

Abstract
NY-ESO-1 is a germ cell antigen aberrantly expressed in different tumor types that elicits strong humoral and cellular immune responses in cancer patients. Monitoring spontaneous CD8+ T cell responses against NY-ESO-1 peptides 157–165 (S9C) and 157–167 (S11L) in a series of HLA-A2+ cancer patients showed that these two peptides had overlapping antigenic profiles and were equally immunogenic. However, discrepancies between S9C and S11L reactivities were observed upon vaccination with both peptides to generate or boost T cell responses to NY-ESO-1 in cancer patients. We here analyze the fine specificity of these responses and describe an HLA-A2-restricted epitope, NY-ESO-1 peptide 159–167 (L9L), which is strongly recognized by CD8+ T cells as a result of peptide vaccination of cancer patients. Responses to L9L were stimulated by S11L and appeared early in the course of vaccination, independently of S9C responses. However, L9L-specific CD8+ T cells failed to recognize tumor cells naturally expressing NY-ESO-1 or B lymphoblastoid cells transduced with NY-ESO-1. Processing of L9L could be rescued after IFN-γ treatment of tumor cells or by dendritic cells pulsed with NY-ESO-1 protein/antibody immune complexes. The present results demonstrate a dual specificity within peptide S11L, with S9C as the natural antigenic tumor epitope, and L9L as a cryptic epitope with dominant immunogenicity upon vaccination that diverts the immune response from tumor recognition. These unanticipated findings raise questions about the use of S11L in the clinic and emphasize the importance of analyzing the fine specificity of vaccine-induced T cell responses in patients as a basis for constructing effective cancer vaccines.
The immune system is able to influence the development of cancer and shape the antigenic profile of emerging cancer cells (1). Vaccination approaches designed to harness this powerful response have flourished in recent years (2–4). With the ever-increasing number of tumor antigens being discovered by T cell epitope cloning (5) and SEREX (6), the field of cancer vaccine development has been placed on a firm foundation. Cancer/testis antigens, a category of gene products defined by their frequent expression in tumors and their restricted expression in normal tissues (germ cells), have emerged as near-ideal candidates for cancer-specific immunotherapies (7, 8). Among them, NY-ESO-1 (9) is of particular interest because of its capacity to elicit both humoral (10) and cellular (11) responses in a high frequency of cancer patients with NY-ESO-1 expression in their tumors.
In 1998, Jäger et al. (11) described three peptides from NY-ESO-1 recognized by CD8+ T cells from a mixed tumor/lymphocyte culture. The three peptides, 155–163 (Q9T), 157–165 (S9C), and 157–167 (S11L), have overlapping sequences and are restricted by the HLA-A2 molecule (Fig. 1). To determine whether these peptides were independent epitopes or not, a study was designed to assess the frequency of specific T cell responses to the three peptides in HLA-A2+ patients with advanced NY-ESO-1+ tumors and to correlate it with the presence of serum antibody to NY-ESO-1 (12). In a series of 36 patients, a substantial proportion had spontaneous and concomitant NY-ESO-1 antibody and CD8+ T cell reactivity against NY-ESO-1 peptides. It appeared that peptide Q9T was not frequently immunogenic, that S9C and S11L had overlapping reactivities, and that in vitro stimulation with either S9C or S11L led to cross-reactive responses.
Figure 1.
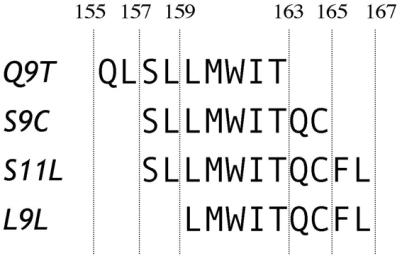
NY-ESO-1 peptides used in this study.
A clinical trial was designed to study the immunogenicity of NY-ESO-1 peptides S9C and S11L along with granulocyte–macrophage colony-stimulating factor as an adjuvant in patients with NY-ESO-1+ tumors (4). Peptide immunization was effective, as NY-ESO-1-specific CD8+ T cell responses to S11L and S9C could be detected in most vaccinated patients without preexisting immunity to NY-ESO-1.
Upon detailed analysis of T cell activity against the immunizing peptides, it appeared that T cell reactivity against S11L appeared earlier in the course of vaccination than against S9C, despite simultaneous administration of the peptides and previous findings of cross-reactivity.
We analyze here the fine specificity of these T cell responses and describe an epitope from NY-ESO-1, 159–167 (L9L), presented by HLA-A2, that appears as a result of vaccination.
Materials and Methods
Patients.
Patients analyzed in this study are described in Table 1. Informed consent was obtained for enrollment in three Institutional Review Board-approved immunization protocols sponsored by the Ludwig Institute for Cancer Research: LUD97–008, which has been described (4), LUD00–019, and LUD97–012. For LUD00–019 and LUD97–008, briefly, HLA-A*0201+ patients were immunized each week intradermally with 100 μg of peptide S9C and S11L in PBS 33% DMSO, and SC injections of granulocyte–macrophage colony-stimulating factor at 75 μg/day were given weekly as an adjuvant for 2 days (LUD00–019) or 6 days (LUD97–008). For LUD97–012, patients received two consecutive weekly intradermal injections of 33 μg of S9C and S11L peptides spaced by 4 weeks, and 20 μg/kg Flt3 ligand for 14 days every month as an adjuvant.
Table 1.
Patient characteristics
| Patient code | Tumor type | NY-ESO-1 mRNA tumor expression* | NY-ESO-1 antibody status* | Preexisting CD8+ T cell responses to NY-ESO-1 peptides | Vaccination protocol with NY-ESO-1 peptides |
|---|---|---|---|---|---|
| NW415 | Melanoma | Positive | Negative | No | LUD97-008 |
| NW886 | Melanoma | Positive | Negative | No | LUD97-008 |
| NW924 | Melanoma | Positive | Positive | Yes | LUD97-008 |
| NW1288 | Gastric | Positive | Positive | Yes | None |
| NW1307 | Schwannoma | Positive | Positive | Yes | None |
| 109/ANS | Melanoma | Positive | Negative | No | LUD97-012 |
| PI-E01 | NSCLC | Positive | Negative | No | LUD00-019 |
| ES-E03 | NSCLC | Positive | Negative | No | LUD00-019 |
NSCLC, non small cell lung cancer.
NY-ESO-1 mRNA tumor expression and serum antibody detected as described (10).
Peptides and Viral Vectors.
Synthetic peptides 157–165 (SLLMWITQC, S9C), 157–167 (SLLMWITQCFL, S11L), and 159–167 (LLMWITQCFL, L9L) were obtained from Multiple Peptide Systems (San Diego) with a purity of >93% as determined by reverse-phase HPLC. Adenovirus recombinant for full-length NY-ESO-1 (Ad/ESO), wild-type vaccinia virus (v.v. WT), and vaccinia virus recombinant for full-length NY-ESO-1 (v.v. ESO) have been described (13).
In Vitro Sensitization with Peptides or Adenoviral Constructs.
CD8+ T lymphocytes were separated from peripheral blood lymphocytes (PBLs) of cancer patients by antibody-coated magnetic beads (Dynabeads; Dynal, Oslo) and seeded into round-bottomed 96-well plates (Corning) at a concentration of 5 × 105 cells per well in RPMI medium 1640 supplemented with 10% human AB serum (NABI, Boca Raton, FL), 2 mM l-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin, and 1% nonessential amino acids. As antigen-presenting cells (APCs), PBLs depleted of CD8+ T cells were either pulsed with 10 μM peptide or infected with Ad/ESO at 1,000 units/cell, overnight at 37°C in 250 μl serum-free medium (X-VIVO-15, Bio-Whittaker). Pulsed or infected APCs were then washed, irradiated, and added to the plates containing CD8+ T cells, at a concentration of 1 × 106 APCs per well. After 8 h, IL-2 (10 units/ml, Roche Molecular Biochemicals) and IL-7 (20 ng/ml, R&D Systems) were added to culture wells, and this step was repeated every 3–4 days, until the cells were harvested for testing.
Tetramer Synthesis and Assay.
HLA-A2 tetrameric complexes were synthesized as described (12, 14). Tetramers were assembled with NY-ESO-1-derived peptides S9C (157–165), S11L (157–167), or L9L (159–167). Presensitized CD8+ T cells in 50 μl PBS containing 3% FCS (Summit Biotechnology, Fort Collins, CO) were stained with phycoerythrin-labeled tetramer for 15 min at 37°C before addition of Tricolor-CD8 mAb (Caltag, South San Francisco) and anti-CD62L mAb (Caltag) for 15 min on ice. After washing, results were analyzed by flow cytometry (FACSCalibur; Becton Dickinson).
Target Cells.
All cells used in this study expressed the HLA-A*0201 molecule. NY-ESO-1-expressing melanoma cell lines SK-MEL-37, MZ-MEL-19, and NW-MEL-38, Epstein–Barr virus (EBV)-transformed B lymphocytes, and the mutant cell line T2 (CEMx721.174.T2) all were cultured in RPMI medium 1640 supplemented with 10% FCS (Summit Biotechnology), 2 mM l-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin, and 1% nonessential amino acids.
For assays with T2 or EBV-B cells, 5 × 105 cells were either pulsed with 10 μM peptide or infected at 30 plaque-forming units/cell with wild-type vaccinia virus (v.v. WT) or vaccinia virus recombinant for NY-ESO-1 (v.v. ESO), in 250 μl serum-free medium overnight (X-VIVO-15). To generate dendritic cells (DCs), CD14+ monocytes were enriched by negative selection with magnetic beads (Dynal) and then incubated in RPMI with 10% FCS, 1,000 units/ml granulocyte–macrophage colony-stimulating factor (PharMingen) and 1,000 units/ml of IL-4 (PharMingen). On day 6, immature DCs were harvested and used for antigen presentation assays.
Immune Complex (IC) Formation and Loading.
NY-ESO-1, MAGE-A1, and MAGE-A3 proteins were expressed in Escherichia coli as histidine-tagged full-length proteins and purified as described (10). The purified proteins were specifically reactive with the corresponding mAbs ES121 (anti-NY-ESO-1 IgG1), MA454 (anti-MAGE-A1 IgG1), and M3H67 (anti-MAGE-A3 IgG1), generated from mice immunized with recombinant proteins (10). Protein (5 μg/ml) and mAb were mixed at a 4:1 molar ratio in serum-free RPMI and incubated at 37°C for 30 min to form ICs, and then added to 1.5 × 105 DCs in RPMI supplemented with 10% FCS, 1,000 units/ml granulocyte–macrophage colony-stimulating factor, and 1,000 units/ml IL-4 for 7 h at 37°C (15).
ELISPOT and CYTOSPOT Assays.
For ELISPOT assays, flat-bottomed, 96-well nitrocellulose plates (MultiScreen-HA; Millipore) were coated with IFN-γ mAb (2 μg/ml, 1-D1K; MABTECH, Stockholm) and incubated overnight at 4°C. After washing with RPMI, plates were blocked with 10% human AB-type serum for 2 h at 37°C. Presensitized CD8+ T cells (5 × 104 and 1 × 104) and 5 × 104 targets cells (peptide-pulsed or v.v. ESO-infected EBV-B, or tumor cells) were added to each well and incubated for 20 h in RPMI medium 1640 without serum. Plates were then washed thoroughly with water containing 0.05% Tween 20 to remove cells, and IFN-γ mAb (0.2 μg/ml, 7-B6–1-biotin; MABTECH) was added to each well. After incubation for 2 h at 37°C, plates were washed and developed with streptavidin-alkaline phosphatase (1 μg/ml; MABTECH) for 1 h at room temperature. After washing, substrate (5-bromo-4-chloro-3-indolyl phosphate/nitroblue tetrazolium; Sigma) was added and incubated for 5 min. After final washes, plate membranes displayed dark-violet spots that were counted under the microscope.
For CYTOSPOT assay (intracellular staining of cytokines), presensitized CD8+ T cells were incubated with target cells at a 1:2 ratio in 300 μl serum-free medium (X-VIVO-15) in the presence of brefeldin A (10 μg/ml; Sigma) for 4.5 h. Cells were fixed, permeabilized (Becton Dickinson), and stained with Tricolor-labeled CD8 mAb (Caltag) and FITC-labeled IFN-γ mAb (PharMingen) at room temperature for 15 min. Results were analyzed by flow cytometry by gating on CD8+ lymphocytes.
Results
Spontaneous T Cell Responses to NY-ESO-1 Peptides.
PBLs from patient NW1307 with preexisting immunity to NY-ESO-1 were presensitized in vitro with peptides S9C or S11L (peptides described in Fig. 1). Peptides S9C and S11L were equally recognized by CD8+ T cells after stimulation with either S9C or S11L, indicating that the two peptides either cross-reacted or shared the same epitope, both as immunogen and antigen (Fig. 2A). The simultaneous S9C and S11L response pattern was characteristic of previous monitoring studies with NY-ESO-1 peptides in nonvaccinated HLA-A2 patients (12).
Figure 2.
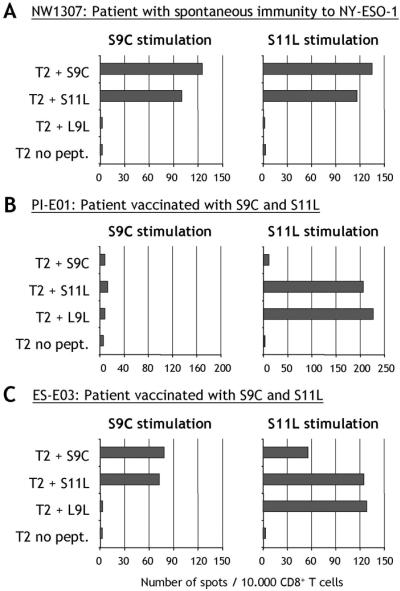
ELISPOT assay measuring IFN-γ secretion by CD8+ T cells in response to NY-ESO-1 peptides in a patient with spontaneous immunity to NY-ESO-1 (A) or in patients vaccinated for 8 weeks with NY-ESO-1 peptides S9C and S11L (B and C). CD8+ T lymphocytes were presensitized in vitro with either S9C (Left) or S11L (Right) and assayed on day 10 against T2 targets pulsed with NY-ESO-1 peptides.
T Cell Responses After Vaccination with NY-ESO-1 Peptides S9C and S11L.
In contrast with these initial observations, discrepancies were found upon monitoring T cell responses of cancer patients immunized with peptides 157–165 (S9C) and 157–167 (S11L), within the context of three independent clinical trials. As a consequence of vaccination, patients with no detectable preexisting immunity to NY-ESO-1 often developed T cell reactivity against S11L in the absence of S9C responses (example for patient PI-E01 in Fig. 2B, cf. ref. 4). In most patients, S9C responses, if any, appeared later than S11L responses during the course of vaccination (4). Because both S9C and S11L were used for immunization, this discrepancy showed that S11L had the capacity to stimulate a specific response, distinct from S9C.
To define this response, peptide 159–165, L9L, was synthesized as a second nonamer peptide included in the sequence of S11L (Fig. 1). Fig. 2B shows that patient PI-E01, who responded to S11L but not S9C stimulation, could recognize L9L by ELISPOT after S11L stimulation. Similarly, patient ES-E03 also showed reactivity to the L9L peptide after stimulation with S11L, but not stimulation with S9C (Fig. 2C). Thus, it appeared S11L and L9L had a similar antigenic profile in these vaccinated patients.
To analyze this L9L response further, PBLs from several patients were presensitized and expanded in vitro with peptides S9C, S11L, and L9L separately, or with naturally processed NY-ESO-1 expressed from recombinant adenovirus (Ad/ESO). Peptide-specific T cells were stained with tetramers of S9C, S11L, or L9L peptides in complex with HLA-A2 recombinant molecules.
Patient NW1288 was representative of nonvaccinated patients with preexisting immunity. CD8+ T cells from this patient showed staining with only the S9C tetramer, after stimulation with S9C, S11L, or Ad/ESO, but not with L9L (Table 2). In this natural setting, L9L reactivity was not seen.
Table 2.
Tetramer staining of patient CD8+ T cells presensitized with NY-ESO-1 peptides
|
In vitro stimulation of T cells with:
|
||||
|---|---|---|---|---|
| S9C | S11L | L9L | Adeno/ESO | |
| Patient NW1288: Preexisting immunity to NY-ESO-1—Not vaccinated | ||||
| S9C-tetramer | 1.48% | 1.58% | 0.04% | 1.01% |
| S11L-tetramer | 0.10% | 0.10% | 0.02% | 0.02% |
| L9L-tetramer | 0.03% | 0.17% | 0.04% | 0.03% |
| Patient NW886: No preexisting immunity to NY-ESO-1—Vaccinated with S9C and S11L | ||||
| S9C-tetramer | 0.04% | 0.06% | ND | 0.13% |
| S11L-tetramer | 0.02% | 0.03% | ND | 0.06% |
| L9L-tetramer | 0.05% | 3.98% | ND | 0.06% |
| Patient NW924: Preexisting immunity to NY-ESO-1—Vaccinated with S9C and S11L | ||||
| S9C-tetramer | 9.72% | 5.88% | ND | 0.43% |
| S11L-tetramer | 0.09% | 0.13% | ND | 0.02% |
| L9L-tetramer | 0.16% | 5.59% | ND | 0.06% |
CD8+ T lymphocytes were presensitized in vitro with S9C, S11L, L9L, or adenovirus recombinant for NY-ESO-1 (Adeno/ESO) and assayed on day 8 (patient NW1288), day 26 (patient NW886), or day 11 (patient NW924). ND, not determined. Bold numbers represent detectable tetramer-positive populations.
Conversely, patient NW886, who had no preexisting immunity to NY-ESO-1 and was vaccinated with peptides S9C and S11L, failed to respond to S9C. Rather, CD8+ T cells from this patient showed staining with only the L9L tetramer (Table 2). There was no reactivity to S9C despite vaccination with this peptide and no staining with S11L-tetramers despite S11L reactivity by ELISPOT (not shown). Cells stained with L9L tetramers were obtained after S11L stimulation, but not after S9C or Ad/ESO stimulation (Table 2).
Similarly, for patient NW924, who had preexisting immunity to NY-ESO-1 and received peptide vaccination, L9L tetramers were also detected in response to S11L stimulation, alongside existing S9C reactivity (Table 2). Stimulation with naturally processed NY-ESO-1 from recombinant adenovirus was not able to recall L9L-specific responses (Table 2).
The time course for the development of L9L reactivity in patient PI-E01 after vaccination with S9C and S11L is shown in Table 3. Reactivity to L9L was not seen before vaccination, but appeared during the course of vaccination. Tetramer results correlated with ELISPOT assays (Table 3).
Table 3.
The response to NY-ESO-1 peptide L9L appears as a result of vaccination with S11L
| Study week | Stimulated with S9C
|
Stimulated with S11L
|
||
|---|---|---|---|---|
| S9C-Tetramer (% CD8+ cells) | ELISPOT T2+S9C (spots/50,000 cells) | L9L-Tetramer (% CD8+ cells) | ELISPOT T2+L9L (spots/50,000 cells) | |
| Prestudy | 0.01% | 6 (2) | 0.02% | 5 (8) |
| Week 2 | 0.00% | 3 (1) | 0.07% | 7 (4) |
| Week 4 | 0.03% | 2 (1) | 0.21% | 42 (35) |
| Week 6 | 0.01% | 9 (11) | 0.44% | 126 (9) |
| Week 8 | 0.03% | 1 (8) | 2.97% | 289 (15) |
CD8+ T cells from patient PI-E01 were presensitized with S9C or S11L and tested at day 10 with tetramers made with S9C or L9L, and by ELISPOT assay with T2 cells pulsed with S9C or L9L (spots for T2 cells without peptide are shown in parentheses). Bold numbers represent detectable increases. No response to S9C was seen in this patient.
A characteristic peptide titration assay with CD8+ T cells from vaccinated patients is shown in Fig. 3. After presensitization with L9L, T cells from patient 109/ANS recognized L9L at peptide concentrations down to 1 nM. Peptide S11L recognition also occurred but at lower avidity (Fig. 3).
Figure 3.
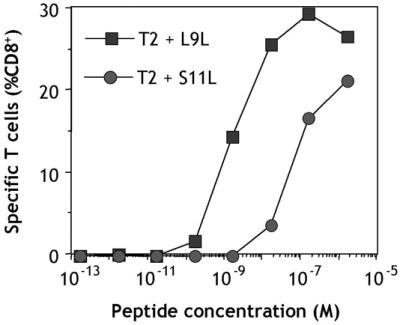
Peptide titration assay with CD8+ T cells from patient 109/ANS immunized with NY-ESO-1 peptides S9C and S11L. PBLs were obtained 85 days after initiation of vaccination. CD8+ T cells were presensitized with L9L and tested on day 11 by CYTOSPOT assay measuring IFN-γ production against T2 cells pulsed with different concentrations of L9L or S11L.
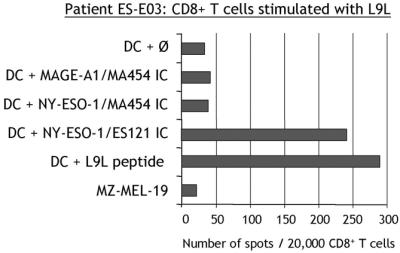
To determine the cross-stimulation and cross-recognition capacity of NY-ESO-1 peptides, a patient with no preexisting immunity to NY-ESO-1, but responding to both S9C and S11L after S9C and S11L vaccination, was analyzed. Peptides S9C, S11L, or L9L were used to presensitize CD8+ T cells from patient ES-E03. All peptide-stimulated T cells could recognize S11L as a target antigen in ELISPOT assays, whereas S9C and L9L reactivities were mutually exclusive (Fig. 4).
Figure 4.
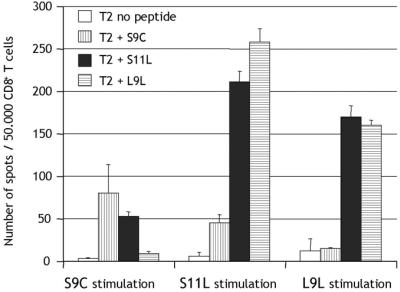
Analysis of cross-stimulation and cross-recognition of NY-ESO-1 peptides S9C, S11L, and L9L. ELISPOT with CD8+ T cells from vaccinated patient ES-E03 presensitized with the different NY-ESO-1 peptides and tested at day 9 against T2 cells pulsed with S9C (vertical stripes), S11L (solid), L9L (horizontal stripes), or without peptide (open).
Patient NW415, who also had no preexisting immunity to NY-ESO-1 and was vaccinated with S9C and S11L, demonstrated a wide range of tetramer staining, especially with the S11L tetramer that was otherwise rarely observed in vaccinated patients (Fig. 5). This pattern indicated that S11L did not require trimming by cellular peptidases to be recognized by CD8+ T cells. Interestingly, costaining with T cell activation marker CD62L (16) indicated that cells stimulated with S11L had three distinct patterns as detected by the three different tetramers (Fig. 5 Center). S9C-specific cells appeared CD62L+ whereas S11L- and L9L-specific cells had respectively intermediate and low CD62L profiles. This finding indicated that a single peptide, S11L, was capable of stimulating up to three distinct T cell populations with different specificities within the same culture, as detected by their various activation stages.
Figure 5.

Pattern of NY-ESO-1-tetramer staining of CD8+ T cells in a vaccinated patient with preexisting spontaneous NY-ESO-1 immunity. CD8+ T cells from patient NW415 were presensitized with S9L (Left), S11L (Center), or L9L (Right) and stained at day 9 with tetrameric complexes of HLA-A2 molecules and S9C (Top), S11L (Middle), or L9L (Bottom) peptide, along with anti-CD62L. Lymphocytes gated for CD8+ expression are shown.
Recognition of Naturally Processed NY-ESO-1 Epitopes.
CD8+ T cells from patients with spontaneous immunity to NY-ESO-1 were able to recognize tumor cells expressing NY-ESO-1, as well as NY-ESO-1 transduced in lymphoblastoid B cells (EBV-B) with recombinant vaccinia virus. Fig. 6A shows this reactivity pattern for NW924 CD8+ T cells stimulated with S9C.
Figure 6.
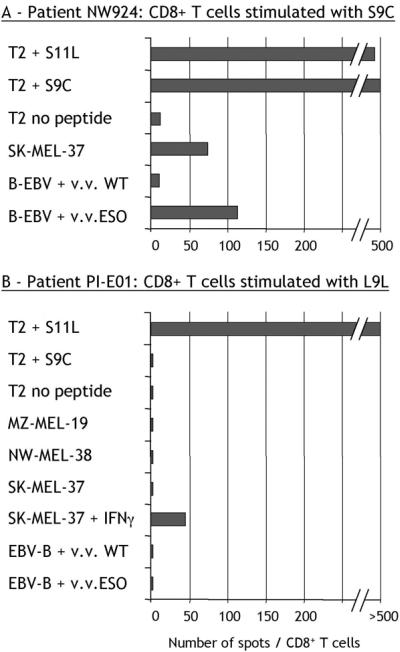
Recognition of naturally processed NY-ESO-1 by L9L-specific CD8+ T cells. ELISPOT assays with CD8+ T cells from NW924, a patient with spontaneous NY-ESO-1 immunity, presensitized with S9C (number of spots/50,000 cells) (A), and CD8+ T cells from PI-E01, a patient with no preexisting NY-ESO-1 immunity vaccinated with S9C and S11L, presensitized with L9L (number of spots/10,000 cells) (B). v.v. WT, wild-type vaccinia virus; v.v. ESO, vaccinia virus recombinant for full-length NY-ESO-1.
In the absence of S9C reactivity, however, this pattern was not seen. CD8+ T cells from patient PI-E01 specific for L9L were incapable of recognizing EBV-B targets infected with vaccinia virus recombinant for NY-ESO-1 (Fig. 6B). Similarly, L9L-specific cells did not produce IFN-γ in response to SK-MEL-37, NW-MEL-38, or MZ-MEL-19 (Fig. 6B), despite NY-ESO-1 and HLA-A2 expression by all of these tumor cell lines, suggesting that L9L is not presented by these melanoma cells.
However, after treatment of SK-MEL-37 with IFN-γ, L9L-specific responses were triggered at a low level (Fig. 6B). Similar observations were made with NW-MEL-38 treated with IFN-γ (not shown). To determine whether IFN-γ treatment was acting by changing the pattern of processing of NY-ESO-1 (17), DCs were used as a source of professional APCs (18). DCs have the capacity to process and present antigens from an exogenous source when given in the form of ICs of protein bound to a specific antibody (15, 19). The full-length NY-ESO-1 protein was incubated with NY-ESO-1-specific mAb ES121 and pulsed on DCs from a healthy HLA-A*0201 donor. Under these conditions, L9L-specific T cells could recognize DCs fed with NY-ESO-1 ICs at levels comparable to DCs pulsed with exogenous L9L peptide (Fig. 7). There was no specific recognition by L9L-specific CD8+ T cells of control ICs formed with MAGE-A1 protein and specific MA454 antibody, or with NY-ESO-1 protein and MAGE-A1-specific antibody MA454 (Fig. 7).
Figure 7.
Recognition of DCs pulsed with NY-ESO-1 ICs by L9L-specific CD8+ T cells. ELISPOT assay with CD8+ T cells from patient ES-E03 presensitized with L9L and tested at day 12 against DCs cultured from HLA-A*0201+ healthy donor NC171.
Together, these data suggest that L9L peptide may be generated by the processing of NY-ESO-1 ICs by DCs, but not constitutively by other APCs, i.e., tumor cells expressing NY-ESO-1 or B lymphoblastoid cells after transfection with NY-ESO-1-coding viral vectors.
Discussion
In this study, we describe T cell recognition in the context of HLA-A*0201 of three overlapping epitopes included within region 157–167 of NY-ESO-1: 157–165 (S9C), 157–167 (S11L), and 159–167 (L9L).
Peptide S11L is recognized by both S9C- and L9L-specific CD8+ T cells in vitro. Cross-reactivity, epitope sharing, or partial peptide degradation may explain this dual antigenicity, because S11L includes both epitopes. As an immunogen, S11L is able to restimulate both S9C- and L9L-specific CD8+ T cells in vitro. Conversely, S9C and L9L reactivities are mutually exclusive, CD8+ T cells recognizing these peptides do not cross-react (Figs. 4 and 5, Table 4).
Table 4.
Summary of cross-stimulation and cross-recognition properties of NY-ESO-1 peptides
| Peptide used as target in T cell assay | Peptide used for in vitro stimulation of T cells
|
||
|---|---|---|---|
| S9C | S11L | L9L | |
| Patients with natural immunity to NY-ESO-1 | |||
| S9C | + | + | − |
| S11L | + | + | − |
| L9L | − | − | − |
| Vaccinated patients responding only to peptide S11L (no preexisting immunity) | |||
| S9C | − | − | − |
| S11L | − | + | + |
| L9L | − | + | + |
| Vaccinated patients responding to both peptides S9C and S11L | |||
| S9C | + | + | − |
| S11L | + | + | + |
| L9L | − | + | + |
Thus far, in the monitoring of HLA-A2 patients for NY-ESO-1 reactivity, peptides S9C and S11L were used to recall and amplify spontaneous responses after a single in vitro stimulation. Patients with natural immunity to NY-ESO-1 are responsive against S9C, and S11L is capable of recalling S9C-specific cells in vitro. Responses specific for S11L are not seen alone in the absence of S9C reactivity. In this natural setting, S11L fails to recall L9L responses (Tables 2 and 4, Fig. 2).
Reactivity against peptide L9L develops as a result of vaccination (Table 3). Patients with NY-ESO-1 immune responses do not appear to respond to L9L spontaneously. As early as 4 weeks after vaccination with peptide S11L, reactivity develops against both S11L and L9L, as detected after a single stimulation in vitro. Peptides S11L and L9L have overlapping specificities, e.g., either cross-react or share the same epitope, although L9L is recognized at greater affinities by T cells compared with the parental peptide S11L used for immunization. Thus, in the absence of spontaneous immunity to NY-ESO-1, it appears that S11L preferentially induces L9L-specific T cells, rather than S9C-specific T cells. In other words, patients that do not have a spontaneous T cell response to S9C will instead be more likely to develop L9L reactivity as a result of vaccination with S11L. Strong S11L and L9L reactivities emerge earlier than S9C reactivity in patients vaccinated with both S9C and S11L. Even in patients with natural S9C reactivity, L9L-specific responses appear as a result of vaccination with S11L.
Within the NY-ESO-1 sequence, peptide L9L has the strongest binding capacity to HLA-A2 as indicated by predictive algorithms (20). Affinity and stability of peptides to HLA molecules have been shown to correlate with their intrinsic immunogenicity (21, 22). This finding could explain the preferential immunogenicity of L9L after S11L immunization.
However, prediction software for constitutive proteasome cutting does not suggest a cleavage signal after residue 167, indicating that S11L and L9L should not be produced from NY-ESO-1 protein. In contrast, residue 165 is recognized as a cleavage site, which would result in the S9C peptide production (23). Thus, the absence of L9L-specific T cell recognition of NY-ESO-1, either from tumor cells or recombinant viral vectors, could be explained on the basis of the natural processing of NY-ESO-1 in these cells. Even though T cells against L9L appear fully activated and have high affinity, they do not recognize NY-ESO-1-expressing tumor cells or B cells expressing the full-length NY-ESO-1 gene.
We have, however, found that L9L-specific T cells can recognize NY-ESO-1 presented by DCs after processing of NY-ESO-1 ICs, or by tumor cells after IFN-γ treatment. A common feature of these APCs is the up-regulation of proteasome-binding subunits affecting the processing of antigens, either after maturation of DCs with IC (15, 18, 19) or by IFN-γ treatment (17). Peptide L9L could thus be a new candidate in the category of epitopes resulting from the action of the immunoproteasome, as recently described for a peptide processed from MAGE-A3 (24). Further studies need to be carried out with purified immunoproteasome or transfected cell models to confirm this hypothesis.
In conclusion, synthetic peptide S11L used in vaccination provides a strong anchor residue in the last position for binding to HLA-A2, which may preferentially recruit a T cell repertoire for L9L rather than S9C as the best-fitted epitope within S11L. Because this epitope is not naturally processed/presented by NY-ESO-1-expressing tumor cells, use of S11L in vaccine strategies needs to be questioned. However, under certain circumstances, some tumors may express immunoproteasome rather than the constitutive proteasome if IFN-γ is present in the local environment (25), and could thus become a target for L9L-specific T cells. Another possible derivative effect of S11L is that it may offer a helper effect during vaccination, because its sequence falls within NY-ESO-1 region 157–170 recognized by CD4+ T cells in the context of HLA-DP4 (26). Although shorter than classical class II restricted peptides, S11L represents a strong immunogenic region that could contribute to a favorable environment for the development of tumor-specific T cells.
This study reveals an unexpected complexity of CD8+ T cell responses to a single NY-ESO-1 peptide, S11L. This peptide contains a cryptic epitope with dominant immunogenicity upon vaccination, which unlike subdominant epitopes described in mice tumor models (27), may skew the immune response away from tumor recognition (28). This finding would not have been evident from monitoring the spontaneous immune reactions to NY-ESO-1, and it emphasizes the importance of analyzing vaccinated patients to uncover the full repertoire of peptide epitopes recognized by T cells.
Abbreviations
- PBL
peripheral blood lymphocyte
- EBV
Epstein–Barr virus
- DC
dendritic cell
- IC
immune complex
- APC
antigen-presenting cell
References
- 1.Shankaran V, Ikeda H, Bruce A T, White J M, Swanson P E, Old L J, Schreiber R D. Nature (London) 2001;410:1107–1111. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- 2.Marchand M, van Baren N, Weynants P, Brichard V, Dreno B, Tessier M H, Rankin E, Parmiani G, Arienti F, Humblet Y, et al. Int J Cancer. 1999;80:219–230. doi: 10.1002/(sici)1097-0215(19990118)80:2<219::aid-ijc10>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 3.Rosenberg S A, Yang J C, Schwartzentruber D J, Hwu P, Marincola F M, Topalian S L, Restifo N P, Dudley M E, Schwarz S L, Spiess P J, et al. Nat Med. 1998;4:321–327. doi: 10.1038/nm0398-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jäger E, Gnjatic S, Nagata Y, Stockert E, Jäger D, Karbach J, Neumann A, Rieckenberg J, Chen Y-T, Ritter G, et al. Proc Natl Acad Sci USA. 2000;97:12198–12203. doi: 10.1073/pnas.220413497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boon T, Old L J. Curr Opin Immunol. 1997;9:681–683. doi: 10.1016/s0952-7915(97)80049-0. [DOI] [PubMed] [Google Scholar]
- 6.Pfreundschuh M. Cancer Chemother Pharmacol. 2000;46:S3–S7. doi: 10.1007/pl00014046. [DOI] [PubMed] [Google Scholar]
- 7.van der Bruggen P, Traverseri C, Chomez P, Lurquin C, De Plaen E, Van den Eynde B, Knuth A, Boon T. Science. 1991;254:1643–1650. doi: 10.1126/science.1840703. [DOI] [PubMed] [Google Scholar]
- 8.Chen Y T, Güre A O, Tsang S, Stockert E, Jäger E, Knuth A, Old L J. Proc Natl Acad Sci USA. 1998;95:6919–6923. doi: 10.1073/pnas.95.12.6919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Y-T, Scanlan M J, Sahin U, Türeci Ö, Güre A O, Tsang S, Williamson B, Stockert E, Pfreundschuh M, Old L J. Proc Natl Acad Sci USA. 1997;94:1914–1918. doi: 10.1073/pnas.94.5.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stockert E, Jäger E, Chen Y-T, Scanlan M J, Gout I, Karbach J, Arand M, Knuth A, Old L J. J Exp Med. 1998;187:1349–1354. doi: 10.1084/jem.187.8.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jäger E, Chen Y-T, Drijfhout J W, Karbach J, Ringhoffer M, Jäger D, Arand M, Wada H, Noguchi Y, Stockert E, et al. J Exp Med. 1998;187:265–270. doi: 10.1084/jem.187.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jäger E, Nagata Y, Gnjatic S, Wada H, Stockert E, Karbach J, Dunbar P R, Lee S Y, Jungbluth A, Jäger D, et al. Proc Natl Acad Sci USA. 2000;97:4760–4765. doi: 10.1073/pnas.97.9.4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gnjatic S, Nagata Y, Jäger E, Stockert E, Shankara S, Roberts B L, Mazzara G P, Lee S Y, Dunbar P R, Dupont B, et al. Proc Natl Acad Sci USA. 2000;97:10917–10922. doi: 10.1073/pnas.97.20.10917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Altman J D, Moss P A H, Goulder P J R, Barouch D H, McHeyzer-Williams M G, Bell J I, McMichael A J, Davis M M. Science. 1996;274:94–96. [PubMed] [Google Scholar]
- 15.Nagata Y, Ono S, Matsuo M, Gnjatic S, Valmori D, Ritter G, Garrett W, Old L J, Mellman I. Proc Natl Acad Sci USA. 2002;99:10629–10634. doi: 10.1073/pnas.112331099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stamenkovic I. Curr Opin Hematol. 1995;2:68–75. doi: 10.1097/00062752-199502010-00010. [DOI] [PubMed] [Google Scholar]
- 17.Gaszynska M, Rock K L, Goldberg A L. Nature (London) 1993;365:264–267. doi: 10.1038/365264a0. [DOI] [PubMed] [Google Scholar]
- 18.Morel S, Levy F, Burlet-Schiltz O, Brasseur F, Probst-Kepper M, Peitrequin A L, Monsarrat B, Van Velthoven R, Cerottini J C, Boon T, et al. Immunity. 2000;12:107–117. doi: 10.1016/s1074-7613(00)80163-6. [DOI] [PubMed] [Google Scholar]
- 19.Regnault A, Lankar D, Lacabanne V, Rodriguez A, Thery C, Rescigno M, Saito T, Verbeek S, Bonnerot C, Ricciardi-Castagnoli P, Amigorena S. J Exp Med. 1999;189:371–380. doi: 10.1084/jem.189.2.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parker K C, Bednarek M A, Coligan J E. J Immunol. 1994;152:163–175. [PubMed] [Google Scholar]
- 21.Keogh E, Fikes J, Southwood S, Celis E, Chesnut R, Sette A. J Immunol. 2001;167:787–796. doi: 10.4049/jimmunol.167.2.787. [DOI] [PubMed] [Google Scholar]
- 22.van der Burg S H, Visseren M J, Brandt R M, Kast W M, Melief C J. J Immunol. 1996;156:3308–3314. [PubMed] [Google Scholar]
- 23.Nussbaum A K, Kuttler C, Hadeler K P, Rammensee H G, Schild H. Immunogenetics. 2001;53:87–94. doi: 10.1007/s002510100300. [DOI] [PubMed] [Google Scholar]
- 24.Schultz E S, Chapiro J, Lurquin C, Claverol S, Burlet-Schiltz O, Warnier G, Russo V, Morel S, Levy F, Boon T, et al. J Exp Med. 2002;195:391–399. doi: 10.1084/jem.20011974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pisa P, Halapi E, Pisa E K, Gerdin E, Hising C, Bucht A, Gerdin B, Kiessling R. Proc Natl Acad Sci USA. 1992;89:7708–7712. doi: 10.1073/pnas.89.16.7708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zeng G, Wang X, Robbins P F, Rosenberg S A, Wang R F. Proc Natl Acad Sci USA. 2001;98:3964–3969. doi: 10.1073/pnas.061507398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Newmaster R S, Mylin L M, Fu T M, Tevethia S S. Virology. 1998;244:427–441. doi: 10.1006/viro.1998.9148. [DOI] [PubMed] [Google Scholar]
- 28. Makki, A., Weidt, G., Blachere, N. E., Lefrançois, L. & Srivastava, P. K. (2002) www.cancerimmunity.org/v2p4/020304.htm. [PubMed]