

Abstract
ATR (ATM and Rad3-related), a PI kinase-related kinase (PIKK), has been implicated in the DNA structure checkpoint in mammalian cells. ATR associates with its partner protein ATRIP to form a functional complex in the nucleus. In this study, we investigated the role of the ATRIP coiled-coil domain in ATR-mediated processes. The coiled-coil domain of human ATRIP contributes to self-dimerization in vivo, which is important for the stable translocation of the ATR-ATRIP complex to nuclear foci that are formed after exposure to genotoxic stress. The expression of dimerization-defective ATRIP diminishes the maintenance of replication forks during treatment with replication inhibitors. By contrast, it does not compromise the G2/M checkpoint after IR-induced DNA damage. These results show that there are two critical functions of ATR-ATRIP after the exposure to genotoxic stress: maintenance of the integrity of replication machinery and execution of cell cycle arrest, which are separable and are achieved via distinct mechanisms. The former function may involve the concentrated localization of ATR to damaged sites for which the ATRIP coiled-coil motif is critical.
INTRODUCTION
The checkpoint pathway for validating the structure of DNA is an evolutionarily conserved genome surveillance system that guarantees the correct transmission of genetic information to descendants (Sancar et al., 2004). Damage-sensor proteins are used by the pathway to detect DNA damage and to initiate signal transduction cascades. The signal transducers activate p53 and inactivate cyclin-dependent kinases to inhibit the progression of the cell cycle. In higher eukaryotes, a lack of components involved in this regulation generally leads to embryonic lethality (Brown and Baltimore, 2000; de Klein et al., 2000; Liu et al., 2000; Takai et al., 2000; Weiss et al., 2000; Budzowska et al., 2004; Hopkins et al., 2004), which indicates that this checkpoint system is indispensable to the development of multicellular organisms with complex genomes. This is probably because DNA replication in these organisms involves the risk of DNA lesions that should be repaired before the consecutive mitosis.
Recent studies have revealed that the phosphoinositide kinase-related kinase (PIKK) family of proteins has a prominent role in the response to DNA structural abnormalities (Abraham, 2001; Shiloh, 2001). After the exposure to genotoxic stress, a paralogous set of PIKKs, ATM and ATR in mammals, governs critical regulation points through the phosphorylation of numerous proteins involved in checkpoint function and DNA repair, thereby coordinating these processes (Kim et al., 1999). ATM and ATR are believed to act in parallel pathways that respond to different types of DNA stress, including double-strand breaks (DSBs) and replication blockage (Brown and Baltimore, 2003). Based on the observations that ATM and ATR do not require other checkpoint proteins for stress-induced activation, these proteins are assumed to sense the presence of lesions (Zou et al., 2002; Sancar et al., 2004). In fact, evidence indicating the direct association of PIKKs with damaged DNA sites has accumulated (Smith et al., 1999; Suzuki et al., 1999; Unsal-Kacmaz et al., 2002; Dart et al., 2004).
Biochemical studies have addressed the mechanism of DNA damage-induced activation of PIKKs. ATM is held inactive as a dimer under normal conditions and is rapidly activated by intermolecular autophosphorylation followed by dimer dissociation after IR treatment (Bakkenist and Kastan, 2003). By contrast, ATR is constantly associating with the interacting protein ATRIP, but the size of the ATR-ATRIP protein complex is controversial (Cortez et al., 2001; Unsal-Kacmaz and Sancar, 2004; Bomgarden et al., 2004). In yeast, the ATRIP counterparts Rad26 and Ddc2 are essential for the kinase activity of the ATR orthologues both in vivo and in vitro (Wolkow and Enoch, 2002; Takata et al., 2004), suggesting that they form a functional complex. Moreover, a quantitative regulation mechanism exists to maintain the protein levels of ATR and ATRIP (Cortez et al., 2001); this appears to be mediated, at least in part, by negative feedback through the ATRIP carboxyl terminal region (Itakura et al., 2004a). The activation mechanism of the ATR kinase remains to be elucidated, but the recruitment of the ATR-ATRIP complex to single-stranded DNA coated with replication protein A (RPA) appears to be required for activation to occur (Zou and Elledge, 2003). In addition, an RPA-independent mode of ATR activation was recently suggested (Bomgarden et al., 2004; Cortez et al., 2004; Dodson et al., 2004).
In contrast to the highly conserved structures of the ATR orthologues, less similarity has been observed among the ATRIP counterparts (Cortez et al., 2001). The coiled-coil motif, however, is commonly shared among divergent organisms from yeasts to mammals, but its function is poorly understood. In this work, we focus on the function of the coiled-coil domain in the ATRIP protein. We present evidence that ATRIP forms as a homodimer through the coiled-coil domain. Moreover, our results suggest a critical role of the dimerized ATRIP in maintaining the integrity of chromosomes during instances of disturbed DNA replication via the regulation of the intranuclear localization of ATR. The G2/M checkpoint-function of ATR appears to operate in the absence of the normal ATRIP coiled-coil configuration, which indicates that the ATRIP coiled-coil domain is variably required depending upon the type of DNA damage.
MATERIALS AND METHODS
Cell Culture and Reagents
A549 human cells were grown at 37°C in DMEM medium (Sigma, St. Louis, MO) containing 10% fetal bovine serum and 1% penicillin-streptomycin (Invitrogen, Carlsbad, CA). Aphidicolin (Sigma) was added to the medium at a concentration of 10 μg/ml. The cells were irradiated with 10 Gy of X-radiation using X-ray irradiation equipment (MBR-1520A-2, Hitachi Medico, Tokyo, Japan) or with UV radiation using a UV cross-linker (Stratagene, La Jolla, CA). Knockdown of ATR and ATRIP with small interference RNA was performed as described previously (Itakura et al., 2004a).
To construct cells expressing tagged versions of ATRIP, A549 cells were infected with retroviral vectors as described previously (Sekoguchi et al., 2003), and stably integrated cells were selected by adding puromycin (Sigma) to the medium at a concentration of 3 μg/ml.
Antibodies
The following antibodies were used: N-19 (Santa Cruz Biotechnology, Santa Cruz, CA) and ab-2 (Oncogene, Boston, MA) for ATR, M2 (Sigma) for FLAG, 9E10 (Babco, Richmond, CA) and 2272 (New England Biolabs, Beverly, MA) for Myc, 9H8 (NeoMarkers, Fremont, CA) for RPA p34, sc-8408 (Santa Cruz Biotechnology) for Chk1, 2344 and 2341 (New England Biolabs) for phospho-Chk1 Ser 317 and 345, respectively, 3421 for phospho-Rad17 Ser645, 06–182 (Upstate Biotechnology, Lake Placid, NY) for ERK, AC-15 (Sigma) for β-actin, and B-5–1-2 (Sigma) for α-tubulin. Antibodies against ATRIP and phospho-ATRIP Ser68, Ser72 were previously described (Itakura et al., 2004a, 2004b). Rabbit polyclonal antibody against ORC1 (Tatsumi et al., 2003) was kindly provided by Dr. C. Obuse (Kyoto University).
Construction of Plasmids
The pMX-based hATRIP expression plasmids were previously described (Itakura et al., 2004a). ATRIP-LG mutations were introduced using a Quikchange site-directed mutagenesis kit (Stratagene). To insert the FLAG tag into ATRIP, a BamHI site was created immediately before the first ATG codon, and the tag sequence from pCMV-FLAG (Stratagene) was inserted. An ATRIP mutant immune to siRNA-mediated silencing was constructed as described previously (Ball et al., 2005) and was introduced via the pMX-puro retroviral vector. To construct a retroviral vector expressing short hairpin RNA (shRNA) against ATRIP, duplexed oligonucleotide encoding shRNA was cloned into pSHAG-1 (a gift from G. Hannon) harboring the U6 promoter. Then, the fragment corresponding to the U6 promoter and the shRNA was transferred to the retroviral vector pQCXIN (BD Biosciences Clontech, San Jose, CA). The sequence of the oligonucleotide was as follows: 5′-ACA TGC CAT CTA ATA ATC TGT GGA CCT TGA AGC TTG AAG GTT CAC AGG TTA TTG GAT GGC ATG TCA ATT TTT T-3′.
Western Blotting, Immunoprecipitation, and In Vitro Kinase Assay
Western blot analysis and immunoprecipitation were performed as previously described (Itakura et al., 2004a). The in vitro kinase assay for ATR was performed as previously described (Takata et al., 2004). Briefly, the immunoprecipitates were resuspended in 20 μl of kinase buffer (20 mM HEPES, pH 7.4, 50 mM NaCl, 10 mM MgCl2, 10 mM MnCl2, 1 mM dithiothreitol [DTT]) containing cold 5 μM ATP and 10 μCi of [γ-32P]ATP and were incubated at 30°C for 30 min. For use as an exogenous substrate, 1 μg of PHAS-I (Stratagene) was added to the reaction. The Chk1 kinase activity was assessed using a K-LISA Checkpoint activity kit (Calbiochem, La Jolla, CA), using immunoprecipitates with anti-Chk1 antibody (PC423, Calbiochem).
Isolation of Chromatin Fraction
The cells were fractionated as previously described (Mendez and Stillman, 2000). The cells (3 × 106 cells) were resuspended in 200 μl buffer A (10 mM HEPES, pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 1 mM DTT, complete mini-EDTA-free [Roche, Indianapolis, IN], 0.1 mM phenylmethylsulfonyl fluoride, and 0.1% Triton X-100) and incubated for 5 min on ice. The insoluble fraction was collected by low-speed centrifugation (4 min, 1300 × g, 4°C). The soluble fraction was further clarified by high-speed centrifugation (15 min, 20,000 × g, 4°C) to remove cell debris and insoluble aggregates. The insoluble fraction was washed once in buffer A, and then half of the fraction was treated with 0.2 U micrococcal nuclease for 3 min at 37°C in buffer A, and the other half was incubated without nuclease. After washing twice in buffer B (3 mM EDTA, 0.2 mM EGTA, and 1 mM DTT), the final chromatin pellet was resuspended in sample buffer and sheared by sonication.
ATRIP-RPA-single-stranded DNA Interaction
Human RPA heterotrimer was purified from A549 cells using gel filtration and single-stranded DNA (ssDNA) affinity columns. A549 cells expressing ATRIP or ATRIP-LG were lysed in lysis buffer, and the cleared lysates were incubated with Dynabeads-coated 100mer ssDNA with or without RPA for 2 h at 4°C as described previously (Ball et al., 2005).
Mitotic Spreads
Mitotic spreads were prepared as previously described (Nghiem et al., 2001).
Gel Filtration
For gel filtration experiments, the cells were lysed in lysis buffer at 4°C for 30 min. The lysates were passed through a 27-gauge needle five times and clarified by centrifugation at 13,000 × g for 30 min. The supernatants were further clarified through Ultrafree-CL (Millipore, Billerica, MA) and loaded onto a HiLoad 26/60 Superdex 200 prep-grade column (Amersham Biosciences, Piscataway, NJ). The proteins were eluted at 1 ml/min with gel filtration buffer containing 50 mM HEPES, pH 7.4, 150 mM NaCl, and 1 mM EDTA. An eluant volume of 1.5 ml was collected per fraction.
Sequential Labeling of DNA with Chlorodeoxyuridine and Iododeoxyuridine
The in vivo labeling of DNA with chlorodeoxyuridine (CldU) and iododeoxyuridine (IdU) was performed as previously described (Dimitrova and Gilbert, 2000). The cells were grown on slides and were fixed with cold 70% ethanol, stored at 4°C for more than 30 min, and incubated in 1.5 N HCl in 0.5% Triton X-100 for 30 min at RT. The slides were subjected to immunofluorescence using anti-BrdU monoclonal antibodies 2B1 (MBL, Nagoya, Japan; for the detection of IdU) and BU1/75 (for the detection of CldU; AbCam, Cambridge, United Kingdom).
Flow Cytometric Analysis
The cells were collected by trypsinization, washed with phosphate-buffered saline (PBS), fixed with 70% ethanol, and stored at –20°C. Immediately preceding the analysis, the cells were washed with PBS and permeabilized with 0.25% Triton X-100 in PBS for 15 min. After washing with 1% bovine serum albumin in PBS, the cells were incubated with anti-MPM2 antibody (Upstate Biotechnology) for 3 h and then incubated with FITC-conjugated anti-mouse IgG (Sigma) in 10 μg/ml RNase (Wako, Osaka, Japan) for 30 min. The cells were collected by centrifugation, resuspended in 10 μg/ml propidium iodide (Wako), and analyzed using a FACSCalibur flow cytometer (Becton Dickinson, Lincoln Park, NJ).
RESULTS
Dimerization of ATRIP through Its Coiled-coil Domain
The coiled-coil motif is commonly shared among the ATRIP counterparts of divergent organisms from yeasts to mammals. We created a mutant (hereafter referred to as ATRIP-LG) by substituting a glycine for each of four leucine residues that are normally located at residues 123, 144, 185, and 188 in the coiled-coil domain (Figure 1A). These mutations were expected to disrupt the α-helices that are important for the integrity of the coiled-coil and to cause the resultant protein to have a compromised secondary structure as estimated by the Multicoil Scoring Form program (http://multicoil.lcs.mit.edu/cgi-bin/multicoil).
Figure 1.

The coiled-coil motif is essential for dimerization of ATRIP. (A) Schematic of wild-type and coiled-coil-defective mutant (ATRIP-LG) proteins of ATRIP. The gray box indicates the coiled-coil domain of ATRIP. ATRIP-LG, which includes glycine substitutions for leucines 123, 144, 185, and 188, possesses the lowest multicoil score as analyzed by the Multicoil Scoring Form (http://multicoil.lcs.mit.edu/cgi-bin/multicoil). (B) Exogenous expression of ATRIP-LG decreased endogenous ATRIP expression. Lysates from A549 cells infected with retrovirus vector expressing ATRIP-Myc or ATRIP-LG-Myc (LG-Myc) or a control were analyzed by immunoblotting with antibodies against ATR, ATRIP, and β-actin. (C) The binding ability of ATRIP-LG to ATR. A549-derived cells were lysed and subjected to immunoprecipitation using anti-ATR or anti-Myc antibody. After extensive washing, the immunoprecipitates were immunoblotted with anti-ATR and anti-ATRIP antibodies. (D) Dimerization of ATRIP and the requirement of the coiled-coil domain. A549 cells expressing FLAG-ATRIP and ATRIP-Myc or ATRIP-LG-Myc were lysed and subjected to immunoprecipitation using anti-FLAG or anti-Myc antibody. After extensive washing, the immunoprecipitates were immunoblotted with anti-FLAG and anti-Myc antibodies. (E) ATRIP-Myc and ATRIP-LG-Myc elute in different peaks on gel filtration. Lysates of cells expressing ATRIP-Myc or ATRIP-LG-Myc were loaded onto a HiLoad26/60 Superdex 200 prep grade column. Each fraction was immunoblotted with anti-ATR and -ATRIP antibodies. The void and fractions containing protein standards of 660, 440, 158, and 67 kDa are indicated.
To examine the effect of the coiled-coil mutation, we introduced a retrovirus-based expression unit for the Myc-tagged version of the wild-type or the coiled-coil-defective ATRIP protein into human A549 cells. The coiled-coil mutant protein and the wild-type protein were expressed from the same promoter and were comparably present, indicating that the mutation did not affect the stability of the protein (Figure 1B). In addition, ectopic expression of the two ATRIP species resulted in the reduction of endogenous ATRIP protein as previously described (Itakura et al., 2004a); therefore, the occurrence of any dominant effects could be effectively presented by the expression of the mutant protein.
We first performed an immunoprecipitation study with the anti-ATR and anti-Myc antibodies. As shown in Figure 1C, the wild-type ATRIP and the LG-mutant protein associated with endogenous ATR. The mutant protein, however, was coimmunoprecipitated with ATR in a lower yield compared with the wild-type ATRIP. With regularity, a reduced amount of ATR was recovered with the Myc-tagged ATRIP-LG (Figure 1C). Furthermore, when the wild-type ATRIP-Myc was immunologically isolated with the anti-Myc antibody, endogenous ATRIP protein was corecovered as well, except with the ATRIP-LG-Myc (Figure 1C, right). All of these data suggest a dominant effect of ATRIP-LG expression on the stoichiometry of ATR and ATRIP in the complex.
The corecovery of ATRIP-Myc with endogenous ATRIP protein suggests that ATRIP forms a homo-multimer in vivo. To examine this possibility directly, we established cells in which two alternatively tagged ATRIP proteins, i.e., ATRIP-Myc and FLAG-ATRIP, were expressed simultaneously. We observed reciprocal coimmunoprecipitation of the two ATRIP species using anti-Myc and anti-FLAG antibodies, confirming multimerization of the ATRIP proteins (Figure 1D). Moreover, the interaction was completely abolished when ATRIP-LG-Myc was expressed instead of ATRIP-Myc (Figure 1D). Therefore, multimerization of ATRIP requires the normal conformation of the coiled-coil domain.
Next, we analyzed the protein complex containing ATR-ATRIP by gel-filtration column chromatography. ATR was eluted as a protein complex of more than 1000 kDa as previously reported (Figure 1E; Wright et al., 1998; Bomgarden et al., 2004). The majority of the endogenous ATRIP coeluted with ATR; full-length Myc-tagged ATRIP was recovered in fractions with ATR (fractions 30–32, corresponding to a molecular weight of ∼1000 kDa) as well as in fractions without ATR (fractions 34–36, corresponding to a molecular weight of 500–600 kDa). By contrast, ATR-free ATRIP-LG-Myc protein eluted at a size that corresponded to half of the ATRIP-Myc (fractions 40–42, 250–300 kDa). These observations suggest that wild-type ATRIP forms a dimer and that the dimerization is not mediated via an interaction with ATR.
ATRIP Dimer Is Stable after DNA Damage
We have shown that ATRIP is phosphorylated by ATR after exposure to genotoxic stress (Itakura et al., 2004b). The modification does not require dimerization, because ATRIP-LG was phosphorylated effectively after UV treatment (Figure 2A). A recent study showed that the activation of ATM is mediated through autophosphorylation and dimer dissociation (Bakkenist and Kastan, 2003), raising the possibility that ATRIP is activated in a manner similar to that of ATM. However, this is not the case: ATRIP dimerization and its association with ATR did not exhibit any detectable changes after DNA damage (Figure 2B). Furthermore, the phosphorylation-defective ATRIP-68A72A mutant was proficient at dimer formation (Figure 2C). We conclude that ATRIP is stable as a dimer, irrespective of the ATR kinase activity.
Figure 2.
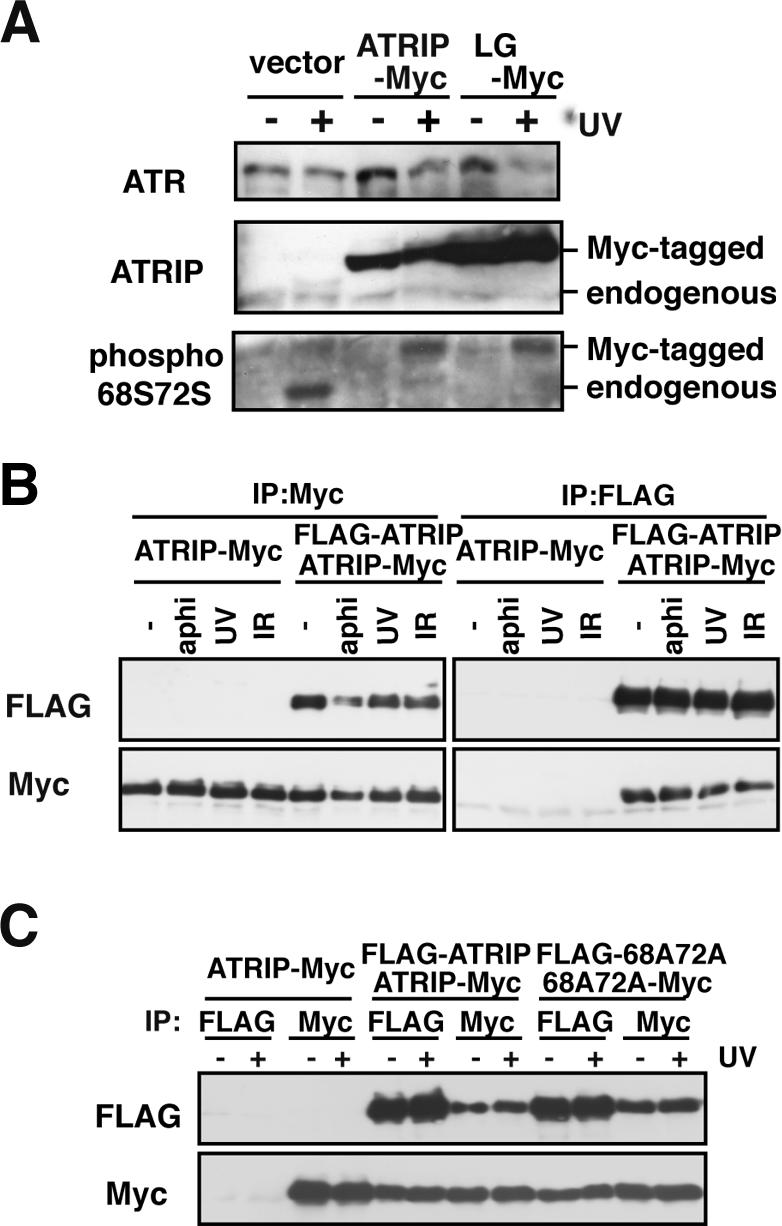
ATRIP dimer is stable after DNA damage. (A) ATRIP-LG is normally phosphorylated by ATR after DNA damage. A549 cells infected with retrovirus harboring ATRIP-Myc or ATRIP-LG-Myc or a control virus were treated with UV (20 J/m2). After 1 h, cells were collected and subjected to immunoblotting using antibodies against ATR, ATRIP and phospho-ATRIP S68S72. (B) Dimerization of ATRIP is unaffected after exposure to genotoxic stresses. ATRIP cells expressing ATRIP-Myc or ATRIP-Myc/FLAG-ATRIP were treated with aphidicolin (10 μg/ml), UV (20 J/m2), or IR (10 Gy). The cell lysates were subjected to immunoprecipitation using antibodies against myc or FLAG, followed by immunoblotting using anti-Myc and anti-FLAG antibodies. (C) Phosphorylation-defective ATRIP is proficient in dimer formation. A549 cells expressing ATRIP-Myc, ATRIP-Myc/FLAG-ATRIP, or ATRIP-S68A,S72A-myc/FLAG-S68A,S72A were untreated or treated with UV (20 J/m2). After 1 h, the cell lysates were subjected to immunoprecipitation, followed by immunoblotting as in B.
ATRIP-LG Mutant Protein Is Defective in Recruitment to Nuclear Foci
We have shown that the amino terminal domain plus the coiled-coil domain of ATRIP is sufficient for its recruitment to sites of DNA damage (Itakura et al., 2004a). We addressed the possibility that the coiled-coil defect would affect the intranuclear dynamics of ATRIP.
The ATR-ATRIP complex is recruited to chromatin in a cell-cycle-dependent manner (Hekmat-Nejad et al., 2000; Dart et al., 2004). To examine whether the coiled-coil conformation is essential for ATRIP to associate with chromatin, we performed subcellular fractionation using differential centrifugation and compared the recovery of ATRIP in the chromatin (P3) fraction. As shown in Figure 3A, part of the endogenous ATRIP protein was recovered in the P3 fraction in an asynchronous condition, as described previously (Bomgarden et al., 2004). The recovery was greatly reduced after cells were arrested at M-phase by the spindle inhibitor TN16, and was significantly increased after treatment with aphidicolin (Figure 3B). We did not find any difference in recovery or nuclease sensitivity between the wild-type and ATRIP-LG mutant protein (Figure 3, B and C). These observations suggest that the coiled-coil mutation does not affect the ability of ATRIP to associate with chromatin. Moreover, the ATRIP-LG protein still retained the ssDNA-interacting activity that was facilitated by RPA (Figure 3D), suggesting that the coiled-coil domain is not required for interaction with RPA-ssDNA.
Figure 3.
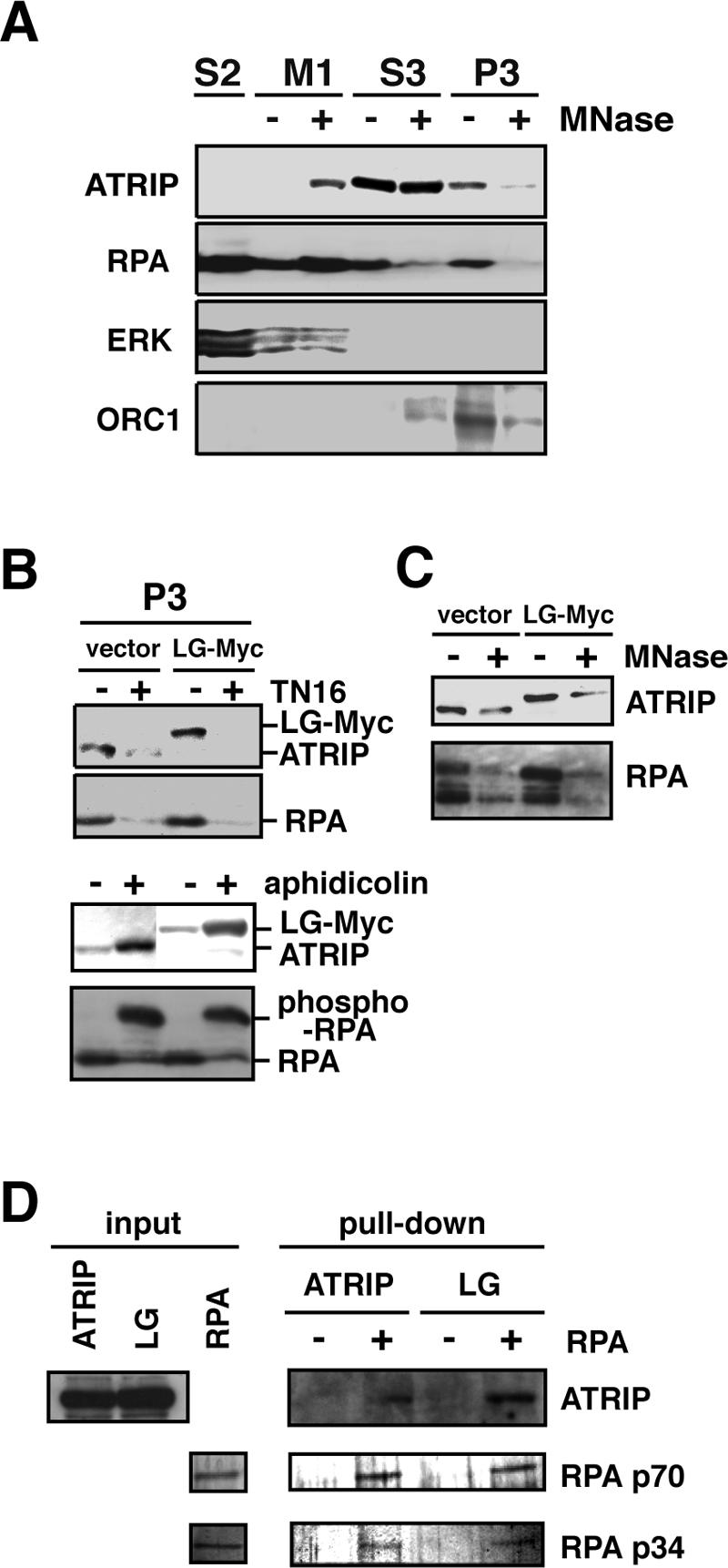
ATRIP protein with a coiled-coil defect retains the ability to bind to chromatin. (A) Chromatin association of ATRIP. A549 cells were subjected to subcellular fractionation (see Materials and Methods), and the fractions were immunoblotted with antibodies against ATRIP, RPA p34, ERK (cytoplasmic marker), and ORC1 (chromatin marker). The fractions are as follows: S2, soluble fraction; M1, micrococcal nuclease fraction; S3, solubilized nuclear fraction; and P3, chromatin-nuclear matrix-bound fraction. (B) ATRIP-LG mutant protein is able to associate with chromatin. A549 cells harboring a control vector or ATRIP-LG-Myc were untreated or treated with 150 ng/ml TN16 for 12 h or 10 μg/ml aphidicolin for 24 h were subjected to subcellular fractionation as in A and were immunoblotted with antibodies against ATRIP and PRA p34. (C) The chromatin association of ATRIP is nuclease-sensitive. The P3 fraction in B was subjected to micrococcal nuclease treatment and was immunoblotted. (D) In vitro association of ATRIP with RPA. Lysates of the A549 cells expressing ATRIP-Myc or ATRIP-LG-Myc were incubated with beads bound to single-stranded DNA that had been coated with RPA or mock coated. After washing, the proteins bound to the beads were subjected to immunoblotting using the anti-Myc antibody. RPA subunits p70 and p34 were visualized by silver staining of the gel.
After DNA lesions are produced in the nucleus, ATR-ATRIP is translocated to form multiprotein complexes with other proteins involved in the repair process and is detected as protein foci under microscopic observation (Wang et al., 2000; Goldberg et al., 2003; Lou et al., 2003; Stewart et al., 2003). We examined the possible contribution of the ATRIP coiled-coil domain to this dynamic behavior.
We established A549 cells expressing a GFP-tagged protein of the wild-type ATRIP or the LG mutant. The amount of endogenous ATRIP protein was reduced as before, owing to the feedback regulation of ATRIP expression in these cells (unpublished data). As shown in Figure 4, these fusion proteins were uniformly dispersed in the nuclei that lacked DNA damage. When the cells were treated with aphidicolin or IR, ATRIP-GFP formed nuclear foci that were costained with RPA (Figure 4, A, C, and D), which is consistent with previous reports (Zou and Elledge, 2003). By contrast, the leucine to glycine mutations diminished the formation of foci and compromised the translocation of endogenous ATR to the foci (Figure 4, B and E). Thus, the coiled-coil domain of ATRIP is important for the recruitment of the ATR-ATRIP complex to nuclear foci after DNA damage. However, RPA foci were developed in the ATRIP-LG-expressing cells (Figure 4A), indicating that the formation of RPA foci occurs upstream of the ATR-ATRIP recruitment.
Figure 4.

ATRIP-LG protein is defective in translocation to DNA damage-induced foci. (A) Diminished ATRIP focus formation by the coiled-coil mutation. A549 cells expressing ATRIP-GFP or ATRIP-LG-GFP were untreated or treated with 10 Gy IR or 10 μg/ml aphidicolin. At 1 h after the irradiation or 24 h after the aphidicolin addition, the cells were fixed and subjected to immunofluorescence using the anti-RPA p34 antibody. (B) Expression of ATRIP-LG diminished focus formation of ATR. The cells were prepared as in A and stained with anti-ATR antibody. (C) Time course of ATRIP focus formation. After 10 Gy irradiation, the cells expressing ATRIP-GFP or ATRIP-LG-GFP were fixed at the indicated times. (D and E) The proportion of cells harboring ATRIP foci as examined by microscopic observation. A549 cells expressing ATRIP-GFP or LG-GFP were untreated or treated with 10 Gy IR or 10 μg/ml aphidicolin. The number of cells harboring ATRIP foci (D) or ATR foci (E) from among 200 nuclei was counted. The data are the averages ± SD of three independent experiments.
Effect of ATRIP-LG on ATR Kinase Activity
The translocation of ATR-ATRIP to the foci after DNA damage has been shown to be dependent on the kinase activity of ATR (Barr et al., 2003). To examine the possibility that the phenotype caused by the disruption of the ATRIP coiled-coil is mediated by a reduction in the ATR activity, we examined the in vitro kinase activity of ATR isolated from cell lysates using anti-ATR antibody. As shown in Figure 5A, the ATR immunocomplex from vector-infected cells possessed caffeine-sensitive phospho-transferring activity against PHAS-1. Moreover, the activity was comparable when ATR was isolated from cells expressing ATRIP-Myc or ATRIP-LG-Myc. As the ATRIP-LG protein was predominantly coimmunoprecipitated with ATR, we concluded that the absence of the coiled-coil structure in the associated ATRIP does not affect ATR kinase activity in vitro.
Figure 5.
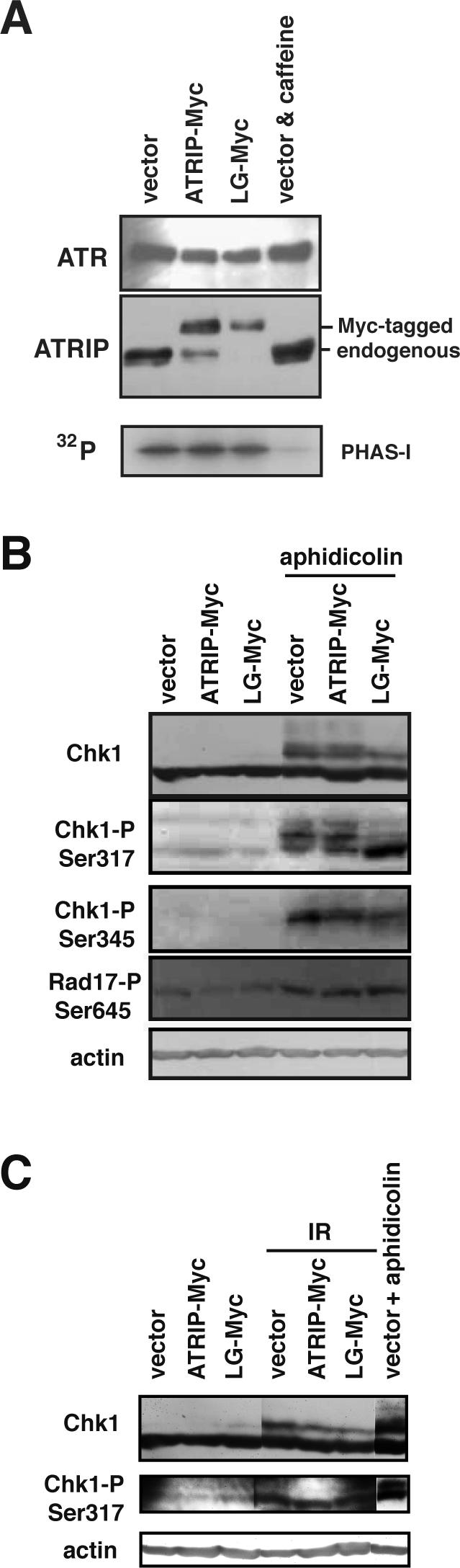
Effect of ATRIP-LG expression on ATR kinase activity in vitro and in vivo. (A) ATRIP-LG did not affect associated ATR kinase activity in vitro. A549 cells expressing ATRIP-Myc or ATRIP-LG-Myc or a vector control were lysed, and the lysates were subjected to immunoprecipitation using anti-ATR antibody. Immunoprecipitates were monitored for their ability to phosphorylate PHAS-1 (Stratagene) or were analyzed by immunoblotting with anti-ATR and anti-ATRIP antibodies. Caffeine was added to a final concentration of 10 mM in the kinase reaction. (B) Modification of ATR target proteins in ATRIP-LG-expressing cells. A549 cells as in A were untreated or treated with aphidicolin for 24 h. The cells were collected, and the lysates were immunoblotted using antibodies against Chk1, phospho-Chk1 Ser317, phospho-Chk1 Ser345, phospho-Rad17 Ser645, and β-actin. (C) Effect of ATRIP-LG expression on IR-induced Chk1 modification. A549 cells were untreated or irradiated with 10 Gy IR and incubated for 1 h, and the lysates were immunoblotted as in B.
Next, we investigated the activation of ATR in vivo by assessing the phosphorylation level of ATR target molecules after DNA damage. Rad17 is a member of a variant clamp loader that is involved in loading the PCNA-like Rad9-Rad1-Hus1 complex, and its damage-induced phosphorylation has been shown to be dependent on ATR (Bao et al., 2001). As shown in Figure 5B, the phosphorylation levels of Rad17-Ser645 after treatment with aphidicolin were comparable between control and ATRIP-LG-infected cells (Figure 5B), suggesting that ATR is normally activated in vivo, even when the ATRIP coiled-coil is defective.
Chk1 is the critical target of ATR kinase in the checkpoint pathway. Ser317 and -345 in Chk1 are important phosphorylation sites, and alanine substitution of those serine residues diminishes the activation of the kinase after exposure to genotoxic stress (Zhao and Piwnica-Worms, 2001). Therefore, we next addressed whether the ATRIP coiled-coil was critical for ATR-mediated activation of Chk1 by examining the phosphorylation status of Chk1 using antibodies that were specific to phospho-Ser317 and -Ser345. In cells that were infected with a control or ATRIP-Myc-expressing vector, Ser317 and -345 were phosphorylated after aphidicolin treatment. By contrast, the expression of ATRIP-LG caused a reduction in the lower mobility species that could be detected with the anti-Ser317 antibody, but the phosphorylation at both serine residues was significantly induced (Figure 5B). Therefore, although ATR is activated after DNA damage in ATRIP-LG cells and can phosphorylate Rad17 and Chk1, the coiled-coil mutation significantly affects the modification at site(s) other than Ser317 and Ser345 of Chk1.
Then, we then examined whether the expression of ATRIP-LG affected the activation of Chk1 kinase after aphidicolin treatment. As shown in Supplementary Figure 1, Chk1 kinase activity assayed in vitro was stimulated after the treatment even in cells expressing the coiled-coil-defective ATRIP protein, suggesting that activation of Chk1 does not require the ATRIP coiled-coil domain. However, the activation appears to be reduced slightly by the expression of ATRIP-LG. Thus, the ATRIP coiled-coil domain may contribute to full activation of Chk1 upon replication blockage.
ATRIP-LG Cells Failed to Resume DNA Replication after Aphidicolin Treatment
Replication blockage causes a decrease in the mitotic cell fraction, which is believed to be attributable, at least in part, to cell cycle arrest via a checkpoint-dependent mechanism. Recent studies have shown that Chk1 is important to the replication checkpoint because it maintains the stability of stalled replication forks (Feijoo et al., 2001; Zachos et al., 2005). Our previous results suggested that the expression of ATRIP-LG affected the Chk1 status; therefore, we examined the cell cycle behavior of ATRIP-LG-expressing cells after their exposure to replication inhibitors.
After aphidicolin or hydroxyurea treatment, cells with G2/M DNA content were reduced because of inhibition of DNA synthesis. When the cells were released from the aphidicolin-induced arrest, wild-type ATRIP-expressing cells efficiently restarted the cell cycle, although greater than 40% of the cells expressing ATRIP-LG persisted with 1C DNA contents (Figure 6A). This suggests that the expression of ATRIP-LG inhibits the recovery of the normal cell cycle from the replication block.
Figure 6.

Cells expressing ATRIP-LG fail to resume DNA replication after aphidicolin treatment. (A) FACS analysis. A549 cells infected with retrovirus harboring ATRIP-Myc or ATRIP-LG-Myc or a control virus were treated with aphidicolin (10 μg/ml) for 24 h and released to aphidicolin-free medium with 150 ng/ml TN16 for 24 h. The cells were fixed, stained with PI, and subjected to FACS analysis. (B) Loss of stalled replication fork integrity in ATRIP-LG-expressing cells. In the left panel, the cells were labeled with IdU for 15 min and subsequently labeled with CldU for 15 min. In the right panel, the sequential labeling was interrupted by aphidicolin treatment for 24 h. After fixation, the incorporated IdU and CldU were detected with mouse 2B1 and rat BU1/75 monoclonal antibodies in combination with Alexa Fluor 594 anti-mouse IgG (red) and Alexa Fluor 488 anti-rat IgG (green), respectively. Dotted circles indicate IdU-positive, CldU-negative cells. (C) The ratio of IdU-positive, CldU-negative cells to IdU-positive cells. The ratio under the ATRIP-knocked down condition is shown as a control.
The budding yeast ortholog of ATR, Mec1, has been shown to be involved in the maintenance of replication forks (Lopes et al., 2001; Tercero and Diffley, 2001; Cha and Kleckner, 2002; Cobb et al., 2003). One explanation for the compromised cell cycle recovery in the ATRIP-LG-expressing cells is the loss of integrity of the DNA replication machinery during aphidicolin treatment. Thus, we further examined in detail the effect of the coiled-coil defective ATRIP on the integrity of stalled replication forks.
We adopted a cytological method using sequential labeling of replicated DNA with IdU and CldU. Without inhibiting the progression of the replication fork, nuclear zones that were labeled by IdU and CldU were adjacent in all cell types. This staining pattern was essentially unaffected in the vector-infected cells even when the sequential labeling was interrupted by aphidicolin treatment for 24 h. By contrast, we observed a significantly higher proportion of ATRIP-LG cells in which CldU was not incorporated into the nuclei despite the initial IdU labeling (Figure 6, B and C). This result suggests that cells expressing the ATRIP-LG mutant are defective at maintaining stalled replication forks, thereby impairing the resumption of DNA replication after replication blockage is relieved.
Aberrant Cell Cycle Behaviors of Cells Expressing ATRIP-LG during Aphidicolin or UV Treatment
The stabilization of stalled replication forks during replication arrest is important for the inhibition of mitosis (Zachos et al., 2005). The loss of viable replication structures causes premature cell cycle progression to the M phase, which is manifested by abnormal chromosomal condensation with extended mitotic spindles. When cells that were treated with siRNA targeted against ATR were incubated with aphidicolin, premature chromosomal condensation proceeded and was evident as abnormally condensed chromatin (Figure 7A), spindle formation (Figure 7B), and increased MPM2-positive cells in the FACS analysis (Figure 7C). Similarly, we found that a significant fraction of ATRIP-LG cells restarted the cell cycle prematurely during the aphidicolin treatment, as measured by the accumulation of cells with M-phase characteristics (Figure 7, A–C). The aberrant cell cycle behavior was also manifested after UV treatment, when the ATRIP-LG-expressing cells showed a higher incidence of premature chromosomal condensation with G1 DNA content and a reduced accumulation of cells with G2 DNA content (Figure 8). These results are consistent with the hypothesis that the expression of ATRIP-LG reduces the integrity of replicating chromosomes upon DNA damage, thereby disrupting the ATR-dependent replication checkpoint.
Figure 7.

Expression of ATRIP-LG promotes premature chromatin condensation (PCC) after aphidicolin treatment. (A) PCC quantified by microscopic observation. A549 cells infected with retrovirus harboring ATRIP-Myc or ATRIP-LG-Myc or a control vector were untreated or treated with 10 μg/ml aphidicolin. After 24 h, the cells were collected by trypsinization, resuspended in KCl for 10 min, and fixed with Carnoy's fixative. A total of 1000 cells was examined by microscopic observation. (B) PCC quantified by spindle formation. A549 grown on slides were untreated or treated with aphidicolin for 24 h. The cells were fixed with 4% paraformaldehyde and stained with anti-α-tubulin antibody and PI. A total of 1000 cells was counted. Green, tubulin; red, PI. (C) PCC quantified by anti-MPM2 antibody. The cells as in A were fixed with 70% ethanol and stained with anti-MPM2 antibody (M phase marker) and PI. A total of 20,000 cells was examined by FACS. In all of the graphs, the data are the averages ± SD of three independent experiments.
Figure 8.

Abnormal S-phase checkpoint in ATRIP-LG-expressing cells after UV treatment. A549 cells infected with retrovirus harboring ATRIP-Myc or ATRIP-LG-Myc or a control virus were treated with UV (20 J/m2). The cells were further incubated with 150 ng/ml TN16, and collected at intervals. The fixed cells were stained with PI and anti-MPM2 antibody, and analyzed by FACS.
Proficient IR-induced G2/M Checkpoint in ATRIP-LG-expressing Cells
The response to IR-induced double-strand breaks involves ATM, which is another PIKK family protein in mammals. A previous study using a Cre/lox-conditional ATR allele revealed that both ATM and ATR play critical roles in the IR-induced checkpoint; they both contribute to the early delay in M-phase entry, but only ATR regulates a majority of the late phase (2–9 h post-IR; Brown and Baltimore, 2003). In agreement with the previous results, we found that repression of ATR using the RNA interference technique caused a defect in the maintenance, but not in the establishment, of G2/M arrest after IR. The coiled-coil mutation in ATRIP did not induce either of the defects: ATRIP-LG cells were arrested in G2/M at 1 h, and the arrest was maintained for 6–24 h (Figure 9 and unpublished data). Consistently, Chk1 modification after IR treatment occurred comparably in ATRIP-LG cells (Figure 5C). These observations suggest that the coiled-coil domain of ATRIP is unimportant for the maintenance of G2/M arrest after IR-induced DNA damage.
Figure 9.

IR-induced G2/M checkpoint is normal in ATRIP-LG-expressing cells. (A) Effect of ATRIP-LG expression on IR-induced G2/M checkpoint. A549 cells expressing ATRIP-Myc or ATRIP-LG-Myc were untreated or irradiated with 10 Gy IR and incubated for 1 or 6 h at 37°C before fixation. The cells were subjected to flow cytometry as in Figure 7C. (B) The proportion of MPM2-positive cells determined by FACS analysis. The data are the averages ± SD of three independent experiments.
DISCUSSION
We previously mapped two functional domains of ATRIP, the amino terminus, associated with intranuclear localization, and the carboxy terminus, associated with the ATR-binding domain (Itakura et al., 2004a). In agreement with our results, a recent report showed that the loss of the ATR-associating domain within ATRIP led to defects in the recruitment of ATR to sites of damage and in checkpoint signaling induced by ATR (Falck et al., 2005). In this study, we show that the coiled-coil, which is the third domain that is evolutionarily conserved among ATRIP counterparts, is critical to the nuclear dynamics of the ATR-ATRIP complex. The coiled-coil-defective ATRIP showed unique characteristics: it associated with chromatin at a normal level, but showed diminished recruitment to sites of DNA damage. Moreover, the mutant lost stalled replication fork integrity, but did not affect the G2/M checkpoint induced by IR. Our results clearly indicate that the coiled-coil domain is critical to the function of the ATR-ATRIP pathway in response to replication stress. Furthermore, a “separation of function” characteristic of the coiled-coil mutant suggests that the ATR-ATRIP complex fulfills two separable functions, i.e., the maintenance of replication forks and execution of cell cycle arrest signals, via distinct mechanisms.
One possible explanation for the phenotype caused by ATRIP-LG expression is the hypomorphic activity of the ATR-ATRIP pathway in cells expressing the endogenous, wild-type ATRIP protein. However, we think that this possibility is less likely, because complete repression of endogenous ATRIP by RNAi did not alter the phenotypes of the ATRIP-LG-expressing cells (Supplementary Figure 2). Moreover, cells of Seckel syndrome patients, who are thought to have a hypomorphic allele of ATR (O'Driscoll et al., 2003), show defects in response to replication stress as well as in the G2/M checkpoint (Alderton et al., 2004). Instead, we prefer a model in which the response to replication stress requires the stable or concentrated localization of ATR-ATRIP to damaged sites and the execution of the cell cycle arrest signal for which concentrated localization may not be essential. This model is consistent with a recent observation showing that the assembly of ATR-ATRIP onto sites of DNA damage is not a prerequisite for ATR activation based on an assay detecting ATR-dependent phosphorylation of Chk1 Ser345 (Ball et al., 2005).
A recent study has shown requirement of the ATRIP coiled-coil domain for ATR-dependent checkpoint signaling, using a mutant ATRIP (ATRIPΔ112–225) in which the domain is completely deleted (Ball and Cortez, 2005). The effects caused by deletion of the coiled-coil domain are essentially similar to those by ATRIP-LG; expression of ATRIPΔ112–225 leads to the loss of ATRIP dimerization and to compromised localization to damage-induced nuclear foci. However, expression of ATRIPΔ112–225 affects DNA damage-induced phosphorylation of Chk1 Ser345, which is contrasted with proficient modification of Chk1 Ser345 in cells expressing ATRIP-LG. This discrepancy may be due to the types of mutations used. It appears that dimerization is not the sole role for the coiled-coil domain of ATRIP (Ball and Cortez, 2005). ATRIPΔ112–225 is assumed to lack all functions related to the domain, whereas ATRIP-LG may still retain some functions that contribute to Chk1 phosphorylation.
Mechanism of Protein Assembly Mediated by ATRIP Coiled-coil
We have shown that the amino-terminal domain plus the coiled-coil domain of ATRIP is sufficient for ATRIP recruitment to sites of DNA damage (Itakura et al., 2004a), which was consistent with the observation that ATRIP without the amino terminus lacked the RPA-ssDNA binding activity and was defective at translocating to sites of DNA damage (Ball et al., 2005). Interestingly, the truncation of the ATRIP amino terminus caused the simultaneous loss of activity for chromatin binding and focus translocation (Ball et al., 2005), whereas the coiled-coil defect led to the loss of only the latter (present study). These observations indicate that chromatin association and accumulation at damage-induced foci may represent different modes of ATR-ATRIP activation. Indeed, differential requirements for recruitment to damaged chromatin and sequestration into foci have been previously shown, in which histone H2AX is essential only for the DNA damage-induced focus formation of several factors including Nbs1 and Brca1 (Celeste et al., 2003).
The precise mechanism underlying ATR-ATRIP focus formation remains unknown. It has been shown that ATRIP-RPA-ssDNA interaction is not sufficient for ATR-ATRIP localization to sites of DNA damage (Ball et al., 2005). Several distinct mechanisms of ATRIP localization have been suggested (Zou and Elledge, 2003; Bomgarden et al., 2004; Unsal-Kacmaz and Sancar, 2004). We hypothesize that chromatin association by the ATR-ATRIP complex, which is not affected by the ATRIP-LG mutation and is probably mediated by ATRIP-RPA-ssDNA interaction, contributes to the initial activation of ATR that leads to the phosphorylation of Ser645 of Rad17 as well as Ser317 and -345 of Chk1. Corroborating this hypothesis is the previous finding that the phosphorylation of Rad17 by ATR, which is proficient in ATRIP-LG cells, occurred on chromatin (Zou et al., 2002). RPA has been shown to be required for recruitment of ATRIP to aphidicolin-induced foci (Itakura et al., 2004a; Ball et al., 2005), and in this study we show that the ATR-ATRIP-LG complex is defective in accumulation at sites of DNA damage in spite of retaining the RPA-interacting activity. Thus, the latter mode of ATR-ATRIP localization, which requires a normal configuration of the ATRIP coiled-coil, may be mediated by stable or enhanced contact with replication components in addition to the interaction with RPA. One candidate for another interacting molecule is MCM7 protein enriched in stalled replication forks (Cortez et al., 2004) because MCM7 is required for stable localization of ATR to damage-induced foci (Tsao et al., 2004).
Function of ATR-ATRIP upon Replication Stress
ATR has been shown to be important for cellular responses to genotoxic stress during S and G2/M phases (Dimitrova and Gilbert, 2000; Tibbetts et al., 2000; Heffernan et al., 2002), but the exact role of ATR in the S-phase checkpoint is still elusive. The loss of ATR induces chromosomal breaks after release from aphidicolin treatment (Casper et al., 2002). Chk1 is phosphorylated by ATR after exposure to genotoxic stress, including treatment with aphidicolin, HU, or UV irradiation (Zhao and Piwnica-Worms, 2001), and the ATR-mediated phosphorylation of Chk1 is essential for the checkpoint response in Xenopus (Guo et al., 2000). However, ATR knockout cells in mice can arrest the cell cycle after aphidicolin treatment despite the lack of detectable phosphorylation of Chk1 (Brown and Baltimore, 2003). These data suggest that ATR is important for maintaining integrity in the replicating chromosomes, but that ATR is not directly involved in cell cycle arrest in the S phase. A recent study using Chk1-deficient DT40 chicken cells suggests that Chk1 maintains the replication checkpoint indirectly by preserving the integrity of replication structures, rather than regulating it directly (Zachos et al., 2005).
We note that the modification status of Chk1 upon replication blockage is apparently affected by the coiled-coil mutation, and consequently the cells showed a phenotype related to the defective activity of the Chk1 kinase. This implies that some of the events downstream from ATR in the signaling cascade, including Chk1 activation, are dependent on the correct localization of the ATR-ATRIP complex, but ATR appears to be activated, at least partially, unless ATR-ATRIP accumulates at sites of DNA damage.
The checkpoint-dependent phosphorylation of Chk1 is a multistep process that includes autophosphorylation (Kumagai et al., 2004). Moreover, the process involves activation of the ATR-ATRIP complexes at replication forks (Kumagai et al., 2004). Claspin is thought to be a component of replication forks (Lee et al., 2003; Sar et al., 2004), to interact with both ATR and Chk1 and to affect the Chk1 status (Jeong et al., 2003; Kumagai and Dunphy, 2003; Kumagai et al., 2004) in Xenopus. Mrc1, a yeast homolog of Claspin, interacts directly with the DNA replication machinery (Katou et al., 2003) and is also important for the Mec1-dependent activation of downstream effectors upon replication stress (Alcasabas et al., 2001; Tanaka and Russell, 2001). The concentrated localization of ATR-ATRIP to stalled replication sites, which requires the ATRIP coiled-coil, may be involved in S-phase-specific activation of the ATR-Claspin-Chk1 pathway, but further studies are necessary to elucidate the mechanism.
G2/M Function of ATR
In contrast to several of the defects in response to replication stress, the IR-induced G2/M checkpoint is little affected by the expression of ATRIP-LG. This suggests that the accumulation of the ATR-ATRIP complex at foci is not necessarily essential for its function in the G2/M checkpoint. As shown in this study, IR-induced modification of Chk1 appears to differ from that which occurs upon replication blockage; interestingly, our results suggest that the normal coiled-coil configuration of ATRIP is required only for the latter process. Our study sheds light on the functional difference of the ATR-ATRIP complex in signal transduction to downstream effectors such as Chk1, after distinct types of DNA damage.
Supplementary Material
Acknowledgments
We thank C. Obuse for providing the anti-ORC1 antibody; G. Hannon for the pSHAG-1 plasmid; Makoto Nakanishi and Kenji Shimada for helpful discussion; Y. Tanaka for technical assistance; and members of the Department of Geriatric Medicine, National Institute for Longevity Sciences, for their suggestions and comments. This work was supported in part by grants from the Ministry of Education, Culture, Sports, Science, and Technology and the Ministry of Health, Labor, and Wealth of Japan, the Public Trust Haraguchi Memorial Cancer Research Fund, and the Kowa Life Science Foundation.
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E05–05–0427) on September 21, 2005.
The online version of this article contains supplemental material at MBC Online (http://www.molbiolcell.org).
References
- Abraham, R. T. (2001). Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 15, 2177–2196. [DOI] [PubMed] [Google Scholar]
- Alderton, G. K., Joenje, H., Varon, R., Borglum, A. D., Jeggo, P. A., and O'Driscoll, M. (2004). Seckel syndrome exhibits cellular features demonstrating defects in the ATR-signalling pathway. Hum. Mol. Genet. 13, 3127–3138. [DOI] [PubMed] [Google Scholar]
- Alcasabas, A. A., Osborn, A. J., Bachant, J., Hu, F., Werler, P. J., Bousset, K., Furuya, K., Diffley, J. F., Carr, A. M., and Elledge, S. J. (2001). Mrc1 transduces signals of DNA replication stress to activate Rad53. Nat. Cell Biol. 3, 958–965. [DOI] [PubMed] [Google Scholar]
- Bakkenist, C. J., and Kastan, M. B. (2003). DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421, 499–506. [DOI] [PubMed] [Google Scholar]
- Ball, H. L., and Cortez, D. (2005). ATRIP oligomerization is required for ATR-dependent checkpoint signaling. J. Biol. Chem. 280, 31390–31396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball, H. L., Myers, J. S., and Cortez, D. (2005). ATRIP binding to replication protein A-single-stranded DNA promotes ATR-ATRIP localization but is dispensable for Chk1 phosphorylation. Mol. Biol. Cell 16, 2372–2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao, S., Tibbetts, R. S., Brumbaugh, K. M., Fang, Y., Richardson, D. A., Ali, A., Chen, S. M., Abraham, R. T., and Wang, X. F. (2001). ATR/ATM-mediated phosphorylation of human Rad17 is required for genotoxic stress responses. Nature 411, 969–974. [DOI] [PubMed] [Google Scholar]
- Barr, S. M., Leung, C. G., Chang, E. E., and Cimprich, K. A. (2003). ATR kinase activity regulates the intranuclear translocation of ATR and RPA following ionizing radiation. Curr. Biol. 13, 1047–1051. [DOI] [PubMed] [Google Scholar]
- Bomgarden, R. D., Yean, D., Yee, M. C., and Cimprich, K. A. (2004). A novel protein activity mediates DNA binding of an ATR-ATRIP complex. J. Biol. Chem. 279, 13346–13353. [DOI] [PubMed] [Google Scholar]
- Brown, E. J., and Baltimore, D. (2000). ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 14, 397–402. [PMC free article] [PubMed] [Google Scholar]
- Brown, E. J., and Baltimore, D. (2003). Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev. 17, 615–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budzowska, M. et al. (2004). Mutation of the mouse Rad17 gene leads to embryonic lethality and reveals a role in DNA damage-dependent recombination. EMBO J. 23, 3548–3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casper, A. M., Nghiem, P., Arlt, M. F., and Glover, T. W. (2002). ATR regulates fragile site stability. Cell 111, 779–789. [DOI] [PubMed] [Google Scholar]
- Celeste, A., Fernandez-Capetillo, O., Kruhlak, M. J., Pilch, D. R., Staudt, D. W., Lee, A., Bonner, R. F., Bonner, W. M., and Nussenzweig, A. (2003). Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat. Cell Biol. 5, 675–679. [DOI] [PubMed] [Google Scholar]
- Cha, R. S., and Kleckner, N. (2002). ATR homolog Mec1 promotes fork progression, thus averting breaks in replication slow zones. Science 297, 602–606. [DOI] [PubMed] [Google Scholar]
- Cobb, J. A., Bjergbaek, L., Shimada, K., Frei, C., and Gasser, S. M. (2003). DNA polymerase stabilization at stalled replication forks requires Mec1 and the RecQ helicase Sgs1. EMBO J. 22, 4325–4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez, D., Glick, G., and Elledge, S. J. (2004). Minichromosome maintenance proteins are direct targets of the ATM and ATR checkpoint kinases. Proc. Natl. Acad. Sci. USA 101, 10078–10083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez, D., Guntuku, S., Qin, J., and Elledge, S. J. (2001). ATR and ATRIP: partners in checkpoint signaling. Science 294, 1713–1716. [DOI] [PubMed] [Google Scholar]
- Dart, D. A., Adams, K. E., Akerman, I., and Lakin, N. D. (2004). Recruitment of the cell cycle checkpoint kinase ATR to chromatin during S-phase. J. Biol. Chem. 279, 16433–16440. [DOI] [PubMed] [Google Scholar]
- de Klein, A., Muijtjens, M., van Os, R., Verhoeven, Y., Smit, B., Carr, A. M., Lehmann, A. R., and Hoeijmakers, J. H. (2000). Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr. Biol. 10, 479–482. [DOI] [PubMed] [Google Scholar]
- Dimitrova, D. S., and Gilbert, D. M. (2000). Temporally coordinated assembly and disassembly of replication factories in the absence of DNA synthesis. Nat. Cell Biol. 2, 686–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson, G. E., Shi, Y., and Tibbetts, R. S. (2004). DNA replication defects, spontaneous DNA damage, and ATM-dependent checkpoint activation in replication protein A-deficient cells. J. Biol. Chem. 279, 34010–34014. [DOI] [PubMed] [Google Scholar]
- Falck, J., Coates, J., and Jackson, S. P. (2005). Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 434, 605–611. [DOI] [PubMed] [Google Scholar]
- Feijoo, C., Hall-Jackson, C., Wu, R., Jenkins, D., Leitch, J., Gilbert, D. M., and Smythe, C. (2001). Activation of mammalian Chk1 during DNA replication arrest: a role for Chk1 in the intra-S phase checkpoint monitoring replication origin firing. J. Cell Biol. 154, 913–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg, M., Stucki, M., Falck, J., D'Amours, D., Rahman, D., Pappin, D., Bartek, J., and Jackson, S. P. (2003). MDC1 is required for the intra-S-phase DNA damage checkpoint. Nature 421, 952–956. [DOI] [PubMed] [Google Scholar]
- Guo, Z., Kumagai, A., Wang, S. X., and Dunphy, W. G. (2000). Requirement for Atr in phosphorylation of Chk1 and cell cycle regulation in response to DNA replication blocks and UV-damaged DNA in Xenopus egg extracts. Genes Dev. 14, 2745–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heffernan, T. P., Simpson, D. A., Frank, A. R., Heinloth, A. N., Paules, R. S., Cordeiro-Stone, M., and Kaufmann, W. K. (2002). An ATR- and Chk1-dependent S checkpoint inhibits replicon initiation following UVC-induced DNA damage. Mol. Cell. Biol. 22, 8552–8561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekmat-Nejad, M., You, Z., Yee, M. C., Newport, J. W., and Cimprich, K. A. (2000). Xenopus ATR is a replication-dependent chromatin-binding protein required for the DNA replication checkpoint. Curr. Biol. 10, 1565–1573. [DOI] [PubMed] [Google Scholar]
- Hopkins, K. M., Auerbach, W., Wang, X. Y., Hande, M. P., Hang, H., Wolgemuth, D. J., Joyner, A. L., and Lieberman, H. B. (2004). Deletion of mouse rad9 causes abnormal cellular responses to DNA damage, genomic instability, and embryonic lethality. Mol. Cell. Biol. 24, 7235–7248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itakura, E., Takai, K. K., Umeda, K., Kimura, M., Ohsumi, M., Tamai, K., and Matsuura, A. (2004a). Amino-terminal domain of ATRIP contributes to intranuclear relocation of the ATR-ATRIP complex following DNA damage. FEBS Lett. 577, 289–293. [DOI] [PubMed] [Google Scholar]
- Itakura, E., Umeda, K., Sekoguchi, E., Takata, H., Ohsumi, M., and Matsuura, A. (2004b). ATR-dependent phosphorylation of ATRIP in response to genotoxic stress. Biochem. Biophys. Res. Commun. 323, 1197–1202. [DOI] [PubMed] [Google Scholar]
- Jeong, S. Y., Kumagai, A., Lee, J., and Dunphy, W. G. (2003). Phosphorylated claspin interacts with a phosphate-binding site in the kinase domain of Chk1 during ATR-mediated activation. J. Biol. Chem. 278, 46782–46788. [DOI] [PubMed] [Google Scholar]
- Katou, Y., Kanoh, Y., Bando, M., Noguchi, H., Tanaka, H., Ashikari, T., Sugimoto, K., and Shirahige, K. (2003). S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature 424, 1078–1083. [DOI] [PubMed] [Google Scholar]
- Kim, S. T., Lim, D. S., Canman, C. E., and Kastan, M. B. (1999). Substrate specificities and identification of putative substrates of ATM kinase family members. J. Biol. Chem. 274, 37538–37543. [DOI] [PubMed] [Google Scholar]
- Kumagai, A., and Dunphy, W. G. (2003). Repeated phosphopeptide motifs in Claspin mediate the regulated binding of Chk1. Nat. Cell Biol. 5, 161–165. [DOI] [PubMed] [Google Scholar]
- Kumagai, A., Kim, S. M., and Dunphy, W. G. (2004). Claspin and the activated form of ATR-ATRIP collaborate in the activation of Chk1. J. Biol. Chem. 279, 49599–49608. [DOI] [PubMed] [Google Scholar]
- Lee, J., Kumagai, A., and Dunphy, W. G. (2003). Claspin, a Chk1-regulatory protein, monitors DNA replication on chromatin independently of RPA, ATR, and Rad17. Mol. Cell 11, 329–340. [DOI] [PubMed] [Google Scholar]
- Liu, Q. et al. (2000). Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 14, 1448–1459. [PMC free article] [PubMed] [Google Scholar]
- Lopes, M., Cotta-Ramusino, C., Pellicioli, A., Liberi, G., Plevani, P., Muzi-Falconi, M., Newlon, C. S., and Foiani, M. (2001). The DNA replication checkpoint response stabilizes stalled replication forks. Nature 412, 557–561. [DOI] [PubMed] [Google Scholar]
- Lou, Z., Chini, C. C., Minter-Dykhouse, K., and Chen, J. (2003). Mediator of DNA damage checkpoint protein 1 regulates BRCA1 localization and phosphorylation in DNA damage checkpoint control. J. Biol. Chem. 278, 13599–13602. [DOI] [PubMed] [Google Scholar]
- Mendez, J., and Stillman, B. (2000). Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol. Cell. Biol. 20, 8602–8612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nghiem, P., Park, P. K., Kim, Y., Vaziri, C., and Schreiber, S. L. (2001). ATR inhibition selectively sensitizes G1 checkpoint-deficient cells to lethal premature chromatin condensation. Proc. Natl. Acad. Sci. USA 98, 9092–9097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Driscoll, M., Ruiz-Perez, V. L., Woods, C. G., Jeggo, P. A., and Goodship, J. A. (2003). A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat. Genet. 33, 497–501. [DOI] [PubMed] [Google Scholar]
- Sancar, A., Lindsey-Boltz, L. A., Unsal-Kaccmaz, K., and Linn, S. (2004). Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 73, 39–85. [DOI] [PubMed] [Google Scholar]
- Sar, F., Lindsey-Boltz, L. A., Subramanian, D., Croteau, D. L., Hutsell, S. Q., Griffith, J. D., and Sancar, A. (2004). Human claspin is a ring-shaped DNA-binding protein with high affinity to branched DNA structures. J. Biol. Chem. 279, 39289–39295. [DOI] [PubMed] [Google Scholar]
- Sekoguchi, E., Sato, N., Yasui, A., Fukada, S., Nimura, Y., Aburatani, H., Ikeda, K., and Matsuura, A. (2003). A novel mitochondrial carnitine-acylcarnitine translocase induced by partial hepatectomy and fasting. J. Biol. Chem. 278, 38796–38802. [DOI] [PubMed] [Google Scholar]
- Shiloh, Y. (2001). ATM and ATR: networking cellular responses to DNA damage. Curr. Opin. Genet. Dev. 11, 71–77. [DOI] [PubMed] [Google Scholar]
- Smith, G. C., Cary, R. B., Lakin, N. D., Hann, B. C., Teo, S. H., Chen, D. J., and Jackson, S. P. (1999). Purification and DNA binding properties of the ataxiatelangiectasia gene product ATM. Proc. Natl. Acad. Sci. USA 96, 11134–11139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart, G. S., Wang, B., Bignell, C. R., Taylor, A. M., and Elledge, S. J. (2003). MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature 421, 961–966. [DOI] [PubMed] [Google Scholar]
- Suzuki, K., Kodama, S., and Watanabe, M. (1999). Recruitment of ATM protein to double strand DNA irradiated with ionizing radiation. J. Biol. Chem. 274, 25571–25575. [DOI] [PubMed] [Google Scholar]
- Takai, H., Tominaga, K., Motoyama, N., Minamishima, Y. A., Nagahama, H., Tsukiyama, T., Ikeda, K., Nakayama, K., and Nakanishi, M. (2000). Aberrant cell cycle checkpoint function and early embryonic death in Chk1(-/-) mice. Genes Dev. 14, 1439–1447. [PMC free article] [PubMed] [Google Scholar]
- Takata, H., Kanoh, Y., Gunge, N., Shirahige, K., and Matsuura, A. (2004). Reciprocal association of the budding yeast ATM-related proteins Tel1 and Mec1 with telomeres in vivo. Mol. Cell 14, 515–522. [DOI] [PubMed] [Google Scholar]
- Tanaka, K., and Russell, P. (2001). Mrc1 channels the DNA replication arrest signal to checkpoint kinase Cds1. Nat. Cell Biol. 3, 966–972. [DOI] [PubMed] [Google Scholar]
- Tatsumi, Y., Ohta, S., Kimura, H., Tsurimoto, T., and Obuse, C. (2003). The ORC1 cycle in human cells: I. Cell cycle-regulated oscillation of human ORC1. J. Biol. Chem. 278, 41528–41534. [DOI] [PubMed] [Google Scholar]
- Tercero, J. A., and Diffley, J. F. (2001). Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature 412, 553–557. [DOI] [PubMed] [Google Scholar]
- Tibbetts, R. S., Cortez, D., Brumbaugh, K. M., Scully, R., Livingston, D., Elledge, S. J., and Abraham, R. T. (2000). Functional interactions between BRCA1 and the checkpoint kinase ATR during genotoxic stress. Genes Dev. 14, 2989–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao, C. C., Geisen, C., and Abraham, R. T. (2004). Interaction between human MCM7 and Rad17 proteins is required for replication checkpoint signaling. EMBO J. 23, 4660–4669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unsal-Kacmaz, K., Makhov, A. M., Griffith, J. D., and Sancar, A. (2002). Preferential binding of ATR protein to UV-damaged DNA. Proc. Natl. Acad. Sci. USA 99, 6673–6678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unsal-Kacmaz, K., and Sancar, A. (2004). Quaternary structure of ATR and effects of ATRIP and replication protein A on its DNA binding and kinase activities. Mol. Cell. Biol. 24, 1292–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y., Cortez, D., Yazdi, P., Neff, N., Elledge, S. J., and Qin, J. (2000). BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. 14, 927–939. [PMC free article] [PubMed] [Google Scholar]
- Weiss, R. S., Enoch, T., and Leder, P. (2000). Inactivation of mouse Hus1 results in genomic instability and impaired responses to genotoxic stress. Genes Dev. 14, 1886–1898. [PMC free article] [PubMed] [Google Scholar]
- Wolkow, T. D., and Enoch, T. (2002). Fission yeast Rad26 is a regulatory subunit of the Rad3 checkpoint kinase. Mol. Biol. Cell 13, 480–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, J. A., Keegan, K. S., Herendeen, D. R., Bentley, N. J., Carr, A. M., Hoekstra, M. F., and Concannon, P. (1998). Protein kinase mutants of human ATR increase sensitivity to UV and ionizing radiation and abrogate cell cycle checkpoint control. Proc. Natl. Acad. Sci. USA 95, 7445–7450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachos, G., Rainey, M. D., and Gillespie, D. A. (2005). Chk1-dependent S-M checkpoint delay in vertebrate cells is linked to maintenance of viable replication structures. Mol. Cell. Biol. 25, 563–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, H., and Piwnica-Worms, H. (2001). ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell. Biol. 21, 4129–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou, L., Cortez, D., and Elledge, S. J. (2002). Regulation of ATR substrate selection by Rad17-dependent loading of Rad9 complexes onto chromatin. Genes Dev. 16, 198–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou, L., and Elledge, S. J. (2003). Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300, 1542–1548. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.