

Abstract
The crystal structure of the ribosomal 50S subunit from Haloarcula marismortui in complex with the transition state analog CCdA-phosphate-puromycin (CCdApPmn) led to a mechanistic proposal wherein the universally conversed A2451 in the ribosomal active site acts as an “oxyanion hole” to promote the peptidyl transferase reaction [Nissen, P., Hansen, J., Ban, N., Moore, P.B., and Steitz, T.A. (2000) Science 289, 920–929]. In the model, close proximity (3 Å) between the A2451 N3 and the nonbridging phosphoramidate oxygen of CCdApPmn suggested that the carbonyl oxyanion formed during the tetrahedral transition state is stabilized by hydrogen bonding to the protonated A2451 N3, the pKa of which must be perturbed substantially. We characterize the contribution of the putative hydrogen bond between the N3 of A2451 and the nonbridging phosphoramidate oxygen by using chemical protection and peptidyl transfer inhibition assays. If this putative hydrogen bond makes a significant thermodynamic contribution, then CCdApPmn-binding affinity to the 50S ribosomal subunit should be strongly pH-dependent, with affinity increasing as the pH is lowered. We report that CCdApPmn binds 50S ribosomes with essentially equal affinity at all pH values between 5.0 and 8.5. These data argue against a mechanism for peptidyl transfer in which a residue with near neutral pKa stabilizes the transition-state oxyanion, at least to the extent that CCdApPmn accurately mimics the transition state.
The ribosome is a molecular machine that assembles polypeptide chains. The addition of an amino acid onto a nascent peptide chain, termed peptidyl transfer, is catalyzed by the 50S ribosomal subunit by using aminoacyl-tRNA and peptidyl-tRNA as substrates. In the course of the reaction, the peptidyl-tRNA, charged with the growing peptide chain, occupies the P site, and an aminoacyl-tRNA, activated with a single amino acid, binds the A site. Peptide bond formation occurs by a transacylation reaction mechanism wherein the α-amino group on the A-site tRNA nucleophilically attacks the ester linkage between the peptide chain and the 3′-hydroxyl of the P-site tRNA. It is expected that the reaction proceeds through a transition state that has a tetrahedral geometry at the carbonyl carbon and includes a negatively charged oxyanion. Collapse of the transition state produces a deacylated P-site tRNA and a peptide chain that is elongated by one amino acid coupled to the A-site tRNA (for review see ref. 1).
Defining how this reaction is catalyzed has been a question of active research for over 30 years. Despite an early and rather indirect indication that a protein side chain might be responsible for catalysis (2, 3), biochemical evidence has identified RNA, which accounts for about two thirds of ribosomal molecular weight (4), as the most likely catalytic component. Highly conserved internal loops of the 23S rRNA domain V have been shown biochemically to interact with the 3′-CCA ends of the A-site and P-site tRNAs (5, 6) as well as aminoacyl residues attached to the P-site tRNA (7). These results showed that rRNA is in close proximity to the nucleophile and leaving group. Other experiments showed that large ribosomal subunit particles stripped of 95% of the ribosomal protein retained catalytic activity, suggesting that 23S and 5S rRNA may constitute the bulk of the peptidyl transferase center (8).
The most unambiguous evidence that the active site of the ribosome is comprised of RNA came from the 2.4-Å crystal structure of the Haloarcula marismortui 50S ribosomal subunit reported by Ban et al. (9) and Nissen et al. (10). The structure of the 50S subunit complexed with the transition-state analog CCdA-phosphate-puromycin (CCdApPmn) was vital to this structural identification. CCdApPmn includes the minimal components of both peptidyl transferase substrates (11). CCdA binds the P site, and puromycin binds the A site (Fig. 1). The α-amino group of puromycin is connected covalently to the 3′ oxygen of CCdA through a phosphoramidate linkage that has a tetrahedral geometry comparable in shape to the peptidyl transfer transition state (11, 12). Within the cocrystal structure only 23S rRNA contacts CCdApPmn. In fact, the nearest protein residue is almost 20 Å removed from the phosphoramidate linkage, which argues that rRNA and not protein catalyzes peptidyl transfer (10).
Figure 1.

Peptidyl transferase transition-state inhibitor and proposed mechanism of transition-state stabilization based on the inhibitor structure. (a) Schematic representation of CCdApPmn in the peptidyl transferase center of the ribosome, indicating the stereochemistry about the phosphoramidate. The reported distance from the N3 of A2451 to the nonbridging O2 of the achiral phosphoramidate linker of CCdApPmn is indicated (10). (b) Mechanism of transition-state oxyanion stabilization by a protonated N3 of A2451 based on the proposal of Nissen et al. (10). The chirality of the transition state is indicated, with presumed stereochemistry and assignments of oxyanion and CH of α-carbon of peptide chain based on the crystallographic model (10). The A-site tRNA is depicted as being charged with tyrosine. In both cases, only the chirality of the tetrahedral center is indicated.
Within the 50S structure, the N3 of the universally conserved A2451 (Escherichia coli numbering) is within hydrogen-bonding distance (3 Å) of the nonbridging phosphoramidate oxygen (designated O2 in the crystal coordinates) of CCdApPmn (Fig. 1a). The implied hydrogen bond occurs despite the fact that neither the O2 oxygen nor the N3 imino group would normally be protonated at pH 5.8, the pH used for crystallization of the 50S subunit (10). Based on the assumption that O2 is analogous to the negatively charged oxyanion formed in the transition state, the close approach of these two groups led Nissen et al. to two conclusions: (i) the N3 pKa of A2451 is perturbed toward neutrality in the transition state, and (ii) the protonated A2451 N3 stabilizes the negative charge on the transition state oxyanion by hydrogen bonding and/or charge neutralization. In this manner A2451 was proposed to serve as the oxyanion hole for the peptidyl transfer reaction (ref. 10; Fig. 1b). This contribution is in addition to its role as a general base for activation of the nucleophilic α-amino group.
Biochemical experiments have attempted to determine the extent to which the A2451 pKa is perturbed in the ground state of the ribosome. The pH dependence of dimethyl sulfate (DMS) reactivity at A2451 suggested that the active-site residue has an unusually high pKa of 7.6 (13). However, subsequent experiments found that DMS modification at A2451 occurred only in inactive E. coli 50S subunits, and no reactivity was observed at any pH after heat activation (14). Furthermore, the DMS modification most likely occurred at the N1 rather than the N3 imino group of A2451 (15). DMS protection experiments performed on ribosomes from several different organisms showed either no reactivity at A2451 or a reactivity pattern inconsistent with a direct pKa effect (15, 16). Overall, these data are more consistent with a pH-dependent conformational change in the peptidyl transfer center that involves A2451 (15), but they do not address whether the N3 pKa is perturbed either in the ground or transition states of the reaction.
At present, the only evidence to support an elevated pKa for A2451 in the transition-state complex is the interatomic distance between the A2451 N3 and the phosphoramidate O2 in the cocrystal structure (10). This distance implies hydrogen-bond formation, but for this to occur the base must be protonated at a pH well above its regular pKa. Thus, the structure makes a biochemical prediction that can be tested experimentally. If the putative hydrogen bond between A2451 and the phosphoramidate contributes to CCdApPmn-binding affinity, then its binding constant should be strongly pH-dependent. The affinity should be higher at acidic pH and become progressively weaker as the pH is raised. We have explored this hypothesis by measuring the pH dependence of CCdApPmn binding. The results argue against transition-state oxyanion stabilization by the peptidyl transferase center of the ribosome insofar as CCdApPmn is an accurate mimic of that transition state.
Materials and Methods
Synthesis of CCdApPmn.
The synthesis of CCdApPmn followed that described by Welch et al. (11) with minor modifications. CCdAp (100 nmol, Dharmacon Research, Lafayette, CO) was coupled to puromycin (12.3 μmol) in the presence of 1-ethyl-3-[3-(dimethylamino)propyl]carbodiimide (EDAC, 50 μmol) buffered with 400 mM Mes at pH 6.0 in an aqueous reaction volume of 125 μl. The reaction was carried out for 4 h at 25°C. The mixture then was diluted to 500 μl with water and added to a 200-μl A-25 Sephadex column. The column was washed with 4 ml of water, followed by 4 ml of 30 mM NH4OAc (pH 6.5), and eluted with 4 ml of 750 mM NH4OAc. CCdApPmn was purified further by HPLC with a C18 column (Rainin Instruments) and eluted with a 100 mM NH4OAc (pH 6.5)/acetonitrile gradient (0–50% acetonitrile) over 80 min. The retention time of CCdApPmn was 38 min. Product formation was confirmed by mass spectrometry (Howard Hughes Medical Institute/Keck Biotechnology Resource Laboratory, Yale University, New Haven, CT) and 31P NMR: theoretical m/z, 1,393.36; actual m/z, 1393.35. 31P NMR: δ, −0.09, 0.14, 5.79 ppm.
Preparation of Ribosomes.
Ribosomal 50S subunits were isolated from early log-phase MRE600 cells and prepared as described by Rheinberger et al. (17) with minor modifications.
Chemical Modification of 23S rRNA.
The 23S rRNA within intact ribosomal particles was modified with DMS. Each reaction contained 50 nM 50S ribosomes (preheated at 42°C for 5 min), 200 mM KCl, 20 mM MgCl2, 50 mM buffer (KOAc, pH 5.0/Mes, pH 5.5, pH 6.0/Mops, pH 6.5, pH 7.0, pH 7.5/Tris-Cl, pH 8.0/Tris-Borate, pH 8.5), 33% methanol, and 0–1,500 nM CCdApPmn in a total volume of 25 μl. DMS was added to ribosomes (1 μl of a 1:10 DMS/ethanol solution prepared immediately before the reaction) and allowed to react at 25°C for 30 min. The RNA was precipitated by adding 2.5 volumes of ethanol and stored at −80°C for 4 h, after which the ribosomes were pelleted by centrifugation. The rRNA was isolated from ribosomal proteins by phenol/chloroform extraction.
Primer Extension and Gel Electrophoresis.
The extent of DMS modification at A2602 was monitored by reverse transcription from a 5′-32P-radiolabeled DNA primer complementary to nucleotides 2,639–2,656. The DNA primer (50,000 cpm) and 23S rRNA (10 nM) were heat-denatured at 90°C for 2 min in annealing buffer (50 mM Tris, pH 8.3/50 mM NaCl/10 mM DTT) in a 5-μl total reaction volume and slow-cooled to 42°C. avian myeloblastosis virus reverse transcriptase (2 units, Roche Applied Science, Indianapolis), dNTPs (80 μM final concentration each), and MgCl2 (10 mM final concentration) were added, and the reaction was incubated for 30 min at 42°C. Adenosine ladders were obtained by adding a final concentration of 5 μM ddUTP to reverse transcription of unmodified 23S rRNA. Reactions were quenched by the addition of an equal volume of formamide loading buffer (95% formamide/25 mM EDTA, pH 8.0) and heat-denatured at 90°C for 2 min. Experiments at each pH were run in duplicate.
Primer extension products were resolved by 8% denaturing polyacrylamide electrophoresis and quantitated by using a Molecular Dynamics STORM PhosphorImager. The DMS modification at A2602 results in a reverse-transcriptase stop that is visible as a band on a polyacrylamide sequencing gel occurring 1 nt before the modified position. CCdApPmn binding results in reduced DMS accessibility at A2602, which attenuates the band intensity. The loss in intensity of DMS modification at A2602 (I) as a function of CCdApPmn concentration was used to determine the dissociation constant (Kd) of the inhibitor at each pH by using the equation
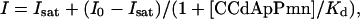 |
1 |
where Isat is the band intensity at saturating inhibitor, and I0 is the band intensity with no inhibitor at a given pH. Band intensities were normalized for loading and overall DMS reactivity within 23S rRNA, and band intensities with inhibitor present ([CCdApPmn] > 0) were normalized relative to I0, the band intensity with no inhibitor ([CCdApPmn] = 0).
Peptidyl Transfer Fragment Assay Inhibition.
Inhibition of peptidyl transfer by CCdApPmn was monitored by using a modified fragment assay that uses CCA-phenylalanyl-caproyl-biotin (CCA-pcb) as the P-site substrate and C-puromycin (CPmn) as the A-site substrate (18). The reaction products are CPmn-phenylalanyl-caproyl-biotin (CPmn-pcb) and CCA, and both products are produced at equivalent rates, although the rate of CCA formation must be corrected for background hydrolysis. Reactions contained 50–75 nM 50S ribosomal subunits (preheated for 5 min at 42°C), 200 mM KCl, 20 mM MgCl2, 50 mM buffer (KOAc, pH 5.0/Mes, pH 5.5, pH 6.0/Mops, pH 6.5, pH 7.0, pH 7.5/Tris-Cl, pH 8.0), 40–500 μM CPmn, 200 nM unphosphorylated CCA-pcb, 2,000–3,000 cpm of 32P-labeled CCA-pcb, 33% methanol, and either 0 or 200 nM CCdApPmn in a total volume of 25 μl. Although methanol is not necessary for the modified fragment assay, it increases the rate at low ribosome and substrate concentrations (K.M.P., unpublished data). Reactions were initiated by the addition of 50S subunits and incubated at 25°C. Reaction time points were obtained by removing 2 μl of the reaction and quenching it with an equal volume of formamide loading buffer (95% formamide/50 mM Tris-sodium phosphate, pH 6.5).
Products were resolved by 12% denaturing polyacrylamide electrophoresis. Gels were run at 4°C by using a low-pH gel running buffer (50 mM Tris/NaHPO3, pH 6.5) to prevent base hydrolysis of CCA-pcb. The fraction reacted was quantitated with a Molecular Dynamics STORM PhosphorImager. These experiments monitored the conversion of CCA-pcb to CCA. Rates were determined by fitting the fraction reacted as a function of time to an exponential function and subtracting rates of background hydrolysis. Ki values were determined by Lineweaver–Burke analysis in which rates of both uninhibited and inhibited reactions were plotted as a function of A-site substrate (CPmn) concentration by using the formula
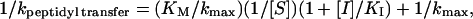 |
2 |
where kpeptidyl transfer is the measured rate, [S] is the A-site substrate concentration, KM refers to the affinity of the A-site substrate for the ribosome, kmax is the maximum rate, [I] is the CCdAPmn concentration, and KI is the inhibition constant. Both KM and kmax were determined by least-squares analysis for the uninhibited ([I] = 0) reactions at each pH and then used to determine KI in the inhibited reactions at that pH. Experiments at each pH value were performed in duplicate. The pH values of all buffers were measured at 25°C, the same temperature as the peptidyl transfer reactions. The efficiency of inhibition was not affected by preincubation of CCdApPmn with 50S subunits for extended periods (data not shown).
Results
We set out to measure the contribution of the putative hydrogen bond between the N3 of A2451 and the O2 nonbridging phosphoramidate oxygen as a means to address if the N3 has a near-neutral pKa in the transition state. At pH values below its pKa, the N3 should be protonated and hydrogen-bond to the phosphoramidate O2 of CCdApPmn, resulting in greater binding affinity. As the pH is increased, the N3 should be protonated less efficiently, and the inhibitor binding affinity should be reduced, eventually resulting in a minimum binding constant. Thus the pKa of N3 in the transition state can be determined by analyzing Kd values for CCdApPmn as a function of pH. Toward this goal, we used both chemical protection and peptidyl transfer inhibition experiments to measure the pH dependence of CCdApPmn affinity.
Chemical Protection of A2602.
The universally conserved residue A2602 is located near the peptidyl transfer center of the ribosome. Within the cocrystal structure, A2602 interacts extensively with the puromycin moiety of CCdApPmn (10). Consistent with this observation, chemical footprinting experiments demonstrated that A2602 shows a decrease in DMS reactivity after binding of A-site tRNA as well as an enhanced DMS reactivity after binding of P-site tRNA (19). DMS footprinting studies showed that A2602 is modestly protected after binding of CCdApPmn (11), perhaps a convolution of enhancement and protection because the transition-state analog contains both A-site and P-site tRNA elements. Fig. 2a shows the roughly 2-fold reduction in A2602 DMS modification at a saturating CCdApPmn concentration. Also shown is a modest stimulation of U2555 toward DMS reactivity, which was noted previously (20). We used the protection of A2602 from DMS modification as a means of determining the Kd of CCdApPmn as a function of pH. Fig. 2b shows two examples of Kd curves for the inhibitor measured at pH 5.0 and 8.5, the extremes of the pH range measured. The curves correspond to a Kd of 220 ± 50 nM for pH 5.0 and a Kd of 220 ± 40 nM for pH 8.5. We also measured CCdApPmn Kd values at pHs between pH 5.0 and 8.5 (Fig. 2c) and found that the CCdApPmn binding affinity is independent of pH within this range, which runs counter to the crystallographic expectation (10).
Figure 2.

pH dependence of CCdApPmn binding based on DMS footprinting. (a) Protection from DMS modification at A2602 by CCdApPmn binding at pH 7.0. 23S rRNA was reverse-transcribed with a radiolabeled DNA primer complementary to nucleotides 2,639–2,656. Lane 1, rRNA adenosine ladder generated by incorporation of ddUTP during reverse transcription; lane 2, reverse transcription of 23S rRNA that was not DMS-modified; lane 3, DMS modification and reverse transcription in the absence of CCdApPmn [note the strong new (+1) reverse-transcription stop that reflects modification of A2602]; lane 4, DMS modification in the presence of 500 nM CCdApPmn and reverse transcription [note the reduction of signal intensity at A2602 and additionally the increase in signal intensity at U2555 (+1)]. (b) Normalized extent of DMS reactivity at A2602 as a function of CCdApPmn at pH 5.0 and 8.5. For each plot the band intensity was corrected for loading and overall DMS reactivity within the 23S rRNA. The Kd value for CCdApPmn in the ribosomal active site was calculated by using Eq. 1. (c) pH dependence of CCdApPmn binding as measured by chemical protection. Each point is an average of two independent experiments. Standard deviations are indicated with error bars.
Previous studies also used the modifying agent 1-cyclohexyl-3-(2-morpholinoethyl)carbodiimide metho-p-toluenesulfonate (CMCT) at U2585 to determine the inhibitor binding constant. We measured the binding of CCdApPmn by CMCT footprinting and found the affinity to be essentially invariant between pH 7.5 and 9.1 (data not shown). Unfortunately this technique is not amenable to pH values below 7.5 because of nonreactivity of the uridine at lower pH (21). Nevertheless, within the pH range that could be measured, the CMCT data are in agreement with that obtained from DMS modification experiments.
Inhibition of Peptidyl Transfer.
As an independent approach to chemical protection, we also measured the CCdApPmn inhibition constant (Ki) as a function of pH. CCdApPmn acts as an inhibitor of peptidyl transfer by competing with both A-site and P-site substrates. Under competitive conditions at pH 8.0, CCdApPmn was shown to have a Ki of 90 ± 4 nM, which correlated with the Kd of 70 ± 20 nM measured by chemical protection under similar conditions (11).
We determined the Ki of CCdApPmn as a function of pH by monitoring inhibition of the modified fragment assay on 50S subunits (18). Over a range of pH values from 5.0 to 8.0, the rates of peptidyl transfer were determined as a function of A-site substrate (CPmn) concentration for both inhibited (200 nM CCdApPmn) and uninhibited (no CCdApPmn) modified fragment assays. Fig. 3a shows the inhibitory effect that the addition of CCdApPmn has on peptidyl transfer, and Fig. 3b is a representative example of competitive inhibition by CCdApPmn at pH 7.0. The inhibition of peptidyl transfer in Fig. 3b corresponds to a Ki value of 170 ± 10 nM. We used peptidyl transfer inhibition to measure binding affinities from pH 5.0 to 8.0 and again found that the Ki values did not change with pH (Fig. 3c). The Ki values are relatively constant (125–175 nM) between pH 6.0 and 8.0. At pH values of 5.5 and 5.0 the Ki values increase ≈3-fold to 220 and 410 nM, respectively. This change in Ki at low pH is in the direction opposite to that predicted by the model. The measured Ki values are in reasonable agreement with Kd values measured by DMS protection (see above) as well as the previous Ki measurement at pH 8.0 (90 ± 4 nM) (11). The chemical protection (Kd) and peptidyl transfer inhibition (Ki) studies overlap in pH and together span the range from pH 5.0 to 8.5. Within this range we observe no trend toward increased CCdApPmn affinity for the ribosome at progressively acidic pH values.
Figure 3.

pH dependence of CCdApPmn binding based on inhibition of the modified fragment assay. (a) Peptidyl transfer fragment assay inhibition by CCdApPmn at pH 7.0. Reactions contain 62 μM CPmn in the presence (200 nM) or absence of CCdApPmn. Also shown is the extent of background hydrolysis of the radiolabeled substrate CCA-pcb in the absence of CPmn. (b) Lineweaver–Burke plot of inhibition of the modified fragment assay at pH 7.0 in the presence or absence of CCdApPmn inhibitor. The concentrations of CPmn were 20, 23, 26, 30, 36, 46, 62, 95, or 200 μM. The Ki value (125 ± 10 nM) was calculated by using Eq. 2. (c) The pH dependence of Ki for CCdApPmn. Each point represents an average of two Lineweaver–Burke plots with two sets of nine CPmn concentrations each. Standard deviations are indicated by error bars.
In light of observations of a pH-dependent conformational change in the ribosomal active site (15), we considered the possibility that an increased affinity between A2451 N3 and the CCdApPmn phosphoramidate O2 oxygen at low pH might be counterbalanced by a corresponding decrease in affinity of the CCdA and CPmn portions of the inhibitor. We found that the affinities of both substrates are essentially invariant from pH 5.5 to 8.0 (data not shown), which would seem to exclude this possibility. Thus, the hypothesis tested, namely that the N3 of A2451 hydrogen-bonds with a phosphoramidate oxygen of CCdApPmn in a pH-dependent manner, is not supported by the experimental data.
Discussion
Role of A2451 N3 as a Possible Oxyanion Hole.
These experiments set out to address a specific question with regard to the proposed ribosomal peptidyl transferase reaction mechanism: Does A2451 N3 act as the oxyanion hole to stabilize the transition state? To act in such a manner the pKa of A2451 N3, which normally is highly acidic (pKa ≤ 1), would have to be perturbed toward neutrality, and the interaction with the transition state would be highly pH-dependent. In contrast to this hypothesis, we find that the interaction between the ribosome and the transition-state analog, CCdApPmn, is pH-independent. Our results cast doubt on the thermodynamic and mechanistic significance of the proposed hydrogen-bonding interaction between the A2451 N3 and the phosphoramidate O2 modeled in the cocrystal structure.
Although our results do not rule out the possibility that the A2451 N3 pKa is perturbed to some degree, they put an upper limit on the magnitude of the perturbation. Assuming that the trend toward weaker binding is reversed at pH values below 5.0, our results argue that the pKa is no higher than ≈pH 4.5. Such a pKa would result in a very small fraction (<5%) of protonated N3 residues within the crystal structure at pH 5.8, which argues that the contact modeled in the crystal structure is not indicative of transition-state stabilization.
In order for the N3 of A2451 to have a near-neutral pKa of 7.3–7.7, as measured for the pKa of the peptidyl transferase reaction (2, 3), it would need to be perturbed dramatically. 15N NMR (22, 23) as well as laser Raman spectroscopy (24, 25) studies of adenosine nucleosides and nucleotides showed that N3 is not protonated even at pH 1.0, indicating that its pKa must be lower than 1.0. At 37°C, a shift from ≈1.0 to 7.3 would correspond to an energetic cost of as much as 9 kcal⋅mol−1. It is not evident from the structure how the ribosome could accomplish a perturbation of this energetic magnitude. A charge-relay mechanism involving a solvent-inaccessible phosphate and G2447 was proposed to be responsible for the A2451 N3 pKa perturbation, but G2447 mutations expected to disrupt such a charge relay have little effect on ribosome activity in vivo or in vitro (26).
An alternative hypothesis consistent with the crystal structure is that the phosphate O2 of CCdApPmn, and not A2451 N3, is protonated. Because the pKa of the phosphoramidate oxygen is ≈3.1 (27), such an interaction also would have resulted in affinity that increased inversely with pH. The binding studies seem to exclude this possibility as well. The data can be generalized to argue that no functional group with a near-neutral pKa in the ribosome makes a meaningful thermodynamic contribution to CCdApPmn binding.
Quality of CCdApPmn as a Mimic of the Ribosome Transition State?
CCdApPmn has been used to provide both structural evidence for and biochemical evidence against the proposal that A2451 N3 acts as an oxyanion hole. This evidence, both pro and con, is relevant only as far as CCdApPmn is a good mimic of the transition state. Although the phosphoramidate geometry of CCdApPmn is tetrahedral, and the bond length from phosphorus to a nonbridging oxygen (1.5 Å) is nearly the same as the bond length from carbon to oxygen (1.4 Å) (28), the charge of the phosphoramidate is delocalized between the two nonbridging oxygens. In contrast, the actual transition state contains a single charged oxyanion. The difference in charge distribution between the analog and the transition state may result in a crystal structure conformation about the phosphoramidate that is somewhat different from what occurs during peptidyl transfer.
Another important difference between the transition state and its phosphoramidate mimic is the absence of a 2′-OH on the terminal adenosine of CCdApPmn. Barta et al. suggested that the terminal dA of the transition-state analog would cause it to be a poor mimic of the peptidyl transfer transition state.¶ In the crystal structure, the deoxyadenosine C2′ of CCdApPmn is within 2.8 Å of the phosphoramidate O2. Were the 2′-OH present on the adenosine as it is in a P-site tRNA, it would cause significant steric clash with the phosphoramidate O2 as the complex is currently modeled. The interactions about the transition state during peptidyl transfer therefore are likely to be different from those modeled in the cocrystal structure.
In addition to subtle ways in which CCdApPmn may not mimic the tetrahedral transition state, mechanistic proposals based on its structure must contend also with stereochemical ambiguity. Peptidyl transfer by the ribosome proceeds through a chiral tetrahedral transition state, whereas the phosphoramidate of CCdApPmn is achiral. As such, it was necessary to assign either the phosphoramidate O1 or O2 as the oxyanion, whereas the other was designated as the α-carbon of the peptide chain (Fig. 1 a and b). Based on the geometry of the inhibitor in the 50S active site, Nissen et al. (10) assigned the oxyanion to be the O2. This assignment led to the proposed interaction between A2451 N3 and the oxyanion. Some consideration must be given to the possibility that the phosphoramidate O1 instead of O2 may correspond to the oxyanion. In this case, A2451 N3 would interact with the α-carbon CH, while the oxyanion would be exposed to solvent (the nearest residue is more than 5 Å away). It would follow from this stereochemical assignment that the ribosome does not explicitly stabilize the oxyanion of the transition state, a model that is consistent with our binding results. Hence, the close distance seen between A2451 N3 and the phosphoramidate O2 may in fact be an energetically unfavorable interaction that is overcome by binding of the analog to both the A site and P site.
Role of A2451 as Possible General Acid or Base.
The proposed role of A2451 as an oxyanion hole is part of a greater overall mechanistic proposal wherein A2451 plays a prominent role in peptide bond formation (10). In this model, the N3 of A2451 acts first as a general base to abstract a proton from the nucleophilic α-amino group or the protonated α-ammonium group‖ of the A site-bound aminoacyl-tRNA. The oxyanion of the tetrahedral transition state then is stabilized by the protonated N3 of A2451. Finally, the A2451 N3 acts as a general acid to transfer its proton to the 3′-oxyanion leaving group of the P-site tRNA, resulting in an amide linkage.
There has been considerable controversy over the role and importance of A2451. Experiments in which A2451 was mutated to another base showed only modest (2–14-fold) decreases in the rate of peptidyl transfer, implying that A2451 does not play a critical role (26, 29). However, these experiments used reconstituted ribosomes, which had very slow rates of peptidyl transfer, and chemistry was not shown to be the rate-limiting step. Therefore, large effects on the faster chemical step by the A2451 mutation may have gone unnoticed. Recent experiments, using a peptidyl transfer assay wherein chemistry is expected to be rate-limiting, show a pH dependence of peptide bond formation that indicates that there is an ionizable group within the ribosome with pKa of 7.4 (30). Mutation of A2451 to U results in a more than a 100-fold decrease in the rate of peptide bond formation coupled with loss of the ribosome's ionizable group. This result argues that A2451 is important for the chemical step of peptide bond formation, possibly through a general base mechanism, although other explanations involving protonation of neighboring residues are possible also (30).
The inhibitor binding data presented in this study do not address the question of whether A2451 or any other base serves as a general base in peptide bond formation. However, the results argue that the pKa measured for the peptidyl transferase reaction does not reflect the pKa of a residue serving as the oxyanion hole. Moreover, these data argue against any residue with a pKa of 5.0 or greater serving as the oxyanion hole, insofar as CCdApPmn is an accurate mimic of the transition state.
The co-crystal structure of CCdApPmn with the 50S subunit corresponds to a “snapshot” at an intermediate stage of peptidyl transfer. Chemical footprinting studies indicate a surprising amount of pH-dependent conformational flexibility at the active site of the ribosome [pKa ≈ 7.6 (15, 16)]. Although our results argue that this flexibility does not contribute to stabilization of the transition state, it is possible that interactions and conformations not seen in the current crystal structure play an important role in catalysis. Structural and biochemical experiments that characterize the ribosome at different stages of peptidyl transfer will be essential to untangle the peptidyl transfer mechanism.
Acknowledgments
We thank Yomi Oyelere and Greg Muth for helpful discussions and Lara Szewczak and Ashley Eversole for critical reading of the manuscript. This work was supported by American Cancer Society Research Scholar Grant to Beginning Investigators 02-052 (to S.A.S.).
Abbreviations
- CCdApPmn
CCdA-phosphate-puromycin
- DMS
dimethyl sulfate
- pcb
phenylalanyl-caproyl-biotin
- CPmn
C-puromycin
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Barta, A., Dorner, S. & Polacek, N. (2001) Science 291, 203 (abstr.).
Berg, J. M. & Lorsch, J. R. (2001) Science 291, 203 (abstr.).
References
- 1.Green R, Noller H F. Annu Rev Biochem. 1997;66:679–716. doi: 10.1146/annurev.biochem.66.1.679. [DOI] [PubMed] [Google Scholar]
- 2.Maden B, Monro R. Eur J Biochem. 1968;6:309–316. doi: 10.1111/j.1432-1033.1968.tb00450.x. [DOI] [PubMed] [Google Scholar]
- 3.Pestka S. Proc Natl Acad Sci USA. 1972;69:624–628. doi: 10.1073/pnas.69.3.624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Noller H F. Annu Rev Biochem. 1984;53:119–162. doi: 10.1146/annurev.bi.53.070184.001003. [DOI] [PubMed] [Google Scholar]
- 5.Samaha R R, Green R, Noller H F. Nature (London) 1995;377:309–314. doi: 10.1038/377309a0. [DOI] [PubMed] [Google Scholar]
- 6.Kim D F, Green R. Mol Cell. 1999;4:859–864. doi: 10.1016/s1097-2765(00)80395-0. [DOI] [PubMed] [Google Scholar]
- 7.Moazed D, Noller H F. Proc Natl Acad Sci USA. 1991;88:3725–3758. doi: 10.1073/pnas.88.9.3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Noller H F, Hoffarth V, Zimniak L. Science. 1992;256:1416–1419. doi: 10.1126/science.1604315. [DOI] [PubMed] [Google Scholar]
- 9.Ban N, Nissen P, Hansen J, Moore P B, Steitz T A. Science. 2000;289:905–920. doi: 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- 10.Nissen P, Hansen J, Ban N, Moore P B, Steitz T A. Science. 2000;289:920–930. doi: 10.1126/science.289.5481.920. [DOI] [PubMed] [Google Scholar]
- 11.Welch M, Chastang J, Yarus M. Biochemistry. 1995;34:385–390. doi: 10.1021/bi00002a001. [DOI] [PubMed] [Google Scholar]
- 12.Hegazi M F, Quinn D M, Schowen R L. In: Transition States of Biochemical Processes. Gandour R D, Schowen R L, editors. New York: Plenum; 1978. [Google Scholar]
- 13.Muth G W, Ortoleva-Donnelly L, Strobel S A. Science. 2000;289:947–950. doi: 10.1126/science.289.5481.947. [DOI] [PubMed] [Google Scholar]
- 14.Bayfield M A, Dahlberg A E, Schulmeister U, Dorner S, Barta A. Proc Natl Acad Sci USA. 2001;98:10096–10101. doi: 10.1073/pnas.171319598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muth G W, Chen L, Kosek A B, Strobel S A. RNA. 2001;7:1403–1415. [PMC free article] [PubMed] [Google Scholar]
- 16.Xiong L, Polacek N, Sander P, Bottger E C, Mankin A. RNA. 2001;7:1365–1369. [PMC free article] [PubMed] [Google Scholar]
- 17.Rheinberger H-J, Geigenmuller U, Wedde M, Nierhaus K H. Methods Enzymol. 1988;164:658–670. doi: 10.1016/s0076-6879(88)64076-6. [DOI] [PubMed] [Google Scholar]
- 18.Schmeing T M, Seila A C, Hansen J L, Freeborn B, Soukup J K, Scaringe S A, Strobel S A, Moore P B, Steitz T A. Nat Struct Biol. 2002;9:225–230. doi: 10.1038/nsb758. [DOI] [PubMed] [Google Scholar]
- 19.Moazed D, Noller H F. Cell. 1989;57:585–597. doi: 10.1016/0092-8674(89)90128-1. [DOI] [PubMed] [Google Scholar]
- 20.Welch M D. Studies of the Structure and Mechanism of Ribosomal Peptidyltransferase. Boulder: Univ. of Colorado; 1996. p. 148. [Google Scholar]
- 21.Naylor R, Gilham P T. Biochemistry. 1966;5:2722–2728. doi: 10.1021/bi00872a032. [DOI] [PubMed] [Google Scholar]
- 22.Gonnella N C, Nakanishi H, Holtwick J B, Horowitz D S, Kanamori K, Leonard N J, Roberts J D. J Am Chem Soc. 1983;105:2050–2055. [Google Scholar]
- 23.Markowski V, Sullivan G R, Roberts J D. J Am Chem Soc. 1977;99:714–717. doi: 10.1021/ja00445a009. [DOI] [PubMed] [Google Scholar]
- 24.O'Connor T, Johnson C, Scovell W M. Biochim Biophys Acta. 1976;447:495–508. doi: 10.1016/0005-2787(76)90086-1. [DOI] [PubMed] [Google Scholar]
- 25.Walker G A, Bhatia S C, Hall J H., Jr J Am Chem Soc. 1987;109:7634–7638. [Google Scholar]
- 26.Thompson J, Kim D F, O'Connor M, Lieberman K R, Bayfield M A, Gregory S T, Green R, Noller H F, Dahlberg A E. Proc Natl Acad Sci USA. 2001;98:9002–9007. doi: 10.1073/pnas.151257098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dean J A. In: Lange's Handbook of Chemistry. Dean J A, editor. New York: McGraw–Hill; 1985. [Google Scholar]
- 28. Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor, R. (1987) J. Chem. Soc. Perkin Trans. 2 S1–S9.
- 29.Polacek N, Gaynor M, Yassin A, Mankin A S. Nature (London) 2001;411:498–501. doi: 10.1038/35078113. [DOI] [PubMed] [Google Scholar]
- 30. Katunin, V. I., Muth, G. W., Strobel, S. A., Wintermeyer, W. & Rodnina, M. V. (2002) Mol. Cell, in press. [DOI] [PubMed]