: Clarendon; 1980. [Google Scholar]
- 35.Braden B C, Souchon H, Eisele J L, Bentley G A, Bhat T N, Navaza J, Poljak R J. J Mol Biol. 1994;243:767–781. doi: 10.1016/0022-2836(94)90046-9. [DOI] [PubMed] [Google Scholar]
- 36.Hu G, Gershon P D, Hodel A E, Quiocho F A. Proc Natl Acad Sci USA. 1999;96:7149–7154. doi: 10.1073/pnas.96.13.7149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Meidine, M. F., Hitchcock, P. B., Kroto, H. W., Taylor, R. & Walton, D. R. M. (1992) J. Chem. Soc. Chem. Commun., 1534–1537.
- 38.Chen R J, Zhang Y, Wang D, Dai H. J Am Chem Soc. 2001;102:3838–3839. doi: 10.1021/ja010172b. [DOI] [PubMed] [Google Scholar]
- 39.Kraulis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 40.Bacon D J, Anderson W F. J Mol Graphics. 1988;6:219–220. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}