

Abstract
Proteins of the Imp4/Brix superfamily are involved in ribosomal RNA processing, an essential function in all cells. We report the first structure of an Imp4/Brix superfamily protein, the Mil (for Methanothermobacter thermautotrophicus Imp4-like) protein (gene product Mth680), from the archaeon M. thermautotrophicus. The amino- and carboxy-terminal halves of Mil show significant structural similarity to one another, suggesting an origin by means of an ancestral duplication. Both halves show the same fold as the anticodon-binding domain of class IIa aminoacyl-tRNA synthetases, with greater conservation seen in the N-terminal half. This structural similarity, together with the charge distribution in Mil, suggests that Imp4/Brix superfamily proteins could bind single-stranded segments of RNA along a concave surface formed by the N-terminal half of their β-sheet and a central α-helix. The crystal structure of Mil is incompatible with the presence, in the Imp4/Brix domain, of a helix–turn–helix motif that was proposed to comprise the RNA-binding moiety of the Imp4/Brix proteins.
Keywords: Imp4 domain, Brix domain, RNA binding, Methanothermobacter, crystal structure
Introduction
Ribosomal biogenesis is a complex process that requires a wealth of trans-acting proteins and small RNAs (Venema & Tollervey, 1999). Many of these factors are involved in the maturation and folding of ribosomal RNAs. The Imp4/Brix domain (Pfam04427, COG2136) was characterized as the defining structural feature of a protein superfamily, which is nearly ubiquitous in archaea and eukaryotes (Eisenhaber et al, 2001; Koonin et al, 2001; Mayer et al, 2001). The Imp4/Brix superfamily includes a set of orthologous, functionally uncharacterized archaeal proteins and five eukaryotic protein families. The archaeal proteins typically contain a core domain of 150–180 residues, whereas the eukaryotic proteins are larger (usually 300–400 residues). The eukaryotic Imp4/Brix proteins studied so far have been shown to have essential roles in ribosomal biogenesis.
Imp4 (Lee & Baserga, 1999) is the prototype member of the first Imp4/Brix eukaryotic family to be identified and has been studied in greater molecular detail than any of its homologues (Lee & Baserga, 1999; Wehner & Baserga, 2002; Granneman et al, 2003; Hsu et al, 2004). Imp4 is part of the U3 small nucleolar ribonucleoprotein (U3 snoRNP; Lee & Baserga, 1999), a complex involved in the early processing of the 18S rRNA (Venema & Tollervey, 1999; Dragon et al, 2002). Genetic depletion of Imp4 results in reduced production of 18S rRNA and therefore affects the biosynthesis of the small ribosomal subunit (Lee & Baserga, 1999). It has been proposed (Wehner & Baserga, 2002) that a short motif, which is conserved in the Imp4/Brix superfamily proteins, was similar to the helix–turn–helix (HTH) motif of the bacterial σ70 family of DNA-binding RNA polymerase subunits. The ability of Imp4 to bind RNA was linked to the presence of this σ70-like motif (Wehner & Baserga, 2002). Furthermore, it was shown that Imp3, an S4-like RNA-binding protein, is necessary for the association of Mpp10 and Imp4 with the U3 snoRNA, suggesting that the RNA-binding domain of Imp4 could directly interact with the pre-rRNA (Wehner et al, 2002). However, a recent study demonstrates that both Imp3 and Imp4 bind in vitro to a 5′ portion of the U3 snoRNA but not to the pre-rRNA (Gérczei & Correll, 2004). The Imp4/Brix domain of Imp4 is sufficient for its binding to the U3 snoRNA, but the full-length Imp4 is required to promote the formation of two hybrid duplexes between the U3 snoRNA and the pre-rRNA (Gérczei & Correll, 2004).
Brix, the prototype member of another eukaryotic family, is localized in the nucleolus and in Cajal bodies, where it associates with the 5S, 5.8S and 28S rRNA, but not with the 18S rRNA (Kaser et al, 2001). Thus, Brix might be involved in the biogenesis of the 60S ribosomal subunit in the nucleolus. In yeast, depletion of the Brx1 protein led to defects in ribosome biogenesis, which explains its essentiality (Kaser et al, 2001). The remaining eukaryotic Imp4/Brix families are typified by the proteins (i) Peter Pan (Migeon et al, 1999) and Ssf1/2 (Yu & Hirsch, 1995), (ii) Rpf1 (ribosome production factor 1), and (iii) Rpf2 (Wehner & Baserga, 2002).
All sequenced archaeal genomes, with the exception of halobacteria and thermoplasma, encode a single homologue of the eukaryotic Imp4/Brix proteins; none of these proteins has been experimentally characterized. There are substantial differences between archaea and eukaryotes with respect to the processing and modification of rRNAs and the proteins involved therein. However, some similarities have been found, such as the presence in some archaea of small noncoding RNAs and homologues of snoRNA-associated proteins such as the rRNA methylase fibrillarin (cf. Omer et al, 2003).
The gene for the archaeal homologue of Imp4 is part of a predicted superoperon, which also encodes homologues of several subunits of the eukaryotic exosome (Koonin et al, 2001). Recently, a complex of four homologues of exosome subunits has been isolated from the archaeon Sulfolobus solfataricus (Evguenieva-Hackenberg et al, 2003). In spite of the fact that other proteins encoded in the same superoperon, including the Imp4/Brix orthologue, were not found in this putative archaeal exosome, the possibility exists of a transient interaction between them and this RNA-processing machine.
Here, we present the crystal structure of Mil (for Methanothermobacter thermautotrophicus Imp4-like, gene product Mth680; Smith et al, 1997) and show that it consists of two related parts structurally similar to the anticodon-binding domain of class IIa aminoacyl-tRNA synthetases (aaRSs).
Results and Discussion
The Mil protein consists of 155 amino-acid residues (molecular mass 17.6 kDa) and has a theoretical pI of 9.3. Previous analysis (Koonin et al, 2001) and further database searches performed during this work showed that the Mil protein has orthologues in all sequenced archaeal genomes, with the exception of halobacteria, thermoplasma and the ‘minimal' archaeal genome of Nanoarchaeon equitans. The latter three groups of euryarchaea have degraded versions of the exosomal superoperon and the gene encoding the Imp4/Brix protein is among those deleted (Koonin et al, 2001). The archaeal Imp4/Brix proteins show limited but significant similarity to the eukaryotic members of the superfamily, particularly to the Imp4 and Rpf1 families. In addition, we detected significant similarity between the amino-terminal half of the Imp4/Brix domain and a similarly located region of a family of bacterial proteins, which also contain a predicted methyltransferase domain (Fig 1). The alignment in Fig 1 shows greater conservation in the N-terminal part of the aligned regions compared with the carboxy-terminal part; the most notable conserved motif is located at the N-terminus.
Figure 1.

Sequence alignment of Mil, selected archaeal orthologues, representatives of the five families of eukaryotic Imp4/Brix proteins and the N-terminal regions of predicted bacterial methyltransferases. The eukaryotic families are represented by the human members. The alignment was constructed using the MACAW program (Schuler et al, 1991) and manually refined using the structural elements of Mil as a guide. The structural elements are shown above the alignment, and the consensus including residues conserved in 80% of the aligned sequences is shown underneath the alignment (h stands for hydrophobic residues, l for aliphatic residues, s for small residues, o for alcohol residues, p for polar residues, s for small residues, u for ‘tiny' residues and + for positively charged residues). Boxed residues have a similarity score >0.2, using the BLOSUM62 matrix. The purported σ70-like motif (Wehner & Baserga, 2002) is shadowed in the eukaryotic proteins. For each protein, the range of the aligned residues is indicated. The proteins are designated by their abbreviated name and species name, and GenBank accession number. Aful, Archaeoglobus fulgidus; Aper, Aeropyrum pernix; Bant, Bacillus anthracis; Hsap, Homo sapiens; Milo, Mil orthologue; Mjan, Methanocaldococcus jannaschii; Mkan, Methanopyrus kandleri; Mthe, M. thermoautotrophicus; MTR, methyltransferase; Paer, Pyrobaculum aerophylum; Pfur, Pyrococcus furiosus; PPH, Peter Pan homologue; Ssol, S. solfataricus; Tmar, Thermotoga maritima.
Overall structure of Mil
The crystal structure of the Mil protein was determined by multiwavelength anomalous dispersion (MAD) using a Se-Met-substituted protein. Mil has overall dimensions of about 45 Å × 38 Å × 37 Å and shows a class α+β structure (Fig 2A). It consists of a wide, saddle-shaped β-sheet with six central antiparallel strands flanked at either side by two short parallel strands. The concavity of the saddle is filled by two α-helices subtending an angle of ∼70° to each other. A third α-helix is packed against one side of the saddle. Interestingly, the electron density map shows that Cys 96 and Cys 104 are engaged in a disulphide bridge (see supplementary figure online).
Figure 2.

Overall structure of Mil. (A) Two orthogonal views of the structure of Mil in ribbon representation. (B) The two halves of Mil shown in the same orientation after their structural superposition. (C) Structural alignment of the two homologous regions of Mil.
The conserved motifs identified in the Imp4/Brix superfamily alignment are located in the N-terminal βA, α1, the loop separating these two elements and the loop following βB. In the C-terminal part of the molecule, the most prominent conserved feature is the long, hydrophobic βJ (Fig 1). Examination of the Mil structure suggested a similar arrangement of the structural elements in the N- and C-terminal halves of the domain. Indeed, a comparison of these halves of Mil using the DALI server (Holm & Sander, 1999) showed significant similarity (Z-score 4.9), which was apparent in the structural superposition (Fig 2B). All secondary structure elements of the N-terminal region readily superimposed with those of the C-terminal part, with the exception of α2, which had no counterpart in the latter (Fig 2C). The similarity between the N- and C-terminal regions is a general feature of the Imp4/Brix superfamily, as is apparent from the conservation of all structural elements (Fig 1). It is also compatible with the presence, in the predicted bacterial methyltransferases, of a homologous region corresponding to one half of the Imp4/Brix domain, with greater similarity to the conserved, N-terminal part of the latter (Fig 1). These observations suggest that the Imp4/Brix domain evolved by means of duplication of an ancestral module.
Possible RNA-binding sites
The putative σ70-like motif of the eukaryotic Imp4/Brix has been shown to confer RNA-binding properties to these proteins (Wehner & Baserga, 2002). However, the C-terminus of Mil, the region that would contain the σ70-like motif, does not fold into a structure resembling an HTH; on the contrary, it accommodates βJ, the last β-strand of the protein (Figs 1,2). βJ is the longest strand in the structure of Mil and one of the two strands at the centre of the molecule. Therefore, the structural integrity of the Imp4/Brix domain depends on the C-terminal sequence adopting a β-strand conformation. Furthermore, this region unequivocally aligned throughout the Imp4/Brix superfamily (Fig 1; P<10−5 according to the MACAW program), effectively ruling out the presence of an HTH, although not the involvement of this part of the domain in RNA binding. Interestingly, point mutations in residues of human Imp4 that would map in the βE and βJ strands suggest the involvement of these regions in the interaction with Mpp10 (Granneman et al, 2003).
We searched for proteins with a similar fold to Mil using the DALI and CE (Shindyalov & Bourne, 1998) servers. Several significant hits were detected, albeit with relatively low Z-scores. The greatest structural similarity (Z-score 4.2 in CE) was observed between Mil and the anticodon-binding domain of histidyl-tRNA synthetase (Protein Data Bank identification (PDB ID) 1QE0), a class IIa aaRS. The closest structural neighbour reported by DALI (Z-score 3.7) was the accessory subunit of the mitochondrial DNA polymerase (PDB ID 1G5H), a derivative of GlyRS (Wolf & Koonin, 2001). In this case, the aligned region precisely corresponded to the domain of the accessory subunit, which is homologous to the anticodon-binding domain of class IIa aaRSs (Carrodeguas et al, 2001). The part of the Mil structure that aligned with the anticodon-binding domains encompassed the ∼75 N-terminal residues of Mil. Given the internal duplication in the Mil structure (Fig 2B,C), we separately ran the structural similarity search for the C-terminal half of Mil. DALI detected the anticodon-binding domain of GlyRS as the highest-scoring hit, with a Z-score of 2.7, whereas CE did not find any structural neighbour. This is compatible with the origin of the C-terminal portion by means of duplication of an ancestral RNA-binding domain related to the anticodon-binding domain of class IIa aaRSs, followed by greater sequence divergence in this region (Fig 1).
The similarity to the structure of an anticodon-binding domain suggests that at least the N-terminal half of the Imp4/Brix domain is involved in RNA binding (Fig 3). The two structures show remarkable superposition of the Cα trace in the aligned regions, especially for the α1 helix and the part of the β-sheet that includes the βA–βE strands of Mil (Fig 3A). Thus, this β-sheet and the α2 helix would constitute a good docking surface for a single-stranded RNA. Nevertheless, the sequence similarity between the aligned regions of Mil and the anticodon-binding domains is rather low, with 7–12% identity. As a result, their molecular surfaces and charge distributions present marked differences (Fig 3B). In particular, the greater number of positively charged amino acids in Mil and other Imp4/Brix proteins (Fig 1) suggests further, nonspecific contacts with RNA. The more extensive surface of Mil might indicate that it interacts with extended segments of single-stranded RNA, for example as seen in the complex of the poly(A)-binding protein with polyadenylate RNA (Deo et al, 1999) where the RNA binds along a surface formed by β-sheets of consecutive RNP domains. Although the traces of ancestral duplication are clearly seen in the Mil structure (Fig 2B,C), its C-terminal half shows further divergence from the anticodon-binding domains. As a result, the C-terminal half contains no equivalent to the α2 helix and also shows a less charged surface. Nevertheless, experimental evidence (Wehner & Baserga, 2002; Gérczei & Correll, 2004) supports that the C-terminal half of the Imp4/Brix domain does bind RNA. It remains uncertain whether the C-terminal part of the Imp4/Brix proteins contributes to a single RNA-binding site together with the N-terminal portion, or contains an extra RNA-binding site.
Figure 3.

Structural similarity between Mil and anticodon-binding domains. (A) The structures of Mil and the anticodon-binding domain of the prolyl-tRNA synthetase (ProRS) from Thermus thermophilus complexed with its tRNA-Pro(CGG) substrate (PDB ID 1H4S) were superposed. The proteins are represented as Cα traces in green (Mil) and blue (ProRS). Only the aligned residues are shown, namely Met1 to Ile73 for Mil and Val290 to Leu372 for ProRS. The Pro-tRNA (nucleotides 30–40) is in orange. (B) Electrostatic potential of Mil (left) and the anticodon-binding domain of ProRS (right) mapped on their respective molecular surfaces, with charges coloured in the range of +10 kT/e (blue) to −10 kT/e (red). Electrostatic calculations were carried out with the program delphi (Sharp & Honig, 1990) and the figure prepared with Chimera (Huang et al, 1996). A black line marks the boundary between Mil N- and C-terminal halves, as defined in Fig 2B. Both proteins are shown in the same orientation on superposition, as in (A). Nucleotides 30–40 of the Pro-tRNA are represented in orange.
Further work will be required to identify the RNA and protein ligands of Mil and to ascertain their mode of binding. The knowledge of its RNA-binding specificity should help in explaining the biological functions of the archaeal Imp4/Brix proteins. By analogy with eukaryotic homologues, a role of this protein in archaeal ribosomal biogenesis seems possible, with caveats due to the differences in this process between the two kingdoms. Conversely, an exosome-related function is suggested by the localization of the gene for Mil and its orthologues in the same superoperon with the genes for the putative exosome subunits (Koonin et al, 2001). An intriguing possibility is that, in archaea, the exosome function is tightly coupled with ribosomal biogenesis, a hypothesis compatible with the co-immunoprecipitation of the putative exosomal complex with ribosomal subunits (Evguenieva-Hackenberg et al, 2003) and the presence of the gene for ribosomal protein L15 within the exosomal superoperon (Koonin et al, 2001).
Methods
Cloning. The mth680 gene from M. thermautotrophicus was amplified by PCR from genomic DNA with primers 5′-GGC ACG GTC ATA TGC TCC TCA CAA CAT C-3′ (forward) and 5′-CTG AGA CCA AGC TTA CTC ACC GAC CCT GAA ATC-3′ (reverse). The resulting product was cloned into the pET-28a expression vector (Novagen, Madison, WI, USA), by using NdeI and HindIII restriction sites, to produce plasmid pYCL01. The clone was sequenced in the expression vector and found to be identical to the published sequence. Expression from pYCL01 yields a recombinant protein including the full-length Mil (residues 1–155) plus a 20-amino-acid-long N-terminal extension with a (His)6 tag.
Protein expression and purification. An overnight culture of pYCL01-transformed Escherichia coli BL21-Rosetta cells was used to inoculate fresh Luria–Bertani medium. The cells were grown at 37°C until OD600 ∼0.6 and then moved at 16°C and induced for overnight expression with 1 mM isopropyl-β-D-thiogalactoside. Similar conditions, except for the E. coli strain (B834) and the growing medium, were used to obtain a Se-Met-substituted protein. The bacterial pellet was resuspended and sonified in 50 mM Hepes (pH 7.5), 0.5 M NaCl and protease inhibitors. The lysate was cleared by centrifugation and the supernatant applied to Ni2+-loaded HisTrap columns (Amersham, Biosciences, Chalfont St Giles, UK). Fractions containing the Mil protein were pooled and further purified by gel filtration in a Superdex75 column equilibrated against 25 mM Hepes (pH 7.5) and 0.5 M NaCl. The protein was subsequently concentrated to ∼11.5 mg/ml.
Protein crystallization. Crystals of Mil were obtained by the hanging-drop vapour diffusion method at 19°C, using 1+1 μl (tagged protein+reservoir) with reservoir conditions of 20% PEG 3350 and 0.1 M Hepes (pH 7.5). Drops including these crystals were re-equilibrated for 2 h against reservoirs containing 20% PEG 3350, 0.1 M Hepes (pH 7.5) and 2.1 M NaCl to improve crystal diffraction quality by dehydration. The crystals were then transferred to liquid nitrogen. Most crystals diffracted to ∼7 Å, but one crystal from the Se-Met preparation diffracted to ∼2 Å. This crystal belongs to the P21 space group (Table 1) and has two molecules in the asymmetric unit, with a calculated solvent content of ∼38%.
Table 1.
Crystallographic statistics
|
Data collection |
|
|
|
| Space group |
P21 |
||
| Unit cell |
a=33.7 Å, b=77.5 Å, c=61.0 Å, β=100.01° |
||
| |
Peak |
Inflection |
Remote |
| Resolution (Å)a |
59.9–2.0 (2.11–2.0) |
60.0–2.0 (2.11–2.0) |
60.1–2.0 (2.11–2.0) |
| Wavelength (Å) |
0.97935 |
0.97950 |
0.97565 |
| Completeness (%)a |
100 (100) |
100 (100) |
100 (100) |
| Anomalous completeness (%)a |
99.6 (99.5) |
99.7 (99.5) |
99.7 (99.7) |
| Multiplicityb |
3.9 (1.9) |
3.9 (1.9) |
3.9 (1.9) |
| Unique reflections |
20,785 |
20,867 |
20,938 |
| <I>/σ(I)a |
8.6 (2.4) |
9.1 (2.0) |
9.2 (1.9) |
|
Rmeas (%)a,c |
16.0 (59.6) |
14.7 (69.8) |
14.7 (77.7) |
|
Phasing |
|
|
|
| Figure of meritd |
|
0.277 (0.628) |
|
|
Refinement |
|
|
|
| Rcryst/Rfree (%)e |
|
19.8/24.0 |
|
|
Geometry |
|
|
|
| R.m.s.d.f bonds/angles (Å, deg) | 0.014/1.40 | ||
aValues in parentheses are for the highest resolution shell.
bValues in parentheses are for the anomalous multiplicity.
c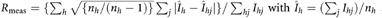 and where nh is the multiplicity of reflection h.
and where nh is the multiplicity of reflection h.
dFirst value is after Shelxe and the value in parentheses after Resolve.
e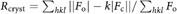 , where Fo and Fc are observed and calculated structure factors, respectively. Rfree is Rcryst calculated for a test set of randomly selected 5% of the data.
, where Fo and Fc are observed and calculated structure factors, respectively. Rfree is Rcryst calculated for a test set of randomly selected 5% of the data.
fRoot mean square deviations.
Crystal structure determination and refinement. The structure of Mil was solved by MAD using three wavelengths. Diffraction data were processed in Mosflm/Scala (CCP4, 1994) and fed into hkl2map (Pape & Schneider, 2004), a graphic interface for the Shelxc/d/e programs. Shelxd (Uson & Sheldrick, 1999) correctly found 7 out of the 14 possible selenium sites. Shelxe (Sheldrick, 2002) assigned the hand and produced a first set of phases, giving an initial map in which protein/solvent boundaries and secondary structure features could be resolved. These phases were improved and a first model fitted in the resulting electron density map using Resolve (Terwilliger, 2003). The new phases were fed into ARP/wARP (Perrakis et al, 1999). Cross-examination of the Resolve and ARP/wARP models allowed the tracing of 155 residues in one of the molecules, and 30 residues in the other one. A copy of the first molecule was placed by molecular replacement in the weaker density of the second one. Model rebuilding was carried out with coot (Emsley & Cowtan, 2004) and refinement with refmac (Murshudov et al, 1997) using the remote wavelength data. The final model includes residues from His 0 to Gly 154 in molecule A and Met 1 to Gly 154 in molecule B. Residues 87–89 and 116–117 in molecule B lacked supporting electron density and were not included in the model. The backbone dihedral angles (φ, Ψ) of 90.2% of the residues fall in the most favoured regions of the Ramachandran plot and the remaining 9.8% in its additionally allowed regions, as defined in procheck (Laskowski et al, 1993). Coordinates and structure factors have been deposited with the PDB ID code 1W94.
Supplementary information is available at EMBO reports online (http://www.nature.com/embor/journal/v6/n2/extref/7400328s1.pdf).
Supplementary Material
Supplementary Information
Acknowledgments
We thank M. Chechik and M. Shevtsov for technical assistance, and Dr J. Chong for providing us with genomic Mth DNA. This work was supported by a Wellcome Trust fellowship to A.A.A. and a Wellcome Prize studentship to D.W. We also thank the Wellcome Trust for the allocation of synchrotron beam time and the staff of the ID23 beamline at the European Synchrotron Radiation Facility (ESRF; Grenoble, France) for their help and support.
References
- Carrodeguas JA, Theis K, Bogenhagen DF, Kisker C (2001) Crystal structure and deletion analysis show that the accessory subunit of mammalian DNA polymerase γ, Pol γB, functions as a homodimer. Mol Cell 7: 43–54 [DOI] [PubMed] [Google Scholar]
- CCP4 (1994) The CCP4 Suite: programs for protein crystallography. Acta Crystallogr D 50: 760–763 [DOI] [PubMed] [Google Scholar]
- Deo RC, Bonanno JB, Sonenberg N, Burley SK (1999) Recognition of polyadenylate RNA by the poly(A)-binding protein. Cell 98: 835–845 [DOI] [PubMed] [Google Scholar]
- Dragon F et al. (2002) A large nucleolar U3 ribonucleoprotein required for 18S ribosomal RNA biogenesis. Nature 417: 967–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhaber F, Wechselberger C, Kreil G (2001) The Brix domain protein family—a key to the ribosomal biogenesis pathway? Trends Biochem Sci 26: 345–347 [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D 60: 2126–2132 [DOI] [PubMed] [Google Scholar]
- Evguenieva-Hackenberg E, Walter P, Hochleitner E, Lottspeich F, Klug G (2003) An exosome-like complex in Sulfolobus solfataricus. EMBO Rep 4: 889–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gérczei T, Correll CC (2004) Imp3p and Imp4p mediate formation of essential U3-precursor rRNA (pre-rRNA) duplexes, possibly to recruit the small subunit processome to the pre-rRNA. Proc Natl Acad Sci USA 101: 15301–15306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granneman S, Gallagher JE, Vogelzangs J, Horstman W, van Venrooij WJ, Baserga SJ, Pruijn GJ (2003) The human Imp3 and Imp4 proteins form a ternary complex with hMpp10, which only interacts with the U3 snoRNA in 60–80S ribonucleoprotein complexes. Nucleic Acids Res 31: 1877–1887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, Sander C (1999) Protein folds and families: sequence and structure alignments. Nucleic Acids Res 27: 244–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu CL, Chen YS, Tsai SY, Tu PJ, Wang MJ, Lin JJ (2004) Interaction of Saccharomyces Cdc13p with Pol1p, Imp4p, Sir4p and Zds2p is involved in telomere replication, telomere maintenance and cell growth control. Nucleic Acids Res 32: 511–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Couch GS, Pettersen EF, Ferrin TE (1996) Chimera: an extensible molecular modeling application constructed using standard components. Pacific Symp Biocomput 1: 724 [Google Scholar]
- Kaser A, Bogengruber E, Hallegger M, Doppler E, Lepperdinger G, Jantsch M, Breitenbach M, Kreil G (2001) Brix from Xenopus laevis and brx1p from yeast define a new family of proteins involved in the biogenesis of large ribosomal subunits. Biol Chem 382: 1637–1647 [DOI] [PubMed] [Google Scholar]
- Koonin EV, Wolf YI, Aravind L (2001) Prediction of the archaeal exosome and its connections with the proteasome and the translation and transcription machineries by a comparative-genomic approach. Genome Res 11: 240–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26: 283–291 [Google Scholar]
- Lee SJ, Baserga SJ (1999) Imp3p and Imp4p, two specific components of the U3 small nucleolar ribonucleoprotein that are essential for pre-18S rRNA processing. Mol Cell Biol 19: 5441–5452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer C, Suck D, Poch O (2001) The archaeal homolog of the Imp4 protein, a eukaryotic U3 snoRNP component. Trends Biochem Sci 26: 143–144 [DOI] [PubMed] [Google Scholar]
- Migeon JC, Garfinkel MS, Edgar BA (1999) Cloning and characterization of peter pan, a novel Drosophila gene required for larval growth. Mol Biol Cell 10: 1733–1744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Omer AD, Ziesche S, Decatur WA, Fournier MJ, Dennis PP (2003) RNA-modifying machines in archaea. Mol Microbiol 48: 617–629 [DOI] [PubMed] [Google Scholar]
- Pape T, Schneider TR (2004) HKL2MAP: a graphical user interface for macromolecular phasing with SHELX programs. J Appl Crystallogr 37: 843–844 [Google Scholar]
- Perrakis A, Morris R, Lamzin V (1999) Automated protein model building combined with iterative structure refinement. Nat Struct Biol 6: 458–463 [DOI] [PubMed] [Google Scholar]
- Schuler GD, Altschul SF, Lipman DJ (1991) A workbench for multiple alignment construction and analysis. Proteins 9: 180–190 [DOI] [PubMed] [Google Scholar]
- Sharp KA, Honig B (1990) Electrostatic interactions in macromolecules: theory and applications. Annu Rev Biophys Biophys Chem 19: 301–332 [DOI] [PubMed] [Google Scholar]
- Sheldrick GM (2002) Macromolecular phasing with SHELXE. Z Kristallogr 217: 644–650 [Google Scholar]
- Shindyalov I, Bourne P (1998) Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng 11: 739–747 [DOI] [PubMed] [Google Scholar]
- Smith DR et al. (1997) Complete genome sequence of Methanobacterium thermoautotrophicum ΔH: functional analysis and comparative genomics. J Bacteriol 179: 7135–7755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC (2003) Statistical density modification using local pattern matching. Acta Crystallogr D 59: 1688–1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uson I, Sheldrick G (1999) Advances in direct methods for protein crystallography. Curr Opin Struct Biol 9: 643–648 [DOI] [PubMed] [Google Scholar]
- Venema J, Tollervey D (1999) Ribosome synthesis in Saccharomyces cerevisiae. Annu Rev Genet 33: 261–311 [DOI] [PubMed] [Google Scholar]
- Wehner KA, Baserga SJ (2002) The σ(70)-like motif: a eukaryotic RNA binding domain unique to a superfamily of proteins required for ribosome biogenesis. Mol Cell 9: 329–339 [DOI] [PubMed] [Google Scholar]
- Wehner KA, Gallagher JEG, Baserga SJ (2002) Components of an interdependent unit within the SSU processome regulate and mediate its activity. Mol Cell Biol 22: 7258–7267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf YI, Koonin EV (2001) Origin of an animal mitochondrial DNA polymerase subunit via lineage-specific acquisition of a glycyl-tRNA synthetase from bacteria of the Thermus–Deinococcus group. Trends Genet 17: 431–433 [DOI] [PubMed] [Google Scholar]
- Yu Y, Hirsch JP (1995) An essential gene pair in Saccharomyces cerevisiae with a potential role in mating. DNA Cell Biol 14: 411–418 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information