

Abstract
Previous attempts to investigate the origin of the Icelanders have provided estimates of ancestry ranging from a 98% British Isles contribution to an 86% Scandinavian contribution. We generated mitochondrial sequence data for 401 Icelandic individuals and compared these data with >2,500 other European sequences from published sources, to determine the probable origins of women who contributed to Iceland’s settlement. Although the mean number of base-pair differences is high in the Icelandic sequences and they are widely distributed in the overall European mtDNA phylogeny, we find a smaller number of distinct mitochondrial lineages, compared with most other European populations. The frequencies of a number of mtDNA lineages in the Icelanders deviate noticeably from those in neighboring populations, suggesting that founder effects and genetic drift may have had a considerable influence on the Icelandic gene pool. This is in accordance with available demographic evidence about Icelandic population history. A comparison with published mtDNA lineages from European populations indicates that, whereas most founding females probably originated from Scandinavia and the British Isles, lesser contributions from other populations may also have taken place. We present a highly resolved phylogenetic network for the Icelandic data, identifying a number of previously unreported mtDNA lineage clusters and providing a detailed depiction of the evolutionary relationships between European mtDNA clusters. Our findings indicate that European populations contain a large number of closely related mitochondrial lineages, many of which have not yet been sampled in the current comparative data set. Consequently, substantial increases in sample sizes that use mtDNA data will be needed to obtain valid estimates of the diverse ancestral mixtures that ultimately gave rise to contemporary populations.
Introduction
The settlement of Iceland represented the final phase of a series of range expansions that characterized the human inhabitation of northwestern Europe. Historical records indicate that Iceland was discovered by Vikings just before 870 ad. Between the late 8th century and the mid-13th century ad, the Vikings travelled extensively in their long ships, trading and raiding from Scandinavia to the Baltic region and Russia in the east; to France and Spain in the south; and to England, Ireland, Scotland, and the North Atlantic Islands in the west. The Viking age was the outcome of a number of different historical causes: improved ship-building techniques, land shortage in Scandinavia due to increasing population sizes and primogeniture inheritance laws, increasing political centralization at home, and growing opportunities abroad due to widespread political disintegration in the rest of Europe (Jones 1984; Collins 1991). Iceland had no inhabitants prior to its colonization between 870 and 930 ad. Archaeological studies of building styles and artifactual assemblages from the settlement era in Iceland support assertions made in Icelandic medieval literature that the country was colonized from Scandinavia and from Norse settlement areas in the British Isles (Smith 1995). This is further corroborated by the fact that Icelandic is unequivocally a Norse language and is presumed to be closer to Old Norse than other languages currently spoken in Scandinavia.
The ancestry of the settlers is more controversial. Historical evidence suggests that not all of the settlers in Iceland originated from Scandinavia (Jones 1984). At the very least, it is believed that the settlers included a number of women and slaves from Norse settlements in the British Isles. There are numerous references in Icelandic medieval writings to the keeping of slaves, many of whom were obtained through raids on settlements in the British Isles (Jones 1984). Thus, the Icelandic founding gene pool may have received a substantial maternal contribution from the British Isles. A number of previous studies have attempted to determine the admixture proportions between these potential parental populations, using serological markers. However, the results have been inconsistent, with estimates of ancestry ranging from 98% Celtic (Thompson 1973) to 86% Scandinavian (Wijsman 1984). Other studies have concluded either that the Icelandic population is a genetic outlier within Europe (Cavalli-Sforza et al. 1994) or that classical genetic markers provide insufficient information to draw any definite conclusions about Icelandic ancestry (Tills et al. 1982). To date, the issue of the origins of the Icelanders remains unresolved.
Besides the controversy surrounding the ancestry of the Icelanders, there is also the issue of whether the relatively small size of the Icelandic population has resulted in a significant reduction of genetic diversity, compared with other contemporary European populations. Historians have estimated that the original settlement of Iceland involved 8,000–20,000 individuals over a 60-year period (Steffensen 1975, pp. 446). Thereafter, the population is believed to have increased rapidly, to ∼70,000 by the end of the 12th century, after which it went into a gradual decline, falling to 40,000 at the end of the 18th century. This period of decline was punctuated by a number of abrupt and drastic reductions in population size. The most severe of these demographic bottlenecks occurred during the years 1402–1404, when 45% of the population was eliminated by an outbreak of the pneumonic plague. Other significant decreases in population size included a 35% reduction in 1708 due to a particularly severe smallpox epidemic (leading to an all-time postsettlement minimum of 33,000) and a 20% reduction in 1784–1785 due to widespread famine resulting from a volcanic eruption. From the 19th century onward, the population grew rapidly, to its present size of 270,000. This demographic history is likely to have reduced the genetic diversity introduced by the original settlers. Moreover, since the natural barrier of the North Atlantic has tended to hinder postsettlement immigration to the island, there would have been no mechanism to replenish genetic diversity lost as a consequence of demographic fluctuation. It can thus be assumed that virtually all mtDNA lineages observed in the contemporary Icelandic population are descended from the original set of mtDNA lineages present in the female founders 1,100 years ago.
To resolve some of the issues surrounding the ancestry of the Icelandic female founding population and the consequences of postsettlement population decline, we performed a survey of mtDNA sequence variation in a large sample of contemporary Icelanders. Since the precision of phylogeographic analyses is directly related to sequence length, we chose to generate sequence data for both the first and second hypervariable segments (HVS1 and HVS2) of the control region, as well as three restriction sites. A high mutation rate, coupled with a virtual lack of recombination, has resulted in a diverse set of mtDNA lineages that, by virtue of their clustering into phylogenetic clades (Sykes 1999; Macaulay et al. 1999), provides substantially more information about ancestral origins than does the comparison of allele frequencies of classical genetic markers. We performed a detailed comparison of existing mtDNA sequence data from other European populations, to assess ancestral contributions to the Icelandic gene pool and to examine the relative configuration of genetic diversity in the Icelanders.
Material and Methods
Population Samples
DNA was obtained from the blood of a random sample of 401 maternally unrelated Icelandic males and females. All these individuals were volunteers for control groups used for genetic disease studies by deCODE Genetics, Inc. Overall, the makeup of our sample approximately reflects the geographic pattern of habitation in Iceland. The appropriate informed consent was obtained from each individual in this study. The European HVS1 and HVS2 sequences used for comparative purposes are listed by population, along with their source publications, in tables 1 and 2.
Table 1.
mtDNA HVS1 Sequence Diversity in the Icelanders and Other European Populationsa
| Population (Reference [n])b | SampleSize | No. ofLineages | No. ofVariableSites | GeneDiversity | θπ (SD) | θk (95% CI) | θs (SD) | % ofPrivateLineagesc |
| Turks (Calafell et al. 1996 [27], Comas et al. 1996 [45]) | 72 | 63 | 70 | .995 | 5.28 (2.86) | 237.1 (127.9–464.8) | 14.44 (4.04) | 61.9 |
| Germans (Hofmann et al. 1997 [67], Lutz et al. 1998 [200], Richards et al. 1996 [151]) | 418 | 219 | 104 | .981 | 4.07 (2.25) | 185.1 (151.4–226.3) | 15.73 (3.35) | 58.0 |
| Near East (DiRienzo et al. 1991) | 42 | 37 | 58 | .994 | 6.76 (3.61) | 144.8 (65.9–345.8) | 13.48 (4.18) | 78.4 |
| French (Rousselet and Mangin 1998) | 50 | 42 | 48 | .988 | 4.08 (2.30) | 120.9 (62.0–249.3) | 10.72 (3.29) | 45.2 |
| Norwegians (Opdal et al. 1998) | 216 | 123 | 87 | .952 | 3.68 (2.07) | 117.7 (89.3–155.4) | 14.62 (3.44) | 52.8 |
| Spanish (Corte-Real et al. 1996 [71], Handt et al. 1998 [11], Pinto et al. 1996 [18], Salas et al. 1998 [94]) | 192 | 112 | 88 | .950 | 4.04 (2.24) | 111.6 (83.2–149.8) | 15.09 (3.60) | 50.9 |
| British (Piercy et al. 1993 [100], Richards et al. 1996 [69]) | 167 | 98 | 81 | .964 | 3.84 (2.15) | 98.5 (72.0–135.1) | 14.23 (3.48) | 50.0 |
| Estonians (Sajantila et al. 1996) | 26 | 23 | 30 | .988 | 4.35 (2.47) | 91.3 (34.9–266.1) | 7.86 (2.83) | 30.4 |
| Italians (Francalacci et al. 1996) | 49 | 39 | 53 | .967 | 4.75 (2.62) | 86.3 (46.0–169.0) | 11.89 (3.62) | 64.1 |
| Austrians (Handt et al. 1994 [16], Parson et al. 1998 [101]) | 117 | 73 | 73 | .957 | 4.21 (2.33) | 82.0 (56.4–119.8) | 13.68 (3.55) | 45.2 |
| Canary Islanders (Pinto et al. 1996) | 54 | 41 | 49 | .976 | 5.10 (2.79) | 75.9 (42.6–139.6) | 10.75 (3.25) | 65.9 |
| Russians (Orekhov et al. 1999 [103], Sajantila et al. 1995 [29]) | 132 | 74 | 59 | .967 | 4.03 (2.24) | 68.7 (48.5–97.7) | 10.81 (2.82) | 44.6 |
| Danes (Richards et al. 1996) | 31 | 25 | 28 | .985 | 5.41 (2.98) | 57.7 (26.6–133.5) | 7.01 (2.46) | 32.0 |
| Icelanders (this study [394], Richards et al. 1996 [14], Sajantila et al. 1995 [39]) | 447 | 125 | 71 | .975 | 4.27 (2.34) | 57.2 (45.8–71.3) | 10.63 (2.36) | 55.2 |
| Portuguese (Corte-Real et al. 1996) | 54 | 37 | 38 | .934 | 3.42 (1.97) | 50.5 (29.2–89.0) | 8.34 (2.60) | 35.1 |
| Sardinian (DiRienzo et al. 1991) | 69 | 43 | 48 | .936 | 4.00 (2.24) | 47.8 (29.6–77.7) | 9.99 (2.92) | 51.2 |
| Finns (Kittles et al. 1999 [74], Pult et al. 1994 [23], Richards et al. 1996 [29], Sajantila et al. 1995 [50]) | 176 | 74 | 64 | .957 | 3.61 (2.04) | 47.6 (34.7–64.9) | 11.14 (2.78) | 37.8 |
| Basque (Bertranpetit et al. 1995 [45], Corte-Real et al. 1996 [61]) | 106 | 53 | 49 | .936 | 2.84 (1.67) | 41.5 (28.1–61.2) | 9.36 (2.57) | 32.1 |
| Swedes (Kittles et al. 1999 [28], Sajantila et al. 1996 [32]) | 60 | 37 | 49 | .952 | 4.24 (2.36) | 40.1 (24.1–67.4) | 10.51 (3.13) | 35.1 |
| Swiss (Pult et al. 1994) | 76 | 42 | 38 | .967 | 3.27 (1.89) | 37.8 (24.0–59.6) | 7.75 (2.30) | 26.2 |
| Bulgarians (Calafell et al. 1996) | 30 | 22 | 34 | .977 | 4.13 (2.35) | 35.6 (17.1–77.0) | 8.58 (2.97) | 36.4 |
| Karelians (Sajantila et al. 1995) | 83 | 43 | 39 | .963 | 3.70 (2.09) | 35.2 (22.8–54.4) | 7.82 (2.29) | 32.6 |
| Welsh (Richards et al. 1996) | 92 | 45 | 47 | .926 | 3.14 (1.82) | 34.2 (22.5–51.8) | 9.23 (2.60) | 44.4 |
| Adygei (Macaulay et al. 1999) | 50 | 30 | 36 | .952 | 4.73 (2.61) | 30.7 (17.7–53.8) | 8.04 (2.54) | 46.7 |
| Druze (Macaulay et al. 1999) | 45 | 26 | 30 | .959 | 4.20 (2.36) | 24.8 (13.9–44.5) | 6.86 (2.26) | 50.0 |
| Saami (Sajantila et al. 1995) | 115 | 25 | 29 | .815 | 3.79 (2.13) | 9.6 (5.9–15.0) | 5.45 (1.61) | 40.0 |
Populations are arranged in descending order by θk values.
Additional data from Kirgiztan, the US, and the North Atlantic Islands were obtained from Comas et al. (1998), Handt et al. (1998), and Miller et al. (1996; Mitochondrial DNA Concordance), respectively.
Private lineages are defined as those found only in one of the populations included in the present study.
Table 2.
mtDNA HVS2 Sequence Diversity in the Icelanders and Other European Populationsa
| Population (Reference [n]) | SampleSize | No. ofLineages | No. ofVariableSites | GeneDiversity | θπ (SD) | θk (95% CI) | θs (SD) | % ofPrivateLineagesb |
| Turks (Calafell et al. 1996) | 27 | 22 | 28 | .99 | 3.70 (2.15) | 53.1 (23.1–130.5) | 7.26 (2.62) | 36.4 |
| Germans (Hofmann et al. 1997 [67], Lutz et al. 1998 [197]) | 264 | 82 | 44 | .90 | 2.54 (1.51) | 40.4 (30.5–53.1) | 7.15 (1.80) | 54.9 |
| French (Rousselet and Mangin 1998) | 60 | 31 | 26 | .92 | 2.34 (1.44) | 25.1 (15.1–41.7) | 5.58 (1.81) | 41.9 |
| Austrians (Parson et al. 1998) | 99 | 40 | 31 | .91 | 2.49 (1.50) | 24.5 (16.1–36.9) | 6.00 (1.78) | 27.5 |
| Italians (Francalacci et al. 1996) | 49 | 26 | 26 | .93 | 3.06 (1.80) | 21.7 (12.4–37.9) | 5.83 (1.94) | 38.5 |
| British (Piercy et al. 1993) | 118 | 48 | 42 | .90 | 3.00 (1.75) | 20.2 (20.2–43.2) | 7.86 (2.18) | 39.6 |
| Orkney Islanders (Miller et al. 1996) | 44 | 23 | 25 | .92 | 2.91 (1.73) | 18.7 (10.4–33.6) | 5.75 (1.96) | 17.4 |
| Icelanders (this study) | 346 | 50 | 32 | .89 | 2.20 (1.35) | 15.8 (11.4–21.6) | 4.98 (1.30) | 28.0 |
| Bulgarians (Calafell et al. 1996) | 30 | 17 | 16 | .94 | 2.59 (1.59) | 15.4 (7.7–31.1) | 4.04 (1.56) | 17.6 |
| Saami (Delghandi et al. 1998) | 58 | 6 | 11 | .62 | 1.90 (1.22) | 1.5 (.6–3.3) | 2.38 (.94) | 16.7 |
Populations are arranged in descending order by θk values.
Private lineages are defined as those found only in one of the populations included in the present study.
Markers and Protocols
All mtDNA site numbers referred to in this study are in accordance with the scheme introduced by Anderson et al. (1981). The entire mitochondrial control region was amplified by use of the primers L15999 (5′-CACCATTAGCACCCAAAGCT-3′) and H409 (5′-CTGTTAAAAGTGCATACCGCC-3′). Amplification reactions were performed on 10 ng of template DNA in a 20-μl volume by use of AmpliTaq Gold polymerase (PE Biosystems). The cycle profile started with 95°C for 12 min, followed by 25 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 1 min. Both hypervariable segments were sequenced by use of the BigDye Terminator Cycle Sequencing kit from PE Biosystems on an ABI PRISM 377 (PE Biosystems) DNA sequencer. The primers L15999 (HVS1) and L16498 (HVS2) (5′-CCTGAAGTAGGAACCAGATG-3′) were used for cycle-sequencing reactions. More than 70% of the samples were sequenced for both strands of HVS1 (H16498 5′-CATCTGGTTCCTACTTCAGG-3′), which provided an overlap between sites 16024 and 16394. The cycle-sequencing profile was 25 cycles of 96°C for 10 s, 50°C for 5 s, and 60°C for 4 min. The sequences were aligned and manually checked in SEQUENCHER 3.1 (GeneCodes). Initial sequencing of HVS1 resulted in 55 individuals with high-quality sequence between sites 16028 and 16394. Subsequent runs resulted in reliable sequence data for 339 additional individuals, from site 16028, to or beyond 16519. A total of 346 individuals were sequenced for HVS2 between sites 1 and 297. Discrepancies in the numbers of individuals typed for these markers are the result of a limited supply of DNA from some samples. Three segments of the mitochondrial coding region, containing sites 7028, 9052, and 12308, respectively, were amplified for all 401 individuals (these markers are described in more detail in Torroni et al. 1996). The following primer pairs were used: site 7028 L6909 (5′-AAGCAATATGAAATGATCTG-3′) H7115 (5′-CGTAGGTTTGGTCTAGG-3′), site 9052 L8845 (5′-CCTAGCCATGGCCATCC-3′) H9163 (5′-GGCTTACTAGAAGTGTGAAAAC-3′), and site 12308 L12124 (5′-CTCAACCCCGACATCATTACC-3′) H12309 (5′- ATTACTTTTATTTGGAGTTGCACCAAGATT-3′). Each of the three resulting amplicons were digested with the enzyme appropriate to assay the respective site’s character states (AluI for 7028, HaeII for 9052, and HinfI for 12308). The data produced for this study were deposited in Genbank (under accession numbers (AF236888-AF237289).
A total of 185 maternal relatives were typed as quality controls for each marker for 109 of the individuals included in the study. A comparison of maternal relatives for HVS1 (16028–16394, a total of 288 sequences) and HVS2 (1–297, a total of 266 sequences) revealed five erroneous single-base calls missed during manual checking in SEQUENCHER 3.1. This amounts to a transcription error rate of 2.71×10-5 per base examined. No transcription errors were observed in a specific check of the highly polymorphic site 16519 in 273 sequences.
Summary Statistics and Interpopulation Analysis
To maximize the number of other populations in comparison with the Icelanders, summary statistics and interpopulation genetic distances were calculated for HVS1 between sites 16090 and 16365 and for HVS2 between sites 063 and 297. Gene diversity was estimated as  , where n is the total number of sequences, k the number of distinct lineages, and pi the frequency of the distinct lineages. This index represents the probability that two randomly chosen sequences from a sample would be nonidentical by state.
, where n is the total number of sequences, k the number of distinct lineages, and pi the frequency of the distinct lineages. This index represents the probability that two randomly chosen sequences from a sample would be nonidentical by state.
Mean pairwise differences between sequences (θπ) were calculated as  , where dij is the number of mutational differences between lineages i and j in a sample, k is the number of distinct lineages, and pi and pj are the respective frequencies of lineages i and j. θπ provides a good indication of the overall mutational space covered by a set of genetic lineages.
, where dij is the number of mutational differences between lineages i and j in a sample, k is the number of distinct lineages, and pi and pj are the respective frequencies of lineages i and j. θπ provides a good indication of the overall mutational space covered by a set of genetic lineages.
θs was estimated as 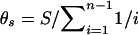 , where S is the number of polymorphic sites in a sample of sequences and n is the number of sequences. θk was estimated by use of the formula
, where S is the number of polymorphic sites in a sample of sequences and n is the number of sequences. θk was estimated by use of the formula  , where k is the number of distinct lineages observed in a sample size of n. Each of these indices, along with their SDs or 95% confidence intervals (CIs), were calculated in the software package ARLEQUIN 1.1 (Schneider et al. 1997). Supplementary programs were written to calculate θk values for large sample sizes and to generate lists of expected numbers of lineages, given a specified θk value for a range of sample sizes.
, where k is the number of distinct lineages observed in a sample size of n. Each of these indices, along with their SDs or 95% confidence intervals (CIs), were calculated in the software package ARLEQUIN 1.1 (Schneider et al. 1997). Supplementary programs were written to calculate θk values for large sample sizes and to generate lists of expected numbers of lineages, given a specified θk value for a range of sample sizes.
The three θ indices referred to above use different aspects of the genetic data to estimate 2Nfeμ (where Nfe represents the female effective-population size and μ the mutation rate) and are based on different assumptions. Because the mtDNA control-region mutation rate should be the same in all populations, differences in θ values reflect differences in female effective-population size (Nfe)—that is, the harmonic mean of the number of women who have transmitted their mitochondrial DNA to female offspring during past generations. θπ estimates the female effective-population size that would have allowed the observed number of pairwise differences between sequences to arise through mutation events in a single population. This means that it tends to reflect the harmonic mean of Nfe over long periods of time and is, in such cases, strongly affected by ancient demographic fluctuations (Rogers and Harpending 1992). The estimators θs and θk, based as they are on the relationship between sample size and the number of polymorphic sites and the number of distinct lineages, respectively, are more sensitive to the effects of lineage sorting during recent demographic history. The validity of these latter two θ estimators is dependent on three important assumptions: (1) that selection is not influencing the locus, (2) that population size has been sufficiently constant to maintain a steady-state distribution of lineages, and (3) that either every new mutation occurs at a unique site—the infinite-sites assumption (applies to θs)—or that each new mutation creates a lineage that is distinguishable by state from all others—the infinite-alleles assumption (applies to θk).
Pairwise genetic distances between populations were calculated by use of ARLEQUIN 1.1 (Schneider et al. 1997) and were represented in two-dimensional space by use of multidimensional scaling analyses in the SPSS 8.0 software package.
Phylogenetic Analysis
A median-joining network was generated to infer phylogenetic relationships between Icelandic mtDNA lineages, by use of the program NETWORK 2.0 (Bandelt et al. 1995; Röhl 1997). To maximize the precision of phylogenetic reconstruction, we used the 297 Icelandic samples sequenced, between sites 16028 and 16519 for HVS1 and between sites 1 and 297 for HVS2, and typed for the three coding region sites at positions 7028, 9052, and 12308. These latter sites have been identified as informative for resolution of the subclusters of lineages defined as haplogroups H, K, and U, respectively (Torroni et al. 1996). No sites were dropped from the analysis (see Macaulay et al. 1999), although insertions and deletions were ignored. Because of the presence of mutation-rate heterogeneity in the control region, a weighting scheme was used whereby sites were divided into three groups—fast, average, and slow—in accordance with the findings of Wakeley (1993) and Hasegawa et al. (1993) and as used by Richards et al. (1998). Seven sites were given low weight (0.5): 16093, 16192, 16311, 16362, 146, 150, and 152, and the three coding-region RFLP markers were given a high weight (2). The remaining sites were assigned a weight of 1.
Results
mtDNA Diversity in Iceland and Europe
Summary statistics for HVS1 lineages in the Icelanders and other European populations are presented in table 1. A total of 125 different HVS1 lineages, characterized by 71 polymorphic sites, were observed in the sample of 447 Icelanders. The standard indices of genetic heterogeneity provide an ambiguous picture of European mtDNA diversity and the relative position of the Icelanders therein. In almost all cases, potential estimation errors due to sampling variance are too great to allow any real confidence in apparent differences between populations. However, if the observed values are taken at face value, it emerges that, in terms of both gene-diversity and mean base-pair differences between sequences (θπ), the Icelanders are among the more diverse populations in Europe. In contrast, a comparison of θ values based on the number of segregating sites (θs) and on the observed number of different lineages (θk) indicates that the Icelanders have recently had a relatively small female effective-population size. In this regard, they group with populations such as the Finns, Basques, Welsh, and Saami.
Table 2 shows the same summary statistics for HVS2 lineages for the Icelanders and a restricted number of European populations. Here we observe 50 different lineages, defined by 32 polymorphic sites, in a total sample of 346 Icelanders. Again, the potential sampling error is great, and few of the observed differences between populations are statistically robust. There are, however, a number of interesting differences between the summary statistics for the two hypervariable segments of the control region. First, the Icelandic HVS2 lineages are more obviously homogeneous for all diversity indices. Second, the θk and θs values are substantially lower for all the populations included in both tables (θk is roughly four times smaller, and θs is roughly two times smaller). This suggests, as others have previously observed, that HVS2 has a lower average mutation rate than HVS1 (Francalacci et al. 1996).
We note that the three θ values exhibit considerable disparity in European populations. θπ shows little correlation with either θk or θs for HVS1 sequences (r=.45 and .21, respectively) but shows higher correlations for HVS2 (r=.70 and .70, respectively). Whereas θk and θs exhibit a strong correlation for both HVS1 and HVS2 (r=.78 and .75, respectively), there is a considerable difference in their magnitudes. This discrepancy, which is high for HVS1 in European populations and is slightly lower in the case of HVS2, indicates the existence of mutational hotspots in the mtDNA control region. The occurrence of back mutations invalidates the infinite-sites assumption that applies to θs but only to a lesser degree than it invalidates the infinite-alleles assumption that applies to θk. This is because, although back mutations do not add to the number of polymorphic sites, they do generate new identifiable lineages when occurring on a novel genetic background created by mutations at other sites. Consequently, in the case of the control region, θk is likely to provide the more reliable estimate of recent female effective-population size. However, since violations of the assumptions about the mutational process in the control region apply equally to different populations, θk and both θs should be valid for interpopulation comparisons.
The populations in tables 1 and 2 are arranged in descending order by their θk values. It is notable that the populations for which HVS1 and HVS2 data are available appear in a similar order in both tables. Thus, whereas the only clear discontinuity is between the Saami and all other populations, the Turks, Germans, French, British, Italians, and Austrians seem to have had relatively large female effective-population sizes, whereas the Icelanders, Bulgarians, and Saami consistently exhibit lower values.
The frequency spectrum of Icelandic mtDNA HVS1 and HVS2 lineages differs from that observed in the nine European populations with sample sizes large enough to allow a reasonable comparison (n>100). After adjustment for sample size, there are fewer lineages in the Icelanders, of which a relatively large proportion are unique. Of the 125 Icelandic HVS1 lineages, only 56 are found elsewhere in Europe, and 29 of these lie outside the European frequency range (as defined when comparative populations were grouped according to the regions indicated on the map in fig. 1). Out of the 36 Icelandic HVS2 lineages shared with other populations, 15 are outside the observed European frequency range.
Figure 1.

Map of Europe, showing the populations included in this study. The colors show a classification of sampled populations into larger geographic regions. This grouping scheme is used in a number of analyses presented in this paper. The arrows show presumed routes of settlement from Scandinavia and the British Isles to Iceland between 870 and 930 ad. The North Atlantic Islands are situated between Iceland, Scotland, and Norway and are (from right to left on the map) the Shetland Islands, the Orkney Islands, and the Faroe Islands.
Interpopulation Differences
One way to quantify the distinctiveness of the frequency distribution of Icelandic mtDNA lineages is to calculate pairwise genetic distances between populations that are based solely on lineage-frequency differences. Figure 2A and 2B shows multidimensional scaling plots for HVS1 and HVS2, respectively, where genetic distance matrices are represented in two-dimensional space. To make sample sizes comparable for these analyses, populations were grouped into the geographical regions displayed in figure 1. For both hypervariable segments of the control region, the peripheral position of the Icelanders highlights the considerable difference between the Icelandic frequency spectrum of lineages and those found in neighboring regions. The Saami exhibit a much larger deviation from the general European distribution of lineages but are excluded from the multidimensional scaling plots, to ensure that the distances between other populations are discernible.
Figure 2.

Representations in two-dimensional space of genetic distances between European geographic regions based on HVS1 and HVS2 lineage frequencies. The correspondence between the distances on two dimensional plots and those in the genetic distance matrices is (A) ∼98% and (B) ∼95%, respectively.
Another way to summarize genetic differences between populations, based on the analysis of molecular variance (AMOVA) method (Excoffier et al. 1992), makes use of the average base-pair differences between the lineages from two populations, corrected for intrapopulation variation. Because this method is slightly less sensitive to lineage-frequency differences, we performed the analyses at the level of individual populations. Genetic distances based on the AMOVA method provide a summary of the differences in the phylogeographic configuration of lineages from a given set of populations—to the extent that pairwise base differences between lineages reflect the actual distance in substitutions between the lineages on the true phylogenetic tree. For HVS1, 98.4% of the genetic variance observed in Europe is contained within individual populations (99.3% if the Saami are excluded), with the same proportions for HVS2 being 97.6% and 99.3%, respectively. These high percentages indicate the absence of phylogeographic structure of mtDNA lineages in the current data set of European populations.
Taken at face value, figure 3A indicates that the Icelanders’ closest genetic neighbors are the Welsh and British populations, whereas the more limited HVS2 data in figure 3B identify the Austrians and Germans as genetic neighbors. In both cases, the Icelanders remain European outliers, although there is a less marked difference in figure 2A and 2B.
Figures 3.

Representations in two-dimensional space of genetic distances between European populations based on an AMOVA. For both A and B, the correspondence between the distances on two-dimensional plots and those in the distance matrices is ∼80%.
Phylogenetic Portrait of Icelandic mtDNA Lineages
Figure 4 shows a median-joining phylogenetic network of the 135 Icelandic lineages in the 297 individuals typed for HVS1 (16028–16519), HVS2 (1–297), and sites 7028, 9052, and 12308. No less than 52 of the 116 sites in the network are represented as having multiple mutation hits, of which sites 152, 16519, 16362, 16093, 195, 16192, 16311, 150, 204, and 073 each require five or more recurrent mutations.
Figure 4.

Median-joining network of Icelandic mtDNA lineages for HVS1 (16028–16519), HVS2 (1–297), and the RFLP markers 7028, 9052, and 12308. Circles are proportional to lineage frequencies, in which the smallest circles represent single-copy lineages and the largest circle represents 21 copies of the same lineage. Bold circle outlines indicate lineages unique to the Icelanders, according to the comparative HVS1 data set described in table 1. Lines represent substitutions and are proportional to the number of substitutions between lineages. Transitions are indicated on the lines by site number; transversions are indicated by site number and base. Reticulations in the network, in which it has been impossible to resolve a recurrent mutation at one or more sites, are represented by parallelogram-like shapes. In such cases, the parallel lines represent possible mutations at the same site. Dotted lines indicate unlikely mutational routes. The haplogroup membership of lineages is indicated by labels at the edges of the network and by the color-coding of circles. The position of the Cambridge reference sequence is indicated by an asterisk.
Although our network conforms broadly to those suggested by previous studies (Torroni et al. 1996; Richards et al. 1998; Macaulay et al. 1999), a number of new details emerge. A loss of an HaeII restriction site at position 9052, usually taken to indicate a lineage’s membership within haplogroup K, is in our data shown to be present on two lineages within haplogroup I. Hence, it is inadvisable to rely solely on the 9052 RFLP marker to assign lineages to haplogroup K. Site 073 in HVS2, another position widely typed as a stable phylogenetic marker, is shown by the network to have undergone five separate mutations (similar results are reported by Izagirre and de la Rúa 1999). This goes against the use of site 073 to assign haplogroup H membership (see Richards et al. 1998). A transition at site 072 in HVS2 is shown to be the founder motif of haplogroup V, of which 16298C identifies only a subcluster of lineages (see Richards et al. 1998; Torroni et al. 1998).
A number of researchers have identified the 16519 site as being too hypervariable to include in phylogenetic analyses (e.g. Forster et al. 1996; Torroni et al. 1998; Macaulay et al. 1999). Curiously, however, both Forster et al. (1996) and Brown et al. (1998) observed that site 16519 was invariable in haplogroups B and X, respectively. Although our data suggest that site 16519 has undergone multiple mutations, it nonetheless seems to be largely fixed for most haplogroups. Thus, with only a few exceptions, all sequences belonging to haplogroups K, U4, U3, I, X, and T in the Icelanders have 16519C, whereas haplogroups V, J, and U5 have 16519T. The only haplogroup with which site 16519 seems to be at variance is H, where roughly half of the lineages have 16519C. An examination of the data presented in Torroni et al. (1996) and Macaulay et al. (1999) reveals the same pattern of association between 16519 and the aforementioned haplogroups. Our data show that many of the lineages with 16519T, which would previously have been classed in haplogroup H, are found in separate clusters supported by additional substitutions: 16304C in lineage cluster H1 and 16311C+7028 in cluster HV2. Lineages belonging to H1 are curiously rare in other European populations, given their abundance in the Icelanders. Although lineages with 16304C are found in a number of populations, only motifs 16304–16274 (Germany) and 16304–16305 (Faroe Islands) show any overlap with the Icelandic H1 cluster.
In general, then, almost all the European haplogroups identified and defined by recent studies (Torroni et al. 1996; Richards et al. 1998; Macaulay et al. 1999) are present in the Icelanders, with the exception of U1, U2, U3, and U6. Table 3 shows the frequencies of haplogroups in Iceland and Europe, along with information about their geographic distribution in Europe. The Icelanders exhibit a typically European pattern of haplogroup frequencies, notwithstanding a relative scarcity of haplogroup H and the slight abundance of haplogroups I, J, and T.
Table 3.
Frequency and Geographic Distribution of European Haplogroups
| % of Icelandic |
Approximate % of European |
||||
| Haplogroup | Sequences(n=325) | Lineages(n=135) | Sequencesa(n=946/n=365)b | Lineagesa(n=396/n=205)c | HighestFrequenciesin Europea |
| Hb | 39.4 | 43.7 | 50 .0 | 38.0 | All |
| I | 5.5 | 5.2 | 2.0 | 3.0 | NW |
| J | 13.5 | 12.6 | 11.0 | 8.3 | All |
| K | 7.7 | 7.4 | 7.0 | 7.1 | All |
| T | 11.7 | 8.1 | 8.0 | 9.1 | All |
| U3b | .0 | .0 | 1 .0 | 1.0 | S |
| U4b | 2.8 | 2.2 | 1 .0 | 2.9 | S |
| U5 | 8.3 | 10.4 | 7.0 | 10.4 | NW and SW |
| U6b | .0 | .0 | 1 .0 | 1.0 | SW |
| V | 2.2 | 3.0 | 4.0 | 3.9 | N and SW |
| W | .3 | .7 | 1.0 | 1.8 | All |
| X | 1.5 | .7 | 2.0 | 3.0 | All |
| Other | 7.4 | 5.9 | 1.7 | 4.0 | |
| Total | 100.0 | 100.0 | 96.7 | 93.5 | |
This information was adapted from an analysis of 942 sequences by Richards et al. (1998). N = north, W = west, and S = south.
Because of the difficulty of assigning lineages to haplogroups on the basis of HVS1 sequences alone, Richards et al. (1998) used a restricted set of 365 sequences (typed for site 73 of HVS2) to determine lineage membership of haplogroups H, V, U3, U4, and U6. This is most probably the reason why the sums of columns 4 and 5 do not add up to 100.
Richards et al. (1998) did not report the no. of distinct lineages in the full and restricted data sets. However, according to our estimates, the full European data set of 942 sequences contained 396 lineages and the restricted data set of 365 sequences contained 205 lineages.
Geographic Origin of Icelandic mtDNA Lineages
A number of Icelandic lineages appear to be geographically informative. The subcluster of haplogroup J in the top left corner of figure 4 (J1b1 according to the terminology of Richards et al. 1998) occurs at a surprisingly high frequency in the Icelanders. In addition to the 22 Icelanders bearing lineages that belong to this cluster, this HVS1 motif has so far only been observed in Northern Ireland (1), the Hebrides (1), England (3), Wales (3), Norway (1), France (1), Germany (1), the United States (7), and Kirgizistan (1). Given the high frequency of this subcluster in Iceland, its unusually restricted northern European geographic distribution, and its prevalence in the British Isles, it is possible that lineages belonging to this subcluster arrived in Iceland 1,100 years ago with females of British origin. The fact that J1b1 lineages with the substitution 16192T have, to date, only been found in Iceland, Northern Ireland, the Hebrides, and the United States lends even more support to this interpretation. The J1b1 lineages from the United States are likely to be descended from Irish, British, and Scandinavian settlers. Another lineage that also supports the historical evidence of gene flow from the British Isles or Scandinavia occurs within haplogroup K and is defined by the substitution 16320T. Although 11 Icelanders carry lineages with this motif, it has, to date, been found only in Norway (2), England (1), Germany (1), and the United States (2).
Interestingly, we find two copies of a lineage from the non-European haplogroup C in the Icelanders. Haplogroup C is common in Asia and South America, but this particular lineage has also been found in Spain—probably as a result of that population’s contact with the New World after the 15th century. However, it seems likely that the Icelandic haplogroup C lineages arrived from Asia via Scandinavia. The single haplogroup-Z lineage also has Asian ties and is not widely observed in Europe (see Schurr et al. 1999). Potential European ancestors of this lineage are only found in the Norwegians, Saami, and Russians (but without the 16362C substitution).
A more quantitative means to assess the possible geographic origins of the lineages currently observed in the Icelanders can be achieved by way of an analysis of lineage sharing. On the basis of the range of published mitochondrial control–region mutation rates (Parsons et al. 1997; Jazin et al. 1998), it is expected that most of the mtDNA lineages presently found in the Icelanders will be identical by state to those carried by female ancestors who arrived at the time of settlement. Icelandic lineages that are not found in putative parental populations can be accounted for by one of the following explanations: (1) they were in the parental population(s) at the time of settlement but have since been lost, (2) they were generated by mutational events in Iceland during the past 1,100 years, or (3) they currently exist in the parental population(s) but have not yet been sampled. We determined the number of Icelandic lineages shared with at least one other European population (hereafter referred to as “shared lineages”). For this analysis, we grouped European populations into eight regions (as shown in fig. 1). An equivalent analysis was performed to account for Icelandic “private” lineages (those that so far have not been observed in other populations), in which potential founder lineages were identified as those differing by the fewest number of substitutions from the Icelandic private lineages. These analyses are based on HVS1 sites 16090–16365 and HVS2 sites 063–297.
Frequency-based admixture analyses are not appropriate in the current study of mtDNA control–region lineages in the Icelanders. Such methods have been shown to provide misleading results in cases in which (1) the parental populations are not easily distinguished with respect to lineage frequencies and (2) the lineage frequencies of one or more of the admixed or parental populations has been subject to stochastic change through genetic drift (Bertorelle and Excoffier 1999). Our results suggest that both of these criteria apply in the case of the Icelanders. Methods that incorporate information about the differences between lineages at the molecular level (e.g., Bertorelle and Excoffier 1999) are no more appropriate, because they assume a more marked phylogeographic structure of mtDNA lineages than can be observed in Europe (see Richards et al. 1998).
Of the 125 HVS1 lineages observed in the Icelanders, 56 are shared with other European populations. In the case of HVS2, the Icelanders share 36 of their 50 lineages with other European populations. Figure 5 shows the pattern of lineage sharing between the Icelanders and these populations grouped into geographic regions, which are ordered in accordance with geographic proximity and historical evidence about participation in the settlement of Iceland. Thus, the fifth column in figure 5 indicates that, although the combined regions of Scandinavia, British Isles, Finland/Estonia, and that of the Saami account for 44 of the 56 shared lineages, an additional 9 shared Icelandic lineages are found in the northwestern Europeans (Germans, Austrians, and the Swiss).
Figure 5.

Bar chart showing the pattern of lineage sharing between the Icelanders and other European populations, classed into eight geographic regions. The full height of each column represents the number of Icelandic lineages that are found in the specified region. The blackened area represents the cumulative increase in the number of Icelandic lineages accounted for when regions are combined sequentially according to geographic proximity to Iceland (from left to right on the figure). This amounts to not counting a shared lineage found in a region after the same lineage has been found in a previous region. Finally, the diagonally shaded area represents the number of Icelandic lineages that are shared only with a single region in the current data set.
The results of this analysis do not seem to fit the simple model of admixture between Scandinavian and British populations. On the basis of the lineage sharing alone, it would be reasonable to postulate that the Icelandic population was founded by a random collection of 9th-century Europeans. When the cumulative increase in the number of accounted lineages is examined, however, it emerges that this is mainly due to the prevalence of a number of common lineages in almost all European regions. However, even with the more parsimonious cumulative approach (see fig. 5 legend), the potential range of source populations for Icelandic lineages remains surprisingly large. Although just <80% of the 56 shared Icelandic HVS1 lineages can be accounted for in Scandinavia, the British Isles, and Finland/Estonia, it is still necessary to look at northwest and southwest Europe to account for them all. For HVS2 lineages, we have a more restricted set of comparative population data, but the pattern is similar.
What of the 69 HVS1 and 14 HVS2 lineages that are found only in the Icelanders? In this case, we are looking for lineages that are ancestral to Icelandic private lineages or, in other words, the lineages that differ by the fewest number of base substitutions. In almost all cases, ancestral lineages were identified that differed by only one substitution. For HVS1, there were six Icelandic private lineages that differed by two substitutions from their nearest ancestral lineages. For HVS2, there was only one such private lineage. In general, more than one different ancestral lineage was identified for any one Icelandic private lineage. These were regarded as equally probable ancestors. We included the Icelanders as a potential source of ancestral lineages, since it is possible that a portion of the private lineages arose through mutation events in Iceland. Figure 6 shows the results of this analysis. Although similar proportions of HVS1 ancestral lineages can be found all over Europe, 57 of the 69 Icelandic private lineages can be accounted for by putative ancestral lineages present in the Icelanders. For 15 of these, potential ancestral lineages are found only in the Icelanders, suggesting perhaps that they are true private lineages, generated by mutation events in Iceland during the past 1,100 years. Of the few ancestral lineages that cannot be found in Iceland, most are found in Scandinavia and the British Isles, although it is still necessary to turn to northwest and southwest Europe, and even to the Near East, to account for all 69 Icelandic private lineages.
Figure 6.

Bar chart showing the geographic distribution of putative ancestral lineages to Icelandic private lineages. The full height of each column represents the number of Icelandic private lineages that can be accounted for by ancestral lineages sampled from the specified region. The blackened area represents the cumulative increase in the number of Icelandic private lineages that can be accounted for by ancestral lineages when regions are combined sequentially according to geographic proximity to Iceland (from left to right on the figure). Hence, once an Icelandic private lineage has been accounted for by an ancestral lineage in one region, it is excluded from the blackened area of columns for subsequent regions. The diagonally shaded area represents the number of Icelandic private lineages that can be accounted for only by an ancestral lineage from a single geographic region in the current data set.
A different picture emerges in the case of Icelandic HVS2 private lineages. Almost all of these lineages can be accounted for in any one of the European regions. Iceland does not provide a greater number of potential ancestor lineages, and none are exclusive to Iceland or any other region. Almost any combination of two regions provides ancestral lineages for all 14 of the HVS2 lineages found only in the Icelanders.
How Many European mtDNA Lineages Have Yet to Be Sampled?
The reliability of our analysis of admixture that is based on lineage sharing is dependent on the assumption that putative parental populations have been adequately sampled. The index of θk models the relationship between a given sample size and the observed number of different lineages. This relationship makes it possible to use the observed values of θk for different populations to predict the expected number of additional lineages that could be identified if the sample size were increased. The relationship between sample size and the expected number of distinct HVS1 lineages was examined for the 10 European populations with sample sizes >100 (others were excluded, to minimize the effect of errors in the estimation of θk).
It is reasonable to ask this question: given our observed θk values, how many more individuals from each of the populations would we need to sample before we have an adequate picture of the variation contained in its mtDNA gene pool (see Ward et al. 1993; Francalacci et al. 1996)? The criterion for when to stop sampling is necessarily arbitrary. Ewens's (1972) sampling formula, used to calculate θk, dictates that the relationship between sample size and the expected number of lineages will be one of diminishing returns. We define the sample-size cutoff point as that when, for repeated incremental increases in sample size of 10, we obtain less than one new lineage. The adjusted sample sizes and expected number of lineages obtained from applying these assumptions to the observed θk values are shown in table 4.
Table 4.
Additional HVS1 Lineages Estimated for Increases in Sample Size on the Basis of θk Values
| Population (θk) | na | kb | Adjustednc | Adjustedkd |
| Germans (185.25) | 418 | 219 | 1,680 | 428 |
| Norwegians (117.73) | 216 | 123 | 1,070 | 273 |
| Spanish (111.56) | 192 | 112 | 1,010 | 258 |
| British (98.52) | 167 | 98 | 900 | 229 |
| Austrian (81.99) | 117 | 73 | 750 | 190 |
| Russian (68.73) | 132 | 74 | 630 | 160 |
| Basque (41.51) | 106 | 53 | 380 | 97 |
| Finns (47.55) | 176 | 74 | 440 | 111 |
| Icelanders (57.25) | 447 | 125 | 530 | 134 |
| Saami (9.55) | 115 | 25 | 100 | 24 |
Actual sample size.
No. of observed lineages.
On the basis of the observed value of θk, sample size was iteratively increased by 10, and the no. of expected lineages was estimated. The number in this column represents the sample size at which <1 new lineage was detected.
No. of expected lineages based on the observed value of θk and the adjusted sample size.
According to table 4, the current sample sizes for the Icelanders and Saami are close to reaching levels of lineage saturation with respect to the criterion specified above. All other populations appear to require 200%–300% increases in sample size to reach equivalent levels of saturation. This finding is supported by Pfeiffer et al.’s (1999) report that the rate of detection of new distinct lineages from a single German village showed no sign of decline, even when 700 individuals had been sampled. Thus, given the number of lineages that presumably have yet to be sampled from the British Isles and Scandinavia, these populations may be sufficient to account for all the lineages currently observed in the Icelanders.
Discussion
Origin of the Icelanders and Their Recent Population History
The analysis of Icelandic mitochondrial lineages in the context of European mtDNA variation provides important information about the genetic history and ancestry of the population. Our findings suggest that the Icelandic mtDNA gene pool contains relatively fewer distinct lineages than most other European populations and that the frequencies of a number of these lineages deviate considerably from those found elsewhere in Europe. This is consistent with current knowledge about Icelandic population history. The Icelanders have been a small and isolated population throughout their history (relative to most European populations). It is also known that variance in fertility was unusually high during recent Icelandic history (Vasey 1996), a factor that would have served to further decrease the female effective-population size and thereby increase levels of genetic drift and the potential for founder effects. This would not only have altered the frequencies of Icelandic mtDNA lineages, it would also have resulted in the extinction of a proportion of the founding lineages from the Icelandic gene pool.
The last 1,100 years of mtDNA evolution in Europe have principally been a history of lineage redistribution, within populations because of drift and between populations because of migration. The settlement of Iceland was equivalent to a migrational sampling event of existing genetic diversity at one or more locations in Europe. The subsequent genetic history of the Icelanders has primarily involved stochastic sampling of these founder lineages between generations (interspersed with a small number of mutation events)—the outcome of which is well reflected by the values of θs and θk. Icelandic HVS1 sequences are characterized by a smaller-than-average number of polymorphic sites and a relatively small number of distinct lineages, reflected in small θs and θk values, respectively. An examination of these indices in European populations (where sample sizes are >99, to minimize the effect of sampling error) revealed that the Icelanders are grouped with the Saami, Basques, and Finns—European populations with relatively small, recent female effective-population sizes. A similar pattern emerged from the HVS2 data set, although a lower HVS2 mutation rate results in lower values for all θ estimators and a higher proportion of Icelandic lineages shared with other European populations.
Nonetheless, the Icelanders do harbor a considerable amount of mtDNA diversity, as is indicated by their gene-diversity and mean pairwise-difference (θπ) values, which are among the highest in Europe for HVS1. Although this appears to conflict with the fact that many of the populations with smaller values for these indices have 50–300-fold larger current and historical population sizes, Pfeiffer et al. (1999) have demonstrated that gene-diversity values for HVS1 sequences in a German population were underestimated in sample sizes <200. Since most of the comparative population samples used in this study are well below this number, many of the gene-diversity values for HVS1 in European populations could be underestimated.
As regards θπ, it is important to bear in mind that the bulk of the mtDNA substitutions in the Icelandic data set arrived in Iceland on the lineages carried by female settlers, and many are likely to have existed even before most present European populations were established. As a result, θπ values primarily provide information about female demographic history at a time long before Iceland was settled. All that the unusually high Icelandic θπ value really tells us is that this population’s lineages occupy a relatively large proportion of the mutational space in the overall European phylogeny, implying that the founding females did not carry a homogeneous set of mtDNA lineages with them to Iceland. This could be taken as evidence against the idea that the Icelanders are exclusively descended from a localized region of one population (such as the west coast of Norway). However, more-extensive regional sampling from Scandinavia and the British Isles is required before any such inferences can be reliably made. Given the number of lineages that have yet to be sampled from each of these regions, it is possible that either might contain a configuration of lineages sufficient to account for almost all of those currently observed in the Icelanders.
As it stands, our analysis of lineage sharing supports the idea that both Scandinavian and British populations could have contributed the majority of the mtDNA lineages that are currently observed in the Icelandic gene pool. Given the present comparative data set, however, it is impossible to identify the parental populations with certainty or to quantify admixture proportions between them. These results are at variance with a recent study of the phenylaline hydroxylase gene in 17 Icelandic phenylketonuria (PKU) patients (Guldberg et al. 1997). That study concluded that a large number of founders from Ireland and Scotland was unlikely, since three of the most common PKU mutations in Ireland and Scotland were not among the nine observed in the Icelandic PKU patients. In fact, an examination of PKU mutation data presented in Eiken et al. (1996), Guldberg et al. (1997), Zschocke et al. (1997), and Tyfield et al. (1997) reveals that the most common Norwegian PKU mutation is not found in the Icelanders. Moreover, although seven of the nine Icelandic mutations are found in Scotland and Ireland (eight if southwest England is included), only five are found in the Norwegians. If mutation frequencies are compared, the Norwegians appear to be more similar to the British populations than either are to the Icelanders. Here again, we are most likely observing the effects of drift on the Icelandic gene pool. Indeed, Guldberg et al. (1997) note that drift is likely to be responsible for the high frequency of one PKU mutation (Y377fsdelT, 42%) that has, to date, been found only in the Icelanders. The PKU data, then, seem to be as inconclusive as the other studied genetic systems about the relative proportions of admixture between Scandinavians and Britons.
It is important to bear in mind that our analysis of mtDNA lineages can provide information only about the geographic origin of the females who colonized Iceland and who have matrilineal descendants in the present population. If historical reports are to be believed and a disproportionate number of the founding females were not Scandinavian (relative to the proportion of founding males), then a moderate discrepancy between autosomal markers and mtDNA lineages is to be expected. In this case, one would expect a disproportionate number of mtDNA lineages to be shared with individuals from the British Isles (and perhaps other European populations). Further studies of autosomal and Y-chromosome molecular markers will have to be undertaken in the relevant populations before firmer assertions can be made about such discrepancies.
Implications for Studies of mtDNA in European Populations
Our phylogenetic analysis of the full Icelandic mtDNA data set revealed one of the most extensive phylogenies of mtDNA lineages presented for a single human population. The character states of HVS1 and HVS2 sequences are phylogenetically well correlated in the Icelandic data, and both regions provide important resolution in the network. We have clarified the phylogenetic context of the highly variable site 16519, which is generally stable in mtDNA haplogroups. The only exception is haplogroup H, which is split in two by this site. On the basis of these findings, it is interesting to speculate whether further analyses of other parts of the mtDNA genome will reveal that haplogroup H is in fact a composite of two or more smaller haplogroups. We have also demonstrated that sites 073 and 9052 are less stable than was suggested by previous studies. Overall, our results lead us to advise against the selective exclusion of sites in phylogenetic analyses with respect to preconceptions about their hypervariability but instead to recommend the use of maximum sequence length in such analyses.
We have demonstrated that there are likely to be numerous mtDNA lineages that have yet to be sampled in most European populations. This deficiency in the European data set presents a problem in the use of mtDNA to address many important questions relating to the continent’s population history. It seems clear, for example, that sample sizes from candidate parental populations would need to be increased substantially to provide a more accurate assessment of the geographic origin of Icelandic mtDNA lineages.
Methods that are based on the identification of founder lineages in parental populations are also affected, such as the use of ρ to date the time frame of genetic divergence between populations (see definition in Forster et al. 1996). In the Icelandic case, ρ would be defined as the average mutational distance between Icelandic lineages and the nearest putative founder lineages observed in Europe. Valid interpretation of this index requires the assumption that all ancestral founding lineages have been observed in the European data and that all novel lineages found in Iceland have been generated by mutations that occurred subsequent to settlement. The parameter ρ provides an average mutational difference of .46 substitutions between Icelandic HVS1 lineages and putative European founder lineages (for the region defined by sites 16090–16365). Given a mutation rate of one transition every 20,186 years (as used by Forster et al. 1996 and Richards et al. 1998), this implies that Icelandic-specific genetic diversity has accumulated over a period of ∼9,646 years—a figure that is almost nine times larger than the historically and archaeologically supported date of 1,100 years. For this estimation, we defined a “founder lineage” as the European lineage that was nearest to a novel Icelandic lineage in terms of observed base-pair differences. Although these mutational differences should be calculated on the basis of a phylogenetic tree (Forster et al. 1996), recurrent mutations at a number of HVS1 sites make this a near-impossible task for such a large data set of nearly 3,000 relatively short European sequences (see Wakeley 1993; Meyer et al. 1999). However, since phylogenetic-based mutation differences can only be larger than raw base-pair differences, our calculations can be viewed as minimum estimates of ρ.
A time frame of 9,646 years for the divergence of the Icelanders from neighboring European populations is clearly a considerable overestimate, presumably because of the large number of founding lineages that have yet to be sampled from European populations. An alternative, but less plausible, explanation for this inflated figure is that the HVS1 average mutation rate has been severely underestimated (see Parsons et al. 1997; Jazin et al. 1998). Either way, this finding has implications for other studies that use estimators like ρ to date genetic divergence between populations. Thus, for example, the paleolithic dates suggested by Richards et al. (1996, 1998) and Sykes (1999) for the separation of the European mtDNA gene pool from that of the Near East may likewise be overestimates. The number of individuals sampled from the Near East is still relatively small, compared with most European populations, particularly given that populations from this region appear to have higher θk values than any European population (see table 1). Judging from this, there should be a substantial number of unsampled mtDNA lineages in the Near East. Even a small increase in the number of lineages shared with European populations is liable to have the effect of reducing the age estimates of haplogroups calculated by Richards et al. (1998). Indeed, a modest increase in sample size (from <100 to 284) from Near Eastern populations has already resulted in a drastic reduction in the supposed European mutational time frame for most of these haplogroups (Sykes 1999). It is clear that a much more extensive sampling of large Eurasian populations is required before any precise and reliable conclusions can be drawn about the genealogical relationships between their mitochondrial gene pools.
Acknowledgments
We thank Dan Bradley, Vincent Macaulay, and the two anonymous reviewers for their constructive comments. A.H. was supported by an ORS award from the Committee of Vice-Chancellors and Principals of Universities and Colleges in the United Kingdom, 1997–1999. We thank E. Bjarnadóttir for her assistance with some of the lab work and D. Guðbjartsson for help in estimating θk confidence intervals.
Electronic-Database Information
Accession numbers and URLs for data in this article are as follows:
- GenBank, http://www.ncbi.nlm.nih.gov/Web/Genbank/index.html (for accession numbers AF236888-AF237289)
- Mitochondrial DNA Concordance, http://shelob.bioanth.cam.ac.uk/mtDNA (see also Miller et al. [1996] below)
References
- Anderson S, Bankier AT, Barrell BG, Debruijn MHL, Coulson AR, Drouin J, Eperon IC, et al (1981) Sequence and organization of the human mitochondrial genome. Nature 290:457–465 [DOI] [PubMed]
- Bandelt HJ, Forster P, Sykes BC, Richards MB (1995) Mitochondrial portraits of human populations using median networks. Genetics 141:743–753 [DOI] [PMC free article] [PubMed]
- Bertorelle G, Excoffier L (1998) Inferring admixture proportions from molecular data. Mol Biol Evol 15:1298–1311 [DOI] [PubMed]
- Bertranpetit J, Sala J, Calafell F, Underhill PA, Moral P, Comas D (1995) Human mitochondrial-DNA variation and the origin of Basques. Ann Hum Genet 59:63–81 [DOI] [PubMed]
- Brown MD, Hosseini SH, Torroni A, Bandelt HJ, Allen JC, Schurr TG, Scozzari R, et al (1998) mtDNA haplogroup X: an ancient link between Europe/Western Asia and North America? Am J Hum Genet 63:1852–1861 [DOI] [PMC free article] [PubMed]
- Calafell F, Underhill P, Tolun A, Angelicheva D, Kalaydjieva L (1996) From Asia to Europe: mitochondrial-DNA sequence variability in Bulgarians and Turks. Ann Hum Genet 60:35–49 [DOI] [PubMed]
- Cavalli-Sforza LL, Menozzi P, Piazza A (1994) The history and geography of human genes. Princeton University Press, Princeton, NJ [Google Scholar]
- Chakraborty R (1986) Gene admixture in human populations: models and predictions. Yearbook Phys Anthropol 29:1–43 [Google Scholar]
- Collins R (1991) Early Medieval Europe 300–1000. Macmillan, London [Google Scholar]
- Comas D, Calafell F, Mateu E, Perez-Lezaun A, Bosch E, Martinez-Arias R, Clarimon J, et al (1998) Trading genes along the silk road: mtDNA sequences and the origin of central Asian populations. Am J Hum Genet 63:1824–1838 [DOI] [PMC free article] [PubMed]
- Corte-Real H, Macaulay VA, Richards MB, Hariti G, Issad MS, Cambon-Thomsen A, Papiha S, et al (1996) Genetic diversity in the Iberian peninsula determined from mitochondrial sequence analysis. Ann Hum Genet 60:331–350 [DOI] [PubMed]
- Delghandi M, Utsi E, Krauss S (1998) Saami mitochondrial DNA reveals deep maternal lineage clusters. Hum Hered 48:108–114 [DOI] [PubMed]
- Di Rienzo A, Wilson AC (1991) Branching pattern in the evolutionary tree for human mitochondrial DNA. Proc Natl Acad Sci USA 88:1597–1601 [DOI] [PMC free article] [PubMed]
- Eiken HG, Knappskog PM, Boman H, Thune KS, Kaada G, Motzfeldt K, Apold J (1996) Relative frequency, heterogeneity and geographic clustering of PKU mutations in Norway. Eur J Hum Genet 4:205–213 [DOI] [PubMed]
- Ewens WJ (1972) The sampling theory of selectively neutral alleles. Theor Popul Biol 3:87–112 [DOI] [PubMed]
- Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491 [DOI] [PMC free article] [PubMed]
- Forster P, Harding R, Torroni A, Bandelt HJ (1996) Origin and evolution of Native American mtDNA variation: a reappraisal. Am J Hum Genet 59:935–945 [PMC free article] [PubMed]
- Francalacci P, Bertranpetit J, Calafell F, Underhill PA (1996) Sequence diversity of the control region of mitochondrial DNA in Tuscany and its implications for the peopling of Europe. Am J Phys Anthropol 100:443–460 [DOI] [PubMed]
- Guldberg P, Zschocke J, Dagbjartsson A, Henriksen KF, Guttler F (1997) A molecular survey of phenylketonuria in Iceland: identification of a founding mutation and evidence of predominant Norse settlement. Eur J Hum Genet 5:376–381 [PubMed]
- Handt O, Meyer S, von Haeseler A (1998) Compilation of human mtDNA control region sequences. Nucleic Acids Res 26:126–129 [DOI] [PMC free article] [PubMed]
- Handt O, Richards M, Trommsdorff M, Kilger C, Simanainen J, Georgiev O, Bauer K, et al (1994) Molecular-genetic analyses of the Tyrolean ice man. Science 264: 1775–1778 [DOI] [PubMed]
- Hasegawa M, Di Rienzo A, Kocher TD, Wilson AC (1993) Toward a more accurate time-scale for the human mitochondrial DNA tree. J Mol Evol 37:347–354 [DOI] [PubMed]
- Hofmann S, Jaksch M, Bezold R, Mertens S, Aholt S, Paprotta A, Gerbitz KD (1997) Population genetics and disease susceptibility: characterization of central European haplogroups by mtDNA gene mutations, correlation with D loop variants and association with disease. Hum Mol Genet 6:1835–1846 [DOI] [PubMed]
- Izagirre N, de la Rúa C (1999) An mtDNA analysis in ancient Basque populations: implications for haplogroup V as a marker for a major Paleolithic expansion from Southwestern Europe. Am J Hum Genet 65:199–207 [DOI] [PMC free article] [PubMed]
- Jazin E, Soodyall H, Jalonen P, Lindholm E, Stoneking M, Gyllensten U (1998) Mitochondrial mutation rate revisited: hot spots and polymorphism. Nat Genet 18:109–110 [DOI] [PubMed]
- Jones G (1984) A history of the Vikings. Oxford University Press, Oxford, UK [Google Scholar]
- Kittles RA, Bergen AW, Urbanek M, Virkkunen M, Linnoila M, Goldman D, Long JC (1999) Autosomal, mitochondrial, and Y chromosome DNA variation in Finland: evidence for a male-specific bottleneck. Am J Phys Anthropol 108:381–399 [DOI] [PubMed]
- Lutz S, Weisser HJ, Heizmann J, Pollak S (1998) Location and frequency of polymorphic positions in the mtDNA control region of individuals from Germany. Int J Legal Med 111:67–77 [DOI] [PubMed]
- Macaulay V, Richards M, Hickey E, Vega E, Cruciani F, Guida V, Scozzari R, et al (1999) The emerging tree of west Eurasian mtDNAs: a synthesis of control-region sequences and RFLPs. Am J Hum Genet 64:232–249 [DOI] [PMC free article] [PubMed]
- Meyer S, Weiss G, von Haesler A (1999) Pattern of nucleotide substitution and rate heterogeneity in the hypervariable regions I and II of human mtDNA. Genetics 152:1103–1110 [DOI] [PMC free article] [PubMed]
- Miller KWP, Dawson JL, Hegelberg E (1996) A concordance of nucleotide substitutions in the first and second hypervariable segments of the human mtDNA control region. Int J Legal Med 109:107–113 (see also Mitochondrial DNA Concordance, in Electronic-Database Information above) [DOI] [PubMed]
- Opdal SH, Rognum TO, Vege A, Stave AK, Dupuy BM, Egeland T (1998) Increased number of substitutions in the D-loop of mitochondrial DNA in the sudden infant death syndrome. Acta Paediatr 87:1039–1044 [DOI] [PubMed]
- Orekhov V, Poltoraus A, Zhivotovsky LA, Spitsyn V, Ivanov P, Yankovsky N (1999) Mitochondrial DNA sequence diversity in Russians. FEBS Letters 445:197–201 [DOI] [PubMed]
- Parson W, Parsons TJ, Scheithauer R, Holland MM (1998) Population data for 101 Austrian Caucasian mitochondrial DNA d-loop sequences: application of mtDNA sequence analysis to a forensic case. Int J Legal Med 111:124–132 [DOI] [PubMed]
- Parsons TJ, Muniec DS, Sullivan K, Woodyatt N, Alliston-Greiner R, Wilson MR, Berry DL, et al (1997) A high observed substitution rate in the human mitochondrial DNA control region. Nat Genet 15:363–368 [DOI] [PubMed]
- Pfeiffer H, Brinkmann B, Hühne J, Rolf B, Morris AA, Steighner R, Holland MM, et al (1999) Expanding the forensic German mitochondrial DNA control region database: genetic diversity as a function of sample size and microgeography. Int J Legal Med 112: 291–298 [DOI] [PubMed]
- Piercy R, Sullivan KM, Benson N, Gill P (1993) The application of mitochondrial DNA typing to the study of white Caucasian genetic identification. Int J Legal Med 106:85–90 [DOI] [PubMed]
- Pinto F, Gonzalez AM, Hernandez M, Larruga JM, Cabrera VM (1996) Genetic relationship between the Canary Islanders and their African and Spanish ancestors inferred from mitochondrial DNA sequences. Ann Hum Genet 60: 321–330 [DOI] [PubMed]
- Pult I, Sajantila A, Simanainen J, Georgiev O, Schaffner W, Paabo S (1994) Mitochondrial DNA sequences from Switzerland reveal striking homogeneity of European populations. Biol Chem Hoppe-Seyler 375:837–840 [PubMed] [Google Scholar]
- Richards MB, Corte-Real H, Forster P, Macaulay V, Wilkinson-Herbots H, Demaine A, Papiha S, et al (1996) Paleolithic and neolithic lineages in the European mitochondrial gene pool. Am J Hum Genet 59:185–203 [PMC free article] [PubMed]
- Richards MB, Macaulay VA, Bandelt HJ, Sykes BC (1998) Phylogeography of mitochondrial DNA in western Europe. Ann Hum Genet 62:241–260 [DOI] [PubMed]
- Rogers AR, Harpending H (1992) Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol 9:552–569 [DOI] [PubMed]
- Röhl A (1997) Network 2.0: a program package for calculating phylogenetic networks. Mathematisches Seminar, University of Hamburg, Hamburg, Germany [Google Scholar]
- Rousselet F, Mangin P (1998) Mitochondrial DNA polymorphisms: a study of 50 French Caucasian individuals and application to forensic casework. Int J Legal Med 111:292–298 [DOI] [PubMed]
- Sajantila A, Lahermo P, Anttinen T, Lukka M, Sistonen P, Savontaus ML, Aula P, et al (1995) Genes and languages in Europe: an analysis of mitochondrial lineages. Genome Res 5:42–52 [DOI] [PubMed]
- Sajantila A, Salem AH, Savolainen P, Bauer K, Gierig C, Paabo S (1996) Paternal and maternal DNA lineages reveal a bottleneck in the founding of the Finnish population. Proc Natl Acad Sci USA 93:12035–12039 [DOI] [PMC free article] [PubMed]
- Salas A, Comas D, Lareu MV, Bertranpetit J, Carracedo A (1998) mtDNA analysis of the Galician population: a genetic edge of European variation. Eur J Hum Genet 6:365–375 [DOI] [PubMed]
- Schneider, S, Kueffer JM, Roessli D, Excoffier L (1997) ARLEQUIN 1.1: software for population genetics data analysis. Genetics and Biometry Laboratory, University of Geneva, Switzerland [Google Scholar]
- Schurr TG, Sukernik RI, Starikovskaya YB, Wallace DC (1999) Mitochondrial DNA variation in Koryaks and Itel'men: population replacement in the Okhotsk Sea Bering Sea region during the Neolithic. Am J Phys Anthropol 108:1–39 [DOI] [PubMed]
- Smith, KP (1995) Landnám: the settlement of Iceland in archaeological and historical perspective. World Archaeol 26:319–347 [Google Scholar]
- Steffensen, J (1975) Menning og meinsemdir: Ritgerðarsafn um mótunarsögu íslenskrar þjóðar og baráttu hennar við hungur og sóttir. Ísafoldarprentsmiðja, Reykjavík, Iceland [Google Scholar]
- Sykes B (1999) The molecular genetics of European ancestry. Philos Trans R Soc Lond B Biol Sci 354:131–138 [DOI] [PMC free article] [PubMed]
- Tyfield LA, Stephenson A, Cockburn F, Harvie A, Bidwell JL, Wood NAP, Pilz DT, et al (1997) Sequence variation at the phenylalanine hydroxylase gene in the British Isles. Am J Hum Genet 60:388–396 [PMC free article] [PubMed]
- Thompson EA (1973) The Icelandic admixture problem. Ann Hum Genet 37:69–80 [DOI] [PubMed]
- Tills D, Warlow A, Kopec AC, Friðriksson S, Mourant AE (1982) The blood groups and other hereditary blood factors of the Icelanders. Ann Hum Biol 9:507–520 [DOI] [PubMed]
- Torroni A, Bandelt HJ, Durbano L, Lahermo P, Moral P, Sellitto D, Rengo C, et al (1998) mtDNA analysis reveals a major late paleolithic population expansion from southwestern to northeastern Europe. Am J Hum Genet 62:1137–1152 [DOI] [PMC free article] [PubMed]
- Torroni A, Huoponen K, Francalacci P, Petrozzi M, Morelli L, Scozzari R, Obinu D, et al (1996) Classification of European mtDNAs from an analysis three European populations. Genetics 144:1835–1850 [DOI] [PMC free article] [PubMed]
- Vasey DE (1996) Population regulation, ecology, and political economy in preindustrial Iceland. Am Ethnologist 23: 366–92 [Google Scholar]
- Wakeley J (1993) Substitution rate variation among sites in hypervariable region 1 of human mitochondrial DNA. J Mol Evol 37: 613–623 [DOI] [PubMed]
- Ward RH, Redd A, Valencia D, Frazier B, Paabo S (1993) Genetic and linguistic differentiation in the Americas. Proc Natl Acad Sci USA 90:10663–10667 [DOI] [PMC free article] [PubMed]
- Wijsman EM (1984) Techniques for estimating genetic admixture and applications to the problem of the origin of the Icelanders and the Ashkenazi Jews. Hum Genet 67:441–448 [DOI] [PubMed]
- Zschocke J, Mallory JP, Eiken HG, Nevin NC (1997) Phenylketonuria and the peoples of Northern Ireland. Hum Genet 100:189–194 [DOI] [PubMed]