

Abstract
Swe1p, the sole Wee1-family kinase in Saccharomyces cerevisiae, is synthesized during late G1 and is then degraded as cells proceed through the cell cycle. However, Swe1p degradation is halted by the morphogenesis checkpoint, which responds to insults that perturb bud formation. The Swe1p stabilization promotes cell cycle arrest through Swe1p-mediated inhibitory phosphorylation of Cdc28p until the cells can recover from the perturbation and resume bud formation. Swe1p degradation involves the relocalization of Swe1p from the nucleus to the mother-bud neck, and neck targeting requires the Swe1p-interacting protein Hsl7p. In addition, Swe1p degradation is stimulated by its substrate, cyclin/Cdc28p, and Swe1p is thought to be a target of the ubiquitin ligase SCFMet30 acting with the ubiquitin-conjugating enzyme Cdc34p. The basis for regulation of Swe1p degradation by the morphogenesis checkpoint remains unclear, and in order to elucidate that regulation we have dissected the Swe1p degradation pathway in more detail, yielding several novel findings. First, we show here that Met30p (and by implication SCFMet30) is not, in fact, required for Swe1p degradation. Second, cyclin/Cdc28p does not influence Swe1p neck targeting, but can directly phosphorylate Swe1p, suggesting that it acts downstream of neck targeting in the Swe1p degradation pathway. Third, a screen for functional but nondegradable mutants of SWE1 identified two small regions of Swe1p that are key to its degradation. One of these regions mediates interaction of Swe1p with Hsl7p, showing that the Swe1p-Hsl7p interaction is critical for Swe1p neck targeting and degradation. The other region did not appear to affect interactions with known Swe1p regulators, suggesting that other as-yet-unknown regulators exist.
INTRODUCTION
Yeast cells deploy a panoply of stress responses to adapt to changes in environmental conditions. Stress responses often involve transient depolarization of the actin cytoskeleton (Chowdhury et al., 1992; Delley and Hall, 1999) in addition to changes in gene expression (Gasch et al., 2000). Bud formation requires a polarized actin cytoskeleton, so that during some stress responses bud formation is temporarily halted. This in turn triggers cell cycle arrest in G2 through the morphogenesis checkpoint (Lew and Reed, 1995; McMillan et al., 1998). The cell cycle arrest is enacted by Swe1p, the sole Wee1-family kinase in S. cerevisiae (Sia et al., 1996).
Swe1p blocks entry into mitosis through inhibitory phosphorylation of a conserved tyrosine residue, Y19, in the cyclin-dependent kinase Cdc28p (Booher et al., 1993). Cdc28p Y19 phosphorylation occurs at low basal levels in unstressed cells, but rises dramatically in response to actin perturbation (Harrison et al., 2001). This response involves at least two pathways: one blocks degradation of Swe1p (Sia et al., 1998), whereas another appears to inhibit Mih1p, the Cdc25-family phosphatase that dephosphorylates Cdc28p at Y19 (Harrison et al., 2001).
Several factors have been shown to participate in targeting Swe1p for degradation in unstressed cells. Met30p is an F-box protein that forms part of an SCF ubiquitin ligase, and Swe1p degradation is blocked in temperature-sensitive met30 mutants, suggesting that SCFMet30 directs Swe1p ubiquitination and consequent destruction (Kaiser et al., 1998). Swe1p degradation also requires the presence of active Clb1p-4p/Cdc28p complexes, suggesting the presence of a feedback loop whereby Swe1p-dependent inhibition of Cdc28p causes stabilization and consequent accumulation of Swe1p (Sia et al., 1998). Finally, Swe1p degradation requires Hsl1p, a Nim1-family protein kinase, and Hsl7p, a methyltransferase that binds directly to both Swe1p and Hsl1p (Ma et al., 1996; Barral et al., 1999; McMillan et al., 1999a; Shulewitz et al., 1999; Lee et al., 2000; Cid et al., 2001).
When it is first synthesized during late G1, Swe1p accumulates predominantly in the nucleus. After bud emergence, a subset of the Swe1p is targeted to the bud side of the mother-bud neck in a septin-dependent manner (Longtine et al., 2000). Cells lacking either Hsl1p or Hsl7p fail to target Swe1p to the neck and fail to degrade Swe1p, suggesting a link between Swe1p neck localization and its degradation (McMillan et al., 1999a; Longtine et al., 2000). Our current hypothesis is that Swe1p cycles between the nucleus and the cytoplasm, and that while Swe1p transits through the cytoplasm, it can be delivered by Hsl7p to the neck, where it becomes marked for destruction. It is not clear exactly how Swe1p becomes marked for destruction, but an obvious possibility is that after neck targeting Swe1p is phosphorylated by neck-resident kinases, in a manner that promotes its subsequent recognition by SCFMet30, which catalyzes Swe1p ubiquitination leading to proteosomal degradation. Candidate neck-localized kinases include Hsl1p (Barral et al., 1999) and the polo-like kinase Cdc5p (Song et al., 2000), which also interacts with Swe1p (Bartholomew et al., 2001).
Many aspects of the scheme outlined above are still quite speculative. In this report, we have examined some aspects of the pathway in more detail. In particular, we found that Met30p was dispensable for Swe1p degradation, that Clb/Cdc28p acts at a step after neck targeting in the Swe1p degradation pathway, and that Clb/Cdc28p directly phosphorylates Swe1p in vitro. We also conducted a screen for functional but nondegradable mutant alleles of SWE1. The 39 mutants emerging from that screen consistently altered just two very small regions of Swe1p, defining Swe1p determinants that are critical for its regulated degradation. One of these determinants was required for effective interaction of Swe1p with Hsl7p, and our analysis strongly supports the importance of Hsl7p interaction and neck targeting in Swe1p degradation. Surprisingly, however, mutations in the other determinant blocked Swe1p degradation without impairing the interaction of Swe1p with any of its known regulators, suggesting that as-yet-unknown factors must also contribute to Swe1p degradation. Because degradation of Wee1-family kinases is also regulated by checkpoints in other species (Michael and Newport, 1998; Raleigh and O'Connell, 2000) and the known factors controlling Swe1p degradation are all highly conserved, our findings may have broad applicability to cell cycle control in eukaryotes.
MATERIALS AND METHODS
Yeast Strains and Plasmids
Standard genetic and molecular biology methods were used to generate all strains and plasmids used in this study. The yeast strains used are listed in Table 1. The oligonucleotides used are listed in Table 2.
Table 1.
Yeast strains used in this study
| Straina | Relevant genotype |
|---|---|
| DLY690 | a cdc24-1 swe1∷LEU2 bar1 |
| DLY3584 | a GAL1-SWE11–510-12myc:LEU2 bar1 |
| DLY4033b | a his3 leu2 lys2 trp1 ura3 |
| DLY4034b | a HSL7-3HA:kan |
| DLY4048 | a/α bar1/bar1 SWE1myc:HIS2/SWE1myc:URA3 SWE1myc:TRP1/trp1 GAL-SIC1∷LEU2/leu2 |
| DLY4599b | a/α HSL7-3HA:kan/HSL7-3HA:kan |
| DLY4266b | a HSL7-3HA:kan GAL1-SWE11–123-12myc:LEU2 |
| DLY4267b | a HSL7-3HA:kan GAL1-SWE11–510-12myc:LEU2 |
| DLY4268b | a HSL7-3HA:kan GAL1-SWE11–310-12myc:LEU2 |
| DLY4269b | a HSL7-3HA:kan GAL1-SWE11–250-12myc:LEU2 |
| DLY4270b | a HSL7-3HA:kan GAL1-SWE1511–819myc:URA3 |
| DLY4271b | a HSL7-3HA:kan GAL1-SWE1757–819myc:URA3 |
| DLY4272b | a HSL7-3HA:kan GAL1-SWE1311–819myc:URA3 |
| DLY4682 | a/α HSL7-3HA:kan/HSL7-3HA:kan GAL-SIC1:LEU2/leu2 |
| DLY5473c | a cdc28-as1 SWE1myc:URA3 |
| JMY1290 | a hsl7ΔURA3 GAL1-MIH1:TRP1 |
| JMY1441 | a SWE1myc:URA3 SWE1myc:TRP1 |
| JMY1477 | a hsl1ΔURA3 SWE1myc:HIS2 SWE1myc:TRP1 bar1 |
| JMY1628 | a swe1ΔLEU2 GAL1-MIH1:TRP1 |
| JMY1680b | a GAL1-SWE1-12myc:URA3 |
| JMY1681b | a GAL1-SWE1Δ1-12myc:URA3 |
| JMY1690 | a swe1∷LEU2 SWE1-12myc:URA3 cdc24-1 |
| JMY1691 | a swe1∷LEU2 SWE1Δ1-12myc:URA3 cdc24-1 |
| JMY1696b | a GAL1-SWE1I806T-12myc:URA3 |
| JMY1697b | a GAL1-SWE1Q807R-12myc:URA3 |
| JMY1718 | a swe1∷LEU2 GAL1-HSL7:LEU2 SWE1-12myc:URA3 cdc24-1 |
| JMY1719 | a swe1∷LEU2 GAL1-HSL7:LEU2 SWE1Δ1-12myc:URA3 cdc24-1 |
| JMY1722 | a swe1∷LEU2 SWE1Q807R-12myc:URA3 cdc24-1 |
| JMY1735 | a swe1∷LEU2 SWE1-12myc:URA3 SWE1-12myc:TRP1 |
| JMY1736 | a swe1∷LEU2 SWE1Δ1-12myc:URA3 SWE1Δ1-12myc:TRP1 |
| JMY1737 | a swe1∷LEU2 SWE1-NES-A-12myc:URA3 SWE1-NES-A-12myc:TRP1 |
| JMY1738 | a swe1∷LEU2 SWE1-NES-I-12myc:URA3 SWE1-NES-I-12myc:TRP1 |
| JMY1739 | a swe1∷LEU2 SWE1Δ1-NES-A-12myc:URA3 SWE1Δ1-NES-A-12myc:TRP1 |
| JMY1740 | a swe1∷LEU2 SWE1Δ1-NES-I-12myc:URA3 SWE1Δ1-NES-I-12myc:TRP1 |
| JMY1742 | a swe1∷LEU2 SWE1Q807R-12myc:URA3 SWE1Q807R-12myc:TRP1 |
| JMY1743 | a swe1∷LEU2 SWE1E797K-12myc:URA3 SWE1E797K-12myc:TRP1 |
| JMY1756 | a swe1∷LEU2 SWE1Q807R-12myc:URA3 GAL1-HSL7:LEU2 cdc24-1 |
| JMY1764 | a swe1∷LEU2 GAL1-SWE1Δ1-12myc:URA3 CDC28Y19F:TRP1 |
| JMY1765 | a swe1∷LEU2 GAL1-SWE1-12myc:URA3 CDC28Y19F:TRP1 |
| JMY1766 | a swe1∷LEU2 GAL1-SWE1I806T-12myc:URA3 CDC28Y19F:TRP1 |
| JMY1768 | a swe1∷LEU2 GAL1-SWE1-12myc:URA3 CDC28Y19F:TRP1 hsl7Δkanr |
| JMY1774 | α met4∷kanr met30∷kanr SWE1myc:HIS2 SWE1myc:TRP1 |
| JMY1777 | a GAL1-MIH1:TRP1 met4∷kanr met30∷kanr |
| JMY1779 | a swe1∷LEU2 GAL1-SWE1-12myc:URA3 CDC28Y19F:TRP1 met4∷kanr met30∷kanr |
| JMY1789b | a GAL1-SWE1-12myc:URA3 CDC28Y19F:TRP1 |
| JMY1792b | a GAL1-SWE1Q807R-12myc:URA3 CDC28Y19F:TRP1 |
| JMY1793b | a GAL1-SWE1-12myc:URA3 CDC28Y19F:TRP1 hsl1Δkanr |
| JMY1805 | α swe1∷LEU2 SWE1-12myc:URA3 SWE1-12myc:TRP1 hsl7Δkanr bar1 |
| JMY1807 | a swe1∷LEU2 SWE1Δ1-12myc:URA3 SWE1Δ1-12myc:TRP1 hsl7Δkanr bar1 |
| JMY1809 | α swe1∷LEU2 SWE1Q807R-12myc:URA3 SWE1Q807R-12myc:TRP1 hsl7Δkanr bar1 |
| JMY1857 | a swe1∷LEU2 GAL1-SWE1Q807R-12myc:URA3 CDC28Y19F:TRP1 |
| RSY016 | a/α GAL1-GST-CDC28:LEU2/GAL1-CLB2:LEU2 |
All strains are in the BF264-15Du (Richardson et al., 1989) background (ade1 his2 leu2-3,112 trp1-1a ura3Δns), except as indicated below.
Strains are in the YEF473A (Bi and Pringle, 1996) background (his3 leu2 trp1 ura3 lys2).
This strain is in the W303 background (ade2-1 trp1-1 leu2-3,112 his3-11 ura3 can1-100).
Table 2.
Oligonucleotides strains used in this study
| Primer | Sequence |
|---|---|
| OCT22 | AGTTCTAAAATTCCCGCATG |
| OCT23 | TTTAAGTGCTAACCCAAAAAATTTTGGCTTAGGTCC |
| OCT24 | GCCAAAATTTTTTGGGTTAGCACTTAAATTAGCTGG |
| OCT25 | TTGTTCACCGTCGACCTCGACGCTGCCGCTACTACCGATATC |
| OJ43 | GGACTTCGAAATGCTGGACACGGAGAACCTCCAGTTTATGGCGCGTTTCGGTGATGAC |
| OJ44 | GTCGTCTTCCTGGATAATAGCACCTGCATTGCGTGTCATTTTTCCTGATGCGGTATTTTCTCCT |
| OJ110 | AGTAAACACACACAGGCGCACACGAGAACAGGCGCGCCATGCGCGTTTCGGTGATGAC |
| OJ111 | GGAATTATATTTGTTTGGTTTAATGGCTTTGGCGCGCCTTTCCTGATGCGGTATTTTCTCCT |
| OJ112 | TCTTCGCCGTTAAATTCCAAAAGGCGTCTAGGCGCGCCGCGCGTTTCGGTGATGAC |
| OJ113 | AAAATTCCCGCATGGGTACCGAAATTTCTTGGGCGCGCCGCGCGTTTCGGTGATGAC |
| OJ114 | GTCGACCTCGACGCTAGCGTAATCTGGAACATCGGCGCGCCTTTCCTGATGCGGTATTTTCTCCT |
| OJ117 | TTTTTGTATGTATGCGTGTG |
| OJ118 | TTTTTCTTTGCGATAACTTG |
| OJ119 | GAAAGAGACAATATCAGTGGT |
| OJ121 | ACGATGGCAGAGGAGAGTGAC |
| OJ122 | CAAGGATGAGGAAAGCGTGGATT |
| OJ148 | GTCGACCTCGACGCTAGCGTAATC |
| OJ149 | GGGGCAACATCTCAAACACATA |
| OJ150 | GTTATTTTGAAAAATGATGATTTATACGGCACGGAC |
| OJ151 | GCCGTATAAATCATCATTTTTCAAAATAACCGAATT |
| OJ167 | ATGGCAACGATAATAATAATGTCA |
| OJ175 | TTCAGAAATCAACTTTTGTTCACCGTCGACAAAAAATTTTGGCTTAGG |
| OJ180 | GTGGAATTGTGCGGACGTTCCTCTTCTTTTATACATATATCAGCTGAAGCTTCGTACGC |
| OJ181 | TATTTGTTGCCGCAGTATATAGTATACAATGCAGAATTCAGCATAGGCCACTAGTGGATCTG |
| OJ207 | GATCCCCGGGAATTGCCATGTCATATAAAAAATTTTGGCTTAGGT |
| TSS1 | GATCTGTCGACTGAGGATCCAAGATGAT |
| TSS2 | CGATCATCTTGGATCCTCAGTCGACA |
| TSS3 | CGATGTCGACTGAGGATCCAAGATGGTAC |
| TSS4 | CATCTTGGATCCTCAGTCGACAT |
| TSS5 | AATTCGGTCGACTGAGGATCCAAGATGGTAC |
| TSS6 | CATCTTGGATCCTCAGTCGACCG |
| TSS7 | AGCTGTCGACTGAGGATCCAAGATG |
| TSS8 | AATTCATCTTGGATCCTCAGTCGAC |
The previously described GAL1:MIH1:TRP1 and SWE1myc:URA3 (McMillan et al., 1998), swe1::LEU2 (Booher et al., 1993), CDC28Y19F:TRP1 and GAL-SIC1::LEU2 (Sia et al., 1998), GAL1-HSL7:LEU2, SWE1myc:TRP1, and SWE1myc:HIS2 (McMillan et al., 1999a), hsl7ΔURA3 (Ma et al., 1996), HSL7-3HA:kan (Longtine et al., 2000), and met4::kanr and met30::kanr (Kaiser et al., 2000) alleles were introduced into yeast by direct transformation. The alleles swe1ΔLEU2 (primers OJ43 and OJ44, deleting the entire open reading frame (ORF) of SWE1 except the first and last 23 codons) and hsl7Δkanr (primers OJ180 and OJ181, deleting the entire HSL7 ORF) were introduced into yeast by a PCR knockout strategy (Wach, 1996), using the templates pRS304 (Sikorski and Hieter, 1989) and pFA6a-kanMX6 (Wach, 1996), respectively.
Strain DLY5473 was made by introducing the SWE1myc:URA3 allele into strain JAU01 (Bishop et al., 2000), a kind gift from J. Ubersax and D. Morgan (University of California, San Francisco, CA).
The construction of several integrating plasmids containing a variety of SWE1 alleles is described below. All of the URA3-marked alleles were integrated at the ura3 locus by transformation with the relevant StuI-digested plasmids, and all of the TRP1-marked alleles were integrated at the trp1 locus by transformation with the relevant Bsu36I-digested plasmids.
Construction of Plasmids Used for SWE1 Mutant Screens
The recipient “gapped” plasmids used for the mutant screens had a pRS316 (Sikorski and Hieter, 1989) backbone (CEN URA3) carrying “gapped” versions of the previously described SWE1myc allele that contains a single HA tag and 12 tandem myc tags at the end of the SWE1 coding sequence (McMillan et al., 1998). The gapped plasmid for the N-terminal screen (pJM1065) was missing sequences from codon 1 to codon 496, and the gapped plasmid for the C-terminal screen (pJM1069) was missing sequences from codon 417 to codon 819 of the 819-codon SWE1 ORF. These plasmids were constructed by replacing the relevant section of SWE1 with a PCR-generated LEU2 cassette flanked by AscI sites, as described below. Digestion of these plasmids with AscI then yielded the gapped plasmids used for the screens.
First, the PstI/BamHI fragment containing SWE1myc from YIplac204SWE1myc (McMillan et al., 1999a) was subcloned into pBLUESCRIPT-KS (Stratagene, La Jolla, CA) to make pJM1068, and then subcloned using XhoI/BamHI into pRS316 (Sikorski and Hieter, 1989) to make pJM1062. The KpnI site in the pJM1062 multiple cloning site was then destroyed by partial digestion with KpnI, blunt ending, and religation to make pJM1064 (in which the KpnI site within SWE1 is now unique). Both pJM1062 and pJM1064 are therefore CEN URA3 plasmids expressing full-length SWE1myc from its own promoter.
A gap repair strategy was then used to replace sections of SWE1 in the above plasmids with the LEU2 cassette. LEU2 was amplified by PCR using pRS305 (Sikorski and Hieter, 1989) as template and the primers OJ110 and OJ111 (for the N-terminal replacement) or OJ112 and OJ114 (for the C-terminal replacement). OJ110 and OJ111 introduce terminal sequences homologous to bases −31 to −1 (5′ end) and 1487–1516 (3′ end) of SWE1 (where +1 is the first base of the ORF), whereas OJ112 and OJ114 introduce terminal sequences homologous to bases 1249–1278 (5′ end) of SWE1 and to the HA tag (3′ end). pJM1062 was digested with BglII, creating a gap within the N-terminal half of SWE1, and the gapped plasmid was cotransformed into yeast together with the first PCR LEU2 cassette above. Gap repair by homologous recombination yielded pJM1065, in which the first 496 codons of SWE1 are replaced by the LEU2 cassette. pJM1064 was digested with KpnI, creating a gap within the C-terminal half of SWE1, and the gapped plasmid was cotransformed into yeast together with the second PCR LEU2 cassette above. Gap repair by homologous recombination yielded pJM1069, in which the last 402 codons of SWE1 are replaced by the LEU2 cassette.
Construction of a New SWE1–12myc Allele
The SWE1myc allele described above contains a single HA tag between the end of the SWE1 coding sequence and the 12 tandem myc tags (McMillan et al., 1998). We constructed a new 12myc-tagged version of SWE1, designated SWE1-12myc, that does not have an HA tag between SWE1 and the myc tags but is otherwise identical to the earlier SWE1myc allele. The sequence at the end of SWE1 in the new allele is: 5′-AAGCCAAAATTTTTTGTCGACGGTGAACAAAAGTTGATTTCTGAAGAAGATTTGAAC-3′, encoding the peptide KPKFFVDGEQKLISEEDLN. The italicized SalI site delineates the boundary between the end of SWE1 and the first myc tag.
The construction of the SWE1-12myc allele was performed as follows. First, a LEU2 PCR cassette amplified as described above using primers OJ113 and OJ114 was cotransformed into yeast with KpnI-digested pJM1064, replacing the final 59 codons of SWE1 with the LEU2 cassette by homologous recombination, yielding pJM1070. Digestion with AscI and religation then removed the cassette, yielding pJM1071. An EcoRV/BamHI fragment from pJM1071 containing the truncated SWE1 and epitope tag sequences was cloned into the XhoI(blunt)/BamHI sites of pRS306 (Sikorski and Hieter, 1989) to make pJM1072. A PCR product fusing the final 90 codons of SWE1 to myc tag sequences (without an intervening HA tag) was generated using YIplac204SWE1myc (McMillan et al., 1999b) as template and primers OJ167 and OJ175, and cotransformed into yeast with AscI-digested pJM1071, restoring the C-terminal SWE1 sequences now fused directly to myc tags via a SalI site by homologous recombination, yielding pJM1091. However, the PCR amplification led to deletion of some of the myc tags. To generate the final SWE1--12myc allele, a HindIII/SalI fragment from pJM1091 containing the C-terminal half of SWE1 was ligated into the corresponding sites in pJM1072, yielding pJM1102 (full-length SWE1-12myc under control of the SWE1 promoter in an integrating URA3-marked plasmid). A similar TRP1-marked plasmid, pJM1115, was constructed by cloning a PstI/BamHI fragment from pJM1102 containing the SWE1-12myc allele into the corresponding sites in pRS304. Finally, a URA3-marked integrating plasmid expressing the SWE1-12myc allele from the GAL1 promoter, pJM1101, was constructed by cloning a HindIII/SalI C-terminal fragment of SWE1 from pJM1091 into the corresponding sites of pDLB955, the plasmid used to make the SWE1myc allele in strain RSY206 (McMillan et al., 1998).
Construction of the SWE1Δ1 Allele
The SWE1Δ1 allele was constructed by a three-way homologous recombination strategy. Two overlapping SWE1 PCR products, containing the Δ1 mutation in the overlapping region, were made using YIplac204SWE1myc (McMillan et al., 1999a) as template and primer pairs OJ121 + OJ150 and OJ122 + OJ151. These products were cotransformed with BglII-digested pJM1062 into yeast, yielding pJM1139, a CEN URA3-marked plasmid expressing SWE1Δ1myc (lacking the sequences coding for R318 to K328) from the SWE1 promoter. The allele was transferred to SWE1 promoter-regulated (pJM1096) and GAL1 promoter-regulated (pJM1099) integrating URA3-marked plasmids by subcloning a HindIII/SalI SWE1Δ1 fragment from pJM1139 into the corresponding sites of pJM1072 and pDLB955, respectively.
To generate versions of SWE1Δ1 containing the new 12myc epitope tag (above), the ClaI/SalI SWE1 fragment from pJM1091 (see above) was used to replace the corresponding fragment in pJM1096 (SWE1Δ1myc) and pJM1099 (GAL1-SWE1Δ1myc), yielding pJM1103 (SWE1Δ1-12myc) and pJM1104 (GAL1- SWE1Δ1-12myc). A similar TRP1-marked plasmid, pJM1116, was constructed by cloning a PstI/BamHI fragment from pJM1103 containing the SWE1Δ1-12myc allele into the corresponding sites in pRS304.
Construction of the SWE1E797K, SWE1I806T, and SWE1Q807R Alleles
The plasmids pJM1165 (E797K), pJM1162 (I806T), and pJM1164 (Q807R) were isolated from the PCR mutagenesis screen and were used as templates for subsequent PCR manipulations. PCR products containing SWE1E797K, SWE1I806T, and SWE1Q807R were amplified using the primers OJ167 and OJ175 and cotransformed into yeast together with AscI-digested pJM1071, yielding pJM1142 (SWE1E797K-12myc), pJM1143 (SWE1I806T-12myc), and pJM1141 (SWE1Q807R-12myc). The presence of the desired mutations (and absence of other mutations) was confirmed by sequencing. Integrating URA3-marked plasmids with these alleles were generated by subcloning ClaI/SalI fragments containing the mutations into the corresponding sites in pJM1102 (SWE1-12myc) or pJM1101 (GAL1-SWE1-12myc), yielding pJM1113 (SWE1E797K-12myc:URA3), pJM1112 (SWE1Q807R-12myc:URA3), and pJM1109 (GAL1-SWE1I806T-12myc:URA3). Similar TRP1-marked plasmids were constructed by transferring the SWE1-containing PstI/BamHI fragments from pJM1113 and pJM1112 into the corresponding sites in pRS304, yielding pJM1123 (SWE1E797K-12myc:TRP1) and pJM1122 (SWE1Q807R-12myc:TRP1).
Construction of the SWE1-NES Alleles
A pair of “forced-localization” NES cassettes encoding two NES motifs (active, NES-A; inactive, NES-I) separated by a T7 epitope tag were previously described (Edgington and Futcher, 2001). These cassettes were introduced in between the Swe1p C terminus and a 12-myc epitope tag by an overlap PCR strategy. Sequences encoding a C-terminal fragment of Swe1p were amplified using pDLB955 as template and primers OCT22 and OCT23, and the forced-localization cassettes were amplified using primers OCT24 and OCT25. Overlap PCR using these fragments as template and primers OCT22 and OCT25 yielded products encoding the C-terminal part of Swe1p fused in frame to the NES cassettes. This product was digested with KpnI and SalI and cloned into the corresponding sites in pDLB955, yielding pDLB1735 (GAL1-SWE1-NES-A-12myc:URA3) and pDLB1736 (GAL1-SWE1-NES-I-12myc:URA3). ClaI/SalI fragments from these plasmids were subcloned into the corresponding sites in pJM1102 (above), yielding pJM1105 (SWE1-NES-A-12myc:URA3) and pJM1106 (SWE1-NES-I-12myc:URA3) driven from the SWE1 promoter. PstI/BamHI fragments from these plasmids were then subcloned into the corresponding sites in pRS304, yielding pJM1117 (SWE1-NES-A-12myc:TRP1) and pJM1118 (SWE1-NES-I-12myc:TRP1).
To generate similar NES-containing versions of SWE1Δ1, we performed a similar set of subcloning manipulations using pJM1103 (SWE1Δ1-12myc) and pRS306 instead of pJM1102 and pRS304 above, yielding pJM1107 (SWE1Δ1-NES-A-12myc:URA3), pJM1108 (SWE1Δ1-NES-I-12myc:URA3), pJM1119 (SWE1Δ1-NES-A-12myc:TRP1), and pJM1120 (SWE1Δ1-NES-I-12myc:TRP1).
Construction of Truncated SWE1myc Alleles
N- or C-terminally truncated derivatives of SWE1 were generated by a two-step strategy. In the first step, internal SWE1 sequences between a pair of selected restriction sites in pBLB955 (GAL1-SWE1myc) were replaced by annealed oligonucleotides with sticky ends compatible with the relevant sites and containing a common core sequence (SalI site, BamHI site, and start codon in a good Kozak context: 5′-GTCGACTGAGGATCCAAGATG-3′, start codon in bold face). The oligonucleotides are listed in Table 2: TSS1 and TSS2 replaced the internal BglII-ClaI fragment, TSS3 and TSS4 replaced the internal ClaI-KpnI fragment, TSS5 and TSS6 replaced the internal EcoRI-KpnI fragment, and TSS7 and TSS8 replaced the internal HindIII-EcoRI fragment. To generate Swe1p fragments lacking N-terminal sequences, BamHI fragments excised from these constructs were cloned into the BamHI site in the GAL1-promoter vector YIpG2 (Stueland et al., 1993), yielding plasmids promoting galactose-inducible expression of myc-tagged Swe1p 311–819 (from the EcoRI site), Swe1p 511–819 (from the ClaI site), and Swe1p 757–819 (from the KpnI site). Plasmids were digested with HpaI to target integration at the leu2 locus. To generate Swe1p fragments lacking C-terminal sequences, the constructs were digested with SalI, gel purified, and religated to circularize the plasmids, removing sequences between the SalI site in the oligonucleotides and the SalI at the beginning of the myc tag sequences, yielding plasmids promoting galactose-inducible expression of myc-tagged Swe1p 1–123 (up to the BglII site), Swe1p 1–250 (up to the HindIII site), Swe1p 1–310 (up to the EcoRI site), and Swe1p 1–510 (up to the ClaI site). Plasmids were digested with StuI to target integration at the ura3 locus.
Two-hybrid Strains and Plasmids
Two-hybrid analysis was performed with the base strain PJ69-4A as described (James et al., 1996). The “bait” (DNA binding domain) vector was pOBD.CYH (Drees et al., 2001) and the “prey” (transcriptional activation domain) vector was pGAD424 (James et al., 1996). Wild-type SWE1 was cloned into pOBD.CYH, yielding pMOS166, as described (Drees et al., 2001). To introduce SWE1Q807R into pOBD.CYH, pMOS166 was digested with KpnI and SalI (generating a “gap” at the 3′ end of SWE1 spanning the sequence of the mutation) and cotransformed into strain DLY4033 together with a SWE1Q807R PCR product generated in two PCR steps using pJM1112 (SWE1Q807R-12myc:URA3, described above) as template. The first step used primers OJ119 and OJ207, and the product was then reamplified using primers OJ119 and BD70R to introduce 3′ sequences homologous to the MCS region of pMOS166 to enhance recombination during the gap repair.
A plasmid (pDLB2191) containing a partial CDC5 ORF (base 1265 to the end, encoding both “polo boxes” but not the kinase domain of Cdc5p) in the prey vector pGAD-C1 (James et al., 1996) was identified in a two-hybrid screen using pMOS166 as bait. The plasmid (YF306) containing full-length CLB2 in the prey vector pGAD-C2 (James et al., 1996) was a kind gift from Fred Cross (Rockefeller University, NY).
GST-HSL7 Expression Plasmid
To express GST-Hsl7p in E. coli, we first cloned sequences encoding the entire HSL7 ORF plus 400 base pairs downstream as a NdeI-SacI fragment into the corresponding sites in pUNI-10 (Liu et al., 1998). The gene was then excised as an EcoRI-SacI fragment and cloned into the corresponding sites of pGEX-KG (Pharmacia, Piscataway, NJ), yielding pDLB2211.
Screen for SWE1 Mutants
Two SWE1 PCR products were generated under mutagenic conditions using Taq DNA polymerase in the presence of 0.2 mM MnCl2 with 2.5 mM dGTP, 2.5 mM dTTP, 0.5 mM dATP, and 0.5 mM dCTP. The PCR template was YIplac204SWE1myc (McMillan et al., 1999a). The PCR primers OJ117 and OJ118 were used to amplify an N-terminal section of SWE1 extending from 147 bases upstream of the start codon to base 1643 (codon 548) of SWE1, which was cotransformed into yeast strain JMY1628 together with AscI-digested pJM1065. The primers OJ148 and OJ149 were used to amplify a C-terminal section of SWE1 extending from base 1144 (codon 382) of SWE1 to the downstream epitope tag, which was cotransformed into yeast strain JMY1628 together with AscI-digested pJM1069. Homologous recombination between the mutagenized PCR products and these gapped plasmids regenerates full-length SWE1myc mutants.
The recipient strain, JMY1628, contains a GAL1:MIH1 allele, resulting in high levels of MIH1 expression on galactose-containing medium but no expression of MIH1 on glucose containing medium. Transformants containing successfully gap-repaired plasmids were selected on galactose medium lacking uracil, and after incubation for 3 d at 30°C, colonies were replica-plated to plates with dextrose medium lacking uracil. After one more day at 30°C, individual colonies were visually examined under a dissecting microscope to identify those that arrested with elongated buds on dextrose but not on galactose medium. Plasmids rescued from the colonies meeting these criteria were retransformed into JMY1628 to confirm that the plasmid was responsible for the phenotype. Plasmids passing this second round of screening were sequenced within the mutagenized region.
Cell Cycle Synchrony, Immunofluorescence, and Microscopy
Cells were synchronized in G1 by pheromone arrest (incubation of MATa bar1 cells growing exponentially in YEPG (1% yeast extract, 2% bacto-peptone, 2% galactose, 0.01% adenine) at 24°C with 100 ng/ml α-factor for 4 h, after which >90% of cells were arrested) and release (cells were harvested by centrifugation, washed once in YEPG, and resuspended in fresh YEPG at 37°C). To visualize the nuclei, aliquots of cells were fixed with 2 volumes of 95% ethanol, washed with H2O, and then resuspended in 0.2 μg/ml 4′,6′-diamidino-2-phenylindole in H2O.
Immunofluorescence localization of Swe1p-myc was performed using a four-antibody sandwich technique as described (Longtine et al., 2000). Cells were viewed on a Axioscope (Carl Zeiss, Inc., Thornwood, NY) equipped with epifluorescence and Nomarski optics and images were captured with a cooled charge-coupled device camera (Princeton Instruments, Princeton, NJ). Microscopic images of whole yeast colonies were similarly captured.
Biochemical Procedures
Procedures for harvesting and lysis of yeast cells, SDS-PAGE, immunoprecipitation, immunoblotting, and pulse-chase analysis of Swe1p stability were as described (McMillan et al., 1999a). Procedures for harvesting and lysis of bacterial cells and purification of GST-tagged proteins were as described (McMillan et al., 1999b; Bose et al., 2001). Binding assays to assess binding of Swe1p to GST-Hsl7p were performed by incubating 250 μg of yeast lysate from strains expressing myc-tagged Swe1p (wild-type or mutant) together with excess (∼40 μg/sample) GST or GST-Hsl7p immobilized on glutathione beads for 30 min at 4°C. Beads were washed with NP40 wash buffer (Bose et al., 2001) three times before analysis by SDS-PAGE and immunoblotting. Kinase assays to assess Swe1p phosphorylation by GST-Cdc28p/Clb2p complexes were performed as described (McMillan et al., 1999b) except that Swe1p-myc beads (immunoprecipitated from 2 mg of yeast lysate and washed with reaction buffer) were added instead of histone H1 as substrate and the reaction was extended to 2 h at 30°C with 25 μCi γ-32P-ATP in each sample. After exposure of dried gels to film, the radioactive Swe1p bands were excised and subjected to partial proteolysis by 50 ng V8 protease, and the resulting peptides were separated on a 12.5% polyacrylamide gel containing 1 mM EDTA (Cleveland et al., 1977).
RESULTS
Role of Met30p in Swe1p Degradation
We previously reported that temperature-sensitive met30-6 mutants blocked both Swe1p degradation in vivo and Swe1p ubiquitination in vitro, suggesting a direct role for SCFMet30 in targeting Swe1p for degradation (Kaiser et al., 1998). We initially wanted to examine Swe1p localization in met30 mutants to determine whether neck targeting was affected. Recent studies have established that the lethality of the met30 mutant is due to its failure to downregulate the transcription factor Met4p (Patton et al., 2000). Thus, met30Δ met4Δ cells are viable (albeit auxotrophic for methionine) and are therefore ideal to assess the in vivo role of Met30p in Swe1p degradation. Swe1p localization in met30Δ met4Δ cells was indistinguishable from that in wild-type cells (Figure 1A). However, we were surprised to find that met30Δ met4Δ mutants did not display significantly elevated levels of Swe1p compared with wild-type cells even in cells undergoing mitosis (see Figure 1 legend). Also unexpectedly, we found that met30Δ met4Δ mutants proliferated normally and did not display elongated buds even when Mih1p was eliminated (Figure 1B). These results prompted us to reexamine the requirement of Met30p for Swe1p degradation, using the met30Δ met4Δ strain. Pulse-chase analysis showed that Swe1p stability was similar in wild-type and met30Δ met4Δ cells (Figure 1, C and D), indicating that Met30p is not in fact required to target Swe1p for degradation. In this experiment the pulse-chase was performed after a shift to 37°C, so that the results would be directly comparable to those we obtained previously with met30-6 mutants (Kaiser et al., 1998). We conclude that the stabilization of Swe1p in met30-6 mutants is indirect, mediated through some action of Met4p.
Figure 1.

Role of Met30p in Swe1p regulation. (A) Localization of myc-tagged Swe1p was assessed by immunofluorescence microscopy. Budded cells were scored according to whether Swe1p was detected only in the nucleus, both in the nucleus and at the neck, or only at the neck, and the proportion of cells showing each distribution was quantitated by counting >200 cells. Analysis of >50 cells in anaphase or telophase showed that although 60% of such hsl1Δ cells had detectable nuclear Swe1p, only 8% of met30Δ met4Δ cells and <1% of wild-type cells had detectable nuclear Swe1p after nuclear division. Strains were JMY1441 (WT), JMY1774 (met30Δ met4Δ), and JMY1477 (hsl1Δ). (B) Strains JMY1290 (hsl7Δ GAL1-MIH1) and JMY1777 (met30Δ met4Δ GAL1-MIH1) were grown in galactose-containing medium and plated onto dextrose containing medium to repress Mih1p expression. Photographs were taken 1 day later. (C) Pulse-chase analysis of Swe1p stability. Strains containing myc-tagged SWE1 expressed from the GAL1 promoter were grown in sucrose-containing medium at 23°C, shifted to 37°C for 2 h, and induced to express Swe1p by addition of galactose. After a 10-min incubation in labeling medium containing 35S-methionine (“pulse”), the cells were washed and resuspended in dextrose-containing medium with added unlabeled methionine at 37°C (“chase”). At the indicated times of chase, aliquots were withdrawn and processed to determine the amount of labeled Swe1p-myc remaining. Strains were JMY1765 (WT), JMY1779 (met30Δ met4Δ), and JMY1768 (hsl7Δ). All strains contained an integrated copy of CDC28Y19F to minimize any effects of Swe1p activity during the experiment. The Swe1p from wild-type cells appears slightly more retarded in its gel mobility in this experiment, but that observation was not reproducible and appears to reflect gel-to-gel variation. (D) Quantitation of the experiment shown in C. (E) Diploid homozygous deletion strains from the Yeast Knockout Collection containing the indicated mutations were grown to exponential phase in YEPD at 30°C, harvested, lysed, and analyzed (100 μg of lysate per lane) by Western blotting to detect Cdc28p Y19 phosphorylation (upper) or total Cdc28p level (lower: the α-PSTAIRE antibody recognizes both Cdc28p and Pho85p. Cdc28p is the upper band.).
If Met30p is not the ubiquitin ligase responsible for the Swe1p polyubiquitination observed previously, then what is? We reasoned that strains unable to degrade Swe1p should contain elevated levels of Cdc28p Y19 phosphorylation, which can be monitored using a phospho-specific antibody (McMillan et al., 1999b). We examined Cdc28p Y19 phosphorylation in strains carrying deletions of each of the 13 genes encoding nonessential F-box proteins. In parallel control strains, swe1Δ mutants exhibited no detectable Cdc28p phosphorylation, whereas mih1Δ and hsl1Δ strains showed significantly elevated Cdc28p phosphorylation, as expected (Figure 1E). However, none of the F-box mutants displayed elevated Cdc28p phosphorylation (Figure 1E). We have also epitope-tagged Swe1p in all but three of those strains, but unlike parallel hsl1Δ controls none of these strains accumulated excess Swe1p (unpublished data). Assuming that all of these strains (obtained via Research Genetics, Inc. from the genome knockout collection) were correctly constructed, the simplest conclusion from this panel is that Swe1p degradation does not require any single nonessential F-box protein.
Role of Clb/Cdc28p in Swe1p Degradation
It is unclear how the requirement for Clb/Cdc28p activity in Swe1p degradation fits into the Swe1p degradation pathway. Phosphorylation of Hsl7p, as detected by a mobility shift of the protein upon SDS-PAGE, occurs in a cell-cycle–regulated manner, from late G1 until telophase (McMillan et al., 1999a), perhaps suggesting that Clb/Cdc28p contributes to some aspect of Hsl1p/7p function. Alternatively, Clb/Cdc28p might be required for a separate step in Swe1p degradation, after Swe1p neck targeting by Hsl1p and Hsl7p. To distinguish between these possibilities, we examined the consequences of overexpressing the Clb/Cdc28p inhibitor Sic1p. The Hsl7p mobility shift provides a convenient indicator of events at the neck, because it is dependent both on Hsl1p kinase activity and on intact septins (our unpublished data). However, we found that the Hsl7p mobility shift was unaffected by Sic1p (Figure 2A) and that Swe1p was still targeted to the neck in cells overexpressing Sic1p (Figure 2B). In both of these experiments the cells arrested with a single nucleus and one or more elongated buds (see Figure 2B inset), indicating that sufficient Sic1p had been made to effectively inhibit Clb/Cdc28p. These findings suggest that Clb/Cdc28p is not required for the aspects of Hsl1p/7p function that we are able to monitor and that Clb/Cdc28p acts after neck targeting in the Swe1p degradation pathway.
Figure 2.

Effect of Clb/Cdc28p inhibition on Hsl7p phosphorylation and Swe1p localization. (A) Strains DLY4599 (HSL7-HA) and DLY4682 (HSL7-HA GAL1-SIC1) were grown in sucrose-containing medium and induced to express Sic1p by addition of galactose. Strain DLY4682 contains multiple (>4) copies of GAL1-SIC1, which causes arrest in G1 with characteristically elongated buds on galactose medium. At the indicated time in galactose, cells were harvested and lysed, and Hsl7p migration on SDS-PAGE was monitored by Western blotting to assess Hsl7p phosphorylation. The upper band is a phosphorylated form of Hsl7p. (B) Strain DLY4048 (SWE1myc GAL1-SIC1) was grown in sucrose-containing medium, and dextrose or galactose were added to repress or induce Sic1p expression, respectively, for 6 h. Localization of myc-tagged Swe1p was assessed by immunofluorescence microscopy. Budded cells were scored as described in Figure 1, and the proportion of cells showing each distribution was quantitated by counting >200 cells. Not included in this analysis were cells (∼20% in each case) that failed to show detectable Swe1p staining. Inset: example of Sic1p-arrested cell with Swe1p localized to the neck.
Swe1p Phosphorylation by Clb/Cdc28p
In other organisms, cyclin/Cdc2 directly phosphorylates Wee1 at multiple sites (Dunphy, 1994), suggesting the simple hypothesis that Clb/Cdc28p directly phosphorylates Swe1p in order to target Swe1p for degradation. Consistent with this hypothesis, we found that Swe1p was readily phosphorylated by Clb2p/Cdc28p in vitro (Figure 3A), and partial digestion of Clb/Cdc28p-phosphorylated Swe1p by V8 protease revealed a complex pattern of phospho-peptides (Figure 3B). In addition, much of the Swe1p in vivo phosphorylation that we detected as a gel mobility shift of Swe1p isolated from yeast cells was eliminated after Cdc28p inhibition (Figure 3C; similar findings were first described by S. Harvey and D. Kellogg, personal communication). Moreover, Swe1p interacted with Clb2p in the two-hybrid assay (Figure 3D; first described by F. Cross, personal communication), although it is not clear whether this interaction reflects the propensity of Swe1p to phosphorylate Clb2p/Cdc28p, or the propensity of Clb2p/Cdc28p to phosphorylate Swe1p, or both. In summary, it is very likely that Clb/Cdc28p phosphorylates Swe1p in S. cerevisiae.
Figure 3.

Swe1p phosphorylation by Cdc28p. (A) GST-Cdc28p/Clb2p complexes were isolated from yeast strain RSY016 and incubated with γ-32P-ATP and Swe1p (JMY1789) or Swe1pQ807R (JMY1792) immunoprecipitated from wild-type or hsl1Δ (JMY1793) cells, as indicated. As a control, immunoprecipitates were prepared in the same way from cells lacking myc-tagged Swe1p (DLY4033: left lane) to ensure that no comigrating Cdc28p substrates were present in the preparation. Phosphorylated proteins were separated by SDS-PAGE and visualized using a phosphorimager, and the total amount of Swe1p-myc was assessed by Western blotting. (B) The phosphorylated Swe1p bands from the gel shown in A were excised and loaded on duplicate lanes in a second gel together with V8 protease. Phosphorylated digestion products were detected using a phosphorimager. Molecular weight markers (kDa) are shown at left. (C) Cdc28p-dependence of Swe1p phosphorylation in yeast was analyzed in strain DLY5473, which contains SWE1myc and carries the analog-inhibitable cdc28-as1 allele. Cells were grown to exponential phase and treated (+) or not treated (−) with 5 μM 1-nm-PP1 (kind gift of D. Morgan) for 1 h to inhibit Cdc28p. Lysates were then processed for Western blotting. (D) Two-hybrid analysis of Swe1p-Clb2p interaction. A HIS3 reporter plasmid was used to monitor interaction. Cells containing the Gal4p activation domain fused to Clb2p and the Gal4p DNA-binding domain fused to Swe1p (wild-type, Q807R, or vector control as indicated) were streaked out on medium containing (+His) or lacking (−His) histidine. The latter plates also contained 30 mM 3-amino-triazole (a His3p inhibitor) to select for cells robustly expressing the reporter gene.
In the course of these studies we noticed that Swe1p isolated from hsl1Δ cells was also a good substrate of Clb2p/Cdc28p in vitro (Figure 3A) and that partial digestion of this phosphoprotein with V8 protease yielded a spectrum of phosphopeptides identical to that seen with Swe1p from wild-type cells (Figure 3B). Thus, it would appear that the phosphorylation of Swe1p by Cdc28p in vitro does not require prior Swe1p neck targeting.
Identification of Stabilized Mutants of SWE1
Given what we know about the Swe1p degradation pathway, we expected that the Swe1p polypeptide would harbor multiple determinants important for Swe1p degradation, including elements that promote Swe1p nuclear export, Swe1p neck targeting, Swe1p phosphorylation, and Swe1p ubiquitination. To identify such presumed determinants, we sought to isolate SWE1 mutants that were functional but escaped degradation.
The phosphatase Mih1p effectively counteracts Swe1p-mediated Cdc28p inhibition, even when Swe1p is stabilized (McMillan et al., 1999a). However, in mih1Δ strains Swe1p stabilization causes G2 arrest associated with the development of characteristically elongated buds (McMillan et al., 1999a). To identify stabilized mutants of Swe1p, we generated SWE1 mutants using error-prone PCR and screened for mutants that caused G2 arrest in a strain that did not express MIH1 (see MATERIALS AND METHODS for details).
We performed two separate screens targeting overlapping N- or C-terminal portions of Swe1p. The phenotypes of representative mutants are illustrated in Figure 4A. Plasmids were recovered from the selected colonies and retransformed into the starting strain to confirm that the phenotype was due to the mutant SWE1. Plasmids yielding a reproducible G2 arrest on dextrose medium (14 from the N-terminal screen and 25 from the C-terminal screen) were sequenced, revealing that most mutants contained multiple changes, as expected for error-prone PCR. Strikingly, however, all 14 of the mutants from the N-terminal screen contained at least one change within a 13-residue stretch (320-LTNSLQQFKDDLY-332) upstream of the catalytic domain (Figure 4B). Moreover, all 25 of the mutants from the C-terminal screen contained an alteration at one of three residues (E797, I806, or Q807) near the very C terminus of Swe1p (Figure 4B). We constructed clean single mutants with these changes and confirmed that they were responsible for the mutant phenotype (see Figure 4 legend). In addition, we constructed a mutant deleting residues 318–328 in the N-terminal domain, and this mutant (Swe1pΔ1) behaved similarly to the others (Figure 4A). Thus, the mutants highlight two small regions that are required for Swe1p downregulation but not for Swe1p function.
Figure 4.

Isolation and characterization of SWE1 mutants. (A) Plasmids expressing the indicated SWE1 alleles expressed from the SWE1 promoter were transformed into strain JMY1628 (swe1Δ GAL1-MIH1), grown on galactose-containing medium, and plated onto dextrose-containing medium to repress Mih1p expression. Photographs were taken 1 day later. N-1 and C-1 mutants were isolated from the initial screen and contained the L324S (N-1) and Q807R (C-1) substitutions as well as others. Mutants containing those same substitutions but no others were generated by site-directed mutagenesis and displayed similar phenotypes, as did an in-frame deletion of residues 318–328 (Δ1). (B) Schematic of Swe1p showing the regions important for Swe1p degradation. The sequences of mutants N1–N7 and C1–C5 show single amino acid substitutions that caused the phenotype illustrated in A when they were the only mutations present. For mutants N8 and N9, the single substitutions had very mild phenotypes, but the indicated double substitutions produced a robust phenotype. All of those mutants were identified in the screen, reconstructed by site-directed mutagenesis (primer sequences available on request) to eliminate any other changes, and tested as in A. For mutants C6–C8, the indicated changes were identified in mutants from the screen and are presumed responsible for the phenotype but have not been reconstructed.
In principle, these mutants could encode either more stable or more active versions of Swe1p. To ask directly whether the mutants encoded stabilized forms of Swe1p, we performed pulse-chase assays on representative mutants from each class. As shown in Figure 5, both classes of mutants were stabilized relative to wild-type Swe1p, and their behavior was comparable to that of wild-type Swe1p in a strain lacking Hsl7p.
Figure 5.

Stability of Swe1p variants. (A) Pulse-chase analysis. Strains containing myc-tagged wild-type or mutant SWE1 expressed from the GAL1 promoter were analyzed as described in Figure 1C, except that cells were grown and treated at a uniform 30°C with no temperature shift. Strains were JMY1765 (GAL1-SWE1myc), JMY1768 (GAL1-SWE1myc hsl7Δ), JMY1764 (GAL1-SWE1-Δ1myc), JMY1766 (GAL1-SWE1-I806Tmyc), and JMY1857 (GAL1-SWE1-Q807Rmyc). All strains contained an integrated copy of CDC28Y19F to minimize any effects of Swe1p activity during the experiment. (B) Quantitation of the experiment shown in A.
In our strain background, hsl7Δ cells do not experience a significant G2 delay during exponential growth, because the presence of active Mih1p phosphatase counteracts the inhibitory effect of the stabilized Swe1p. However, these cells are sensitized to the level of Swe1p, and doubling the SWE1 gene dosage causes a G2 delay associated with mild bud elongation reflecting the degree of Swe1p activity (McMillan et al., 1999a). When two copies of the new SWE1 mutants were introduced into the sensitized hsl7Δ strain, there was no discernible difference between the mutant and wild-type Swe1p in the degree of bud elongation (and by inference in their ability to promote G2 delay; Figure 6). This indicates that the mutants impair a single pathway that targets Swe1p for Hsl7p-dependent degradation.
Figure 6.

Effect of SWE1 mutants in the hsl7Δ background. Strains containing two integrated copies of wild-type or mutant SWE1 as indicated were grown to exponential phase in YEPD at 30°C and photographed. Cell elongation is a measure of the G2 delay introduced by SWE1. Strains were JMY1735 (HSL7 2X WT), JMY1805 (hsl7Δ 2X WT), JMY1807 (hsl7Δ 2X Δ1), and JMY1809 (hsl7Δ 2X Q807R).
Localization of Stabilized Swe1p Variants
Stabilization of Swe1p in strains expressing the mutants could reflect a failure in nuclear export (trapping Swe1p in the nucleus), a failure in neck targeting (so that cytoplasmic Swe1p can return to the nucleus), or a failure to mark Swe1p for destruction once it reaches the neck. To distinguish between these possibilities, we examined the localization of the Swe1p variants encoded by representative mutants from each class. Swe1pΔ1 was present in the nucleus but was no longer detected at the neck (Figure 7A), suggesting a defect in either nuclear export or neck targeting. In contrast, Swe1pQ807R and Swe1pE797K were still localized both to the nucleus and to the neck just like wild-type Swe1p (Figure 7A), suggesting a defect at a later step in the degradation pathway.
Figure 7.

Localization of Swe1p variants. (A) Strains containing two integrated copies of myc-tagged wild-type or mutant SWE1 as indicated were grown to exponential phase in YEPD at 30°C, fixed, and processed to visualize Swe1p by immunofluorescence. Examples of neck-localized Swe1p are indicated by arrowheads. Strains were JMY1735 (WT), JMY1736 (Δ1), JMY1742 (Q807R), and JMY1743 (E797K) (unpublished data). Localization of the I806T mutant was similar to that of the Q807R and E797K mutants. (B) Wild-type or mutant SWE1 as indicated were fused to two tandem NES sequences, and the localization of the Swe1p-NES constructs was assessed as in A. Top panels: control fusions with inactivating mutations in the NES elements. Bottom panels: active NES fusions. Strains were JMY1737 (WT; active NES), JMY1738 (WT; inactive NES), JMY1739 (Δ1; active NES), and JMY1740 (Δ1; inactive NES).
The failure of Swe1pΔ1 to localize to the neck might stem from a specific defect in Hsl1p/7p-dependent neck targeting or from an earlier block in Swe1pΔ1 nuclear export. Because we do not know what pathways are used by Swe1p to enter or leave the nucleus, we cannot directly assay the nuclear export kinetics of Swe1pΔ1 compared with wild-type Swe1p. However, we reasoned that if the only defect in Swe1pΔ1 was an inability to be exported from the nucleus, then it should be possible to restore Swe1pΔ1 neck targeting by forcing it out of the nucleus. To test whether that was the case, we fused two strong nuclear export sequences (NES) derived from PKI to the C terminus of wild-type or mutant Swe1p. As a control we constructed similar fusions with a similar sequence containing inactivating mutations in the NES elements. The control inactive NES did not alter the localization of the Swe1p (either wild-type or Δ1 mutant) to which it was fused (Figure 7B), as expected. Swe1p (either wild-type or Δ1 mutant) fused to the active NES no longer accumulated in the nucleus (Figure 7B), indicating that the appended NES was able to promote effective nuclear export in both cases. However, whereas the wild-type Swe1p-NES was still targeted to the neck, Swe1pΔ1-NES failed to localize to the neck (Figure 7B). This result suggests that the failure of Swe1pΔ1 to accumulate at the neck was due to a defect in neck targeting after nuclear export.
The construction of Swe1p-NES also allowed us to address whether Swe1p nuclear localization was important for Swe1p function. By monitoring the effectiveness of the morphogenesis checkpoint G2 delay in cells treated with the actin-depolymerizing drug latrunculin-B, we found that Swe1p-NES was much less effective at sustaining a checkpoint response than was wild-type Swe1p, indicating that optimal Swe1p function requires Swe1p nuclear localization.
Interaction of Stabilized Swe1p Variants with Hsl7p
Previous studies showed that Swe1p interacts with Hsl7p, both in yeast lysates and in the absence of other yeast proteins (McMillan et al., 1999a; Shulewitz et al., 1999; Cid et al., 2001). To assess whether the stabilized variants could similarly bind to Hsl7p, we made lysates from strains overexpressing myc-tagged wild-type or mutant Swe1p and incubated those lysates with recombinant GST-Hsl7p bound to glutathione beads. Swe1pΔ1 displayed significantly reduced binding to recombinant Hsl7p compared with wild-type Swe1p, Swe1pI806T, or Swe1pQ807R (Figure 8A). This result suggests that the stabilization of Swe1pΔ1 (and by inference that of the other N-terminal mutants) is due to reduced Hsl7p binding, leading to a failure in neck targeting and hence degradation.
Figure 8.

Interaction of Swe1p variants with Hsl7p. (A) Strains containing myc-tagged wild-type or mutant SWE1 expressed from the GAL1 promoter were grown in galactose-containing medium to induce Swe1p expression. Lysates containing Swe1p (Input) were incubated together with beads bound to recombinant bacterially produced GST or GST-Hsl7p as indicated, and the beads were washed and processed to detect bound Swe1p (Bound). Strains were JMY1680 (wild-type), JMY1681 (Δ1), JMY1696 (I806T), and JMY1697 (Q807R). B) cdc24-1 strains containing the indicated SWE1 alleles with or without GAL1-HSL7 were grown at 23°C in galactose-containing medium to induce Hsl7p expression, arrested in G1 with α-factor, and released from the arrest at 37°C to allow cell cycle progression but block bud formation. Aliquots were removed at 30-min intervals, processed to visualize nuclei, and scored to assess the timing of nuclear division. Strains were DLY690 (swe1Δ), JMY1690 (WT−), JMY1718 (WT+), JMY1691 (Δ1−), JMY1719 (Δ1+), JMY1722 (Q807R−), and JMY1756 (Q807R+). (C) Schematic of truncated Swe1p derivatives and summary of Hsl7p binding and localization results. On the diagram of full-length Swe1p, the gray box indicates Swe1p catalytic domain, black bar indicates residues 320–332 identified in the screen as a degradation determinant, and light gray strip at right indicates C-terminal 12myc epitope tag. Hsl7p binding was assessed by coimmunoprecipitation (see part E) and localization was assessed by immunofluorescence. (D) Expression of truncated Swe1p-myc derivatives. Strains DLY4267 (Swe1p1–510), DLY4268 (Swe1p1–310), DLY4269 (Swe1p1–250), DLY4266 (Swe1p1–123), DLY4272 (Swe1p311–819), DLY4270 (Swe1p511–819), and DLY4271 (Swe1p757–819) were induced to express the truncated Swe1p-myc derivatives by growth in galactose-containing medium, and expression was monitored by Western blotting. (E) Coimmunopre-cipitation between Swe1p1–510myc and Hsl7p-HA. Strains DLY4034 (HSL7-HA), DLY4267 (HSL7-HA SWE11–510myc), and DLY3584 (SWE11–510myc) were grown as above and lysed. Immunoprecipitates using α-myc (upper) or anti-HA (lower) antibodies were washed, separated by SDS-PAGE, and immunoblotted for Hsl7p-HA (upper) or Swe1p1–510-myc (lower).
To test whether Swe1pΔ1 was indeed resistant to the action of Hsl7p, we made use of the observation that overexpression of Hsl7p partially overrides the morphogenesis checkpoint G2 delay in cdc24 mutants that fail to form a bud (McMillan et al., 1999a). Whereas Hsl7p overexpression effectively reduced the G2 delay in cdc24 cells containing wild-type Swe1p or Swe1pQ807R, Hsl7p overexpression had no effect on cells containing Swe1pΔ1 (Figure 8B). Together, these data indicate that the N-terminal domain identified in the screen is important for Swe1p interaction with Hsl7p and that this interaction is critical for targeting Swe1p for degradation.
To delineate more precisely the requirements for Swe1p-Hsl7p interaction and neck targeting, we expressed a series of N- or C-terminally truncated Swe1p-myc derivatives in yeast, and assessed their ability to coimmunoprecipitate with Hsl7p-HA as well as their localization (Figure 8, C–E). An N-terminal Swe1p fragment, Swe1p1–510, was able to bind Hsl7p (Figure 8E), indicating that the bulk of the catalytic domain is dispensable for this interaction. In contrast, neither Swe1p1–310 nor Swe1p311–819 was able to bind Hsl7p, consistent with the existence of an interaction domain in the vicinity of residue 310, very close to the degradation determinant (residues 320–332) identified in the screen. Despite its ability to bind to Hsl7p, Swe1p1–510 was not detected at the bud neck, suggesting that Hsl7p interaction is not sufficient to promote effective neck targeting of Swe1p.
Interaction of C-terminal Mutants with Other Swe1p Regulators
The C-terminal mutants identified in our screens do not appear to affect interaction of Swe1p with Hsl7p, they are still downregulated by Hsl7p overexpression, and they are still targeted to the neck. Given our data suggesting that Clb/Cdc28p functions after neck targeting in the Swe1p degradation pathway, we hypothesized that these mutants might be resistant to the action of Clb/Cdc28p. However, we found that Swe1pQ807R interacted as well as wild-type Swe1p with Clb2p (Figure 3D) and that Swe1pQ807R was phosphorylated at least as well as wild-type Swe1p by Clb2p/Cdc28p (Figure 3A). Moreover, partial digestion of Clb/Cdc28p-phosphorylated Swe1p by V8 protease revealed no differences in the pattern of phospho-peptides produced from Swe1pQ807R or from wild-type Swe1p (Figure 3B). Thus, there is no evidence to suggest that the stability of Swe1pQ807R arises from a resistance to phosphorylation by Clb/Cdc28p.
Another regulator that may act on Swe1p is the polo-related kinase Cdc5p, as Swe1p was identified in a two-hybrid screen for proteins that interact with Cdc5p, and overexpression of Cdc5p downregulated Swe1p (Bartholomew et al., 2001). We had identified Cdc5p ourselves in a two-hybrid screen for proteins that interact with Swe1p (unpublished results), but using these constructs we found no difference in the interaction of wild-type Swe1p or Swe1pQ807R with Cdc5p (Figure 9). Thus, there is no evidence to suggest that the stability of Swe1pQ807R arises from a defect in interacting with any of the known regulators of Swe1p.
Figure 9.
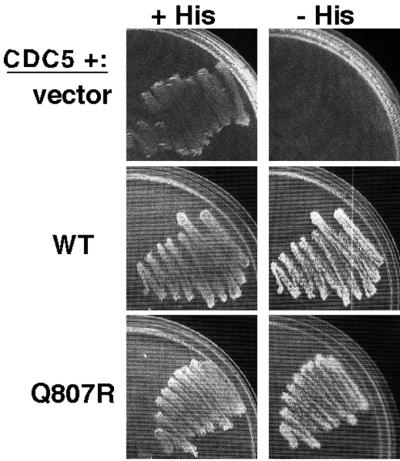
Two-hybrid analysis of Swe1p-Cdc5p interaction. Analysis was performed as described in Figure 3 but with a “prey” plasmid containing C-terminal Cdc5p-encoding sequences.
DISCUSSION
The results presented above have clarified several aspects of the Swe1p degradation pathway in yeast and identified two critical degradation determinants on Swe1p. Implications of these findings for the various steps in the pathway are discussed below.
Nuclear Export of Swe1p
Before bud emergence, Swe1p is concentrated within the nucleus, suggesting that neck targeting after bud emergence involves nuclear export of Swe1p (Longtine et al., 2000). Nuclear export of large proteins (like Swe1p) is mediated by association with “exportins,” most prominently Xpo1p/Crm1p (Gorlich and Kutay, 1999). The Swe1p sequence does not contain an obvious leucine-rich NES of the type that binds Crm1p, but we expected to isolate mutants in our screen that impaired Swe1p-exportin interactions and stabilized Swe1p by trapping it in the nucleus. However, the two domains identified by our 39 mutants appear to define determinants for neck targeting (amino acids 320–332) and subsequent events (amino acids 797–807), rather than nuclear export. There are at least three possible reasons for this surprising absence. First, it is possible that Swe1p harbors two or more redundant NES elements, so that simultaneous inactivating mutations would be required to block export. Second, it is possible that Swe1p nuclear export is important for Swe1p function (as well as for Swe1p degradation) or that the presumed Swe1p NES overlaps with an important functional determinant of Swe1p. Because our screen demanded that the stabilized Swe1p be fully functional, we would not have identified mutations that impaired both nuclear export and function of Swe1p. Third, it is possible that Swe1p nuclear export, as well as neck targeting, is mediated through interaction with Hsl7p. We isolated several mutations defective in Hsl7p interaction and neck targeting, and it is conceivable that these mutations also blocked nuclear export.
Neck Targeting of Swe1p
Neck targeting of Swe1p was previously suggested to be important for Swe1p degradation based on the finding that Hsl1p and Hsl7p (themselves localized to the bud side of the neck) were required for both neck targeting and degradation of Swe1p (McMillan et al., 1999a; Longtine et al., 2000). Moreover, mutations that impaired septin organization reduced or eliminated neck targeting and caused a Swe1p-dependent cell cycle delay, suggesting that neck localization was important for Swe1p degradation (Longtine et al., 2000). We isolated 14 SWE1 alleles bearing mutations in a small region of Swe1p (amino acids 320–332, N-terminal to the catalytic domain). Deletion of this domain impaired Swe1p interaction with Hsl7p, eliminated detectable neck targeting of Swe1p (even if Swe1p was forced out of the nucleus with a strong ectopic NES), and blocked Swe1p degradation. These results strongly support the hypothesis that interaction of Swe1p with Hsl7p is important for Swe1p neck targeting and degradation and identify a key region of Swe1p required for that interaction.
Phosphorylation of Swe1p
Swe1p is phosphorylated at many sites in a cell-cycle–dependent manner (Sia et al., 1998; Sreenivasan and Kellogg, 1999). Our data support the hypothesis that much of that phosphorylation is carried out by Cdc28p, but several unresolved questions remain. Which Cdc28p complexes are responsible for Swe1p phosphorylation in vivo? Where in the cell does that phosphorylation occur? And what role does that phosphorylation play (if any) in Swe1p degradation? We found that Clb/Cdc28p activity was required for a postneck-targeting step in Swe1p degradation, but it is not clear whether that reflects a direct requirement to phosphorylate Swe1p at some specific site(s) or an indirect requirement for Clb/Cdc28p to act on some other Swe1p regulator.
If there were a specific site on Swe1p whose phosphorylation was key to Swe1p degradation, we would expect mutants altering that site to have emerged from our screen. We identified 25 mutants that blocked degradation at a step after Swe1p neck targeting, and each of these altered one of only three residues, none of which are phosphorylatable. Thus, this apparently saturated screen did not identify a key phosphorylation site, perhaps suggesting that Swe1p phosphorylation is not relevant to Swe1p degradation. However, it may be that phosphoryation at any of several Swe1p residues suffices to target Swe1p for degradation, so that no individual site is important enough to stabilize Swe1p when it is mutated.
Ubiquitination of Swe1p
Incubation of Swe1p together with a ubiquitination cocktail and a lysate from wild-type yeast cells yields polyubiquitinated Swe1p, suggesting that Swe1p degradation occurs via the ubiquitin-proteasome pathway (Kaiser et al., 1998). Swe1p was stabilized in met30–6 mutants at restrictive temperature, and in vitro ubiquitination was severely reduced when using lysates from either met30 or cdc34 mutant cells, consistent with the hypothesis that Swe1p ubiquitination was carried out by the ubiquitin ligase SCFMet30 in conjunction with the ubiquitin-conjugating enzyme Cdc34p (Kaiser et al., 1998). However, these data were also consistent with an indirect role for Met30p in Swe1p degradation. Subsequent studies indicated that the only essential function of Met30p was to downregulate the transcription factor Met4p (Patton et al., 2000), and we found that Swe1p was no longer stabilized in met30 met4 double mutants, suggesting that the Swe1p stabilization observed in met30 mutants arose indirectly through some action of the deregulated Met4p in those cells. This led us to suspect that the true ubiquitin ligase responsible might be a distinct SCF complex. There are 16 F-box proteins encoded in the yeast genome (Patton et al., 1998), of which only three (Cdc4p, Met30p, and Ctf13p) are essential for viability. Cdc4p is not required for Swe1p degradation (Kaiser et al., 1998), and Ctf13p is thought to function at the kinetochore rather than as a SCF component. Examination of strains bearing deletions of each of the other F-box-protein encoding genes (part of the Yeast Knockout Collection) revealed that none contained elevated levels of Cdc28p Y19 phosphorylation. It remains possible that one of these strains did contain stabilized Swe1p, but that pleiotropic effects of the mutation (e.g., reduced SWE1 transcription or increased Mih1p activity) precluded detection of a corresponding increase in Cdc28p phosphorylation. However, the simplest interpretation of the result is that Swe1p degradation is not dependent on any single F-box protein.
Another ubiquitin ligase responsible for targeting cell-cycle-regulatory proteins for degradation is the multiprotein anaphase-promoting complex (APC). This is currently thought to come in one of two varieties, carrying either the Cdc20p- or Cdh1p-targeting subunits (Morgan, 1999). Previous findings indicated that Swe1p is rapidly degraded in cells arrested at G2/M by the antimicrotubule drug nocodazole (Sia et al., 1998). Cells arrested in this manner have an activated spindle assembly checkpoint (thought to inactivate Cdc20p) and elevated Clb/Cdc28p activity (thought to inactivate Cdh1p), so it seems unlikely that Swe1p degradation could be mediated by either Cdc20p- or Cdh1p-associated APC. Thus, there is currently no obvious candidate to mediate Swe1p ubiquitination.
Conservation of Swe1p Regulatory Domains
The repeated and independent isolation of mutations affecting each of two small regions of Swe1p in our screen strongly indicates that these regions are critical for Swe1p degradation. To assess how widely conserved these functions might be, we compared the sequence of the regions in Swe1p homologues from various species. Ashbya gossypii is a close relative of S. cerevisiae that grows as a filamentous fungus rather than as a yeast. Both regions identified in our screen were highly conserved in A. gossypii Swe1p, whereas flanking regions were less conserved (Figure 10). Thus, it seems likely that similar interactions are involved in regulating Swe1p in these organisms despite their different modes of growth.
Figure 10.

Sequence comparisons of Swe1p degradation regions among fungal Swe1p homologues. ClustalW alignment of full-length protein sequences was carried out using Macvector software: only the regions of interest are shown. Sequences (from the top) are S. cerevisiae SWE1, A. gossypii SWE1, A. nidulans AnkA, S. pombe wee1, and S. pombe mik1. Numbers indicate first residue shown. Red boxes indicate residues that are identical in three or more homologues. Blue boxes indicate residues that are identical in S. cerevisiae and A. gossypii only. (A) N-terminal Hsl7p-binding region. (B) C-terminal region.
When the analysis was extended to the distantly related fungi Aspergillus nidulans and S. pombe, the N-terminal Hsl7p-binding region did not show significant homology (Figure 10A). This is perhaps indicative that Hsl7p-interaction is not a widely conserved strategy for regulation of Swe1p homologues. Consistent with that hypothesis, the S. pombe Hsl7p homolog Skb1 was reported to have quite distinct effects on cell cycle progression in that organism (Gilbreth et al., 1998). On the other hand, it has also been reported that the human Hsl7p homolog, JBP1, can complement the phenotype of both a S. cerevisiae hsl7Δ mutant (Lee et al., 2000) and a S. pombe skb1Δ mutant (Bao et al., 2001), suggesting that both functions are highly conserved. Because our screen demanded that the SWE1 mutants be fully functional as well as nondegradable, we would not have identified mutants that impaired Hsl7p interaction if they simultaneously impaired Swe1p function. Thus, there may be more highly conserved Hsl7p-interaction domains in Swe1p that were missed in the screen.
In contrast to the N-terminal Swe1p degradation determinant, the C-terminal determinant fell within a 50-residue domain that is highly conserved among the fungal Swe1p homologues (Figure 10B). This domain was not detectably conserved in animal or plant Swe1p homologues, suggesting that its function may be specific to fungi. Interestingly, the three specific residues targeted by our screen were by no means the most highly conserved within the domain (Figure 10B), and it is curious that other residues within this domain were not identified in the saturating screen. One possible explanation for this observation is that mutation of the most conserved residues would have caused misfolding of the domain, possibly destabilizing or inactivating the protein. It may be that only mutations affecting the domain's function in Swe1p degradation without causing overall domain misfolding would have survived our screening procedure. If so, then these mutants may specifically target contact sites for Swe1p regulators acting downstream of neck targeting.
CONCLUSION
The studies presented here have identified two determinants of Swe1p that target it for degradation and have significantly revised our understanding of the Swe1p degradation pathway in S. cerevisiae. The importance of the Swe1p-Hsl7p interaction for both neck targeting and degradation of Swe1p was established, but the events that follow neck targeting remain mysterious, and the ubiquitin ligase previously thought to act on Swe1p was not required for Swe1p degradation. The identification of a previously unappreciated domain at the Swe1p C terminus required for its degradation provides an entry point for seeking the Swe1p-interacting factors that act after neck targeting to degrade Swe1p.
ACKNOWLEDGMENTS
We thank Fred Cross, David Morgan, Peter Kaiser, Steve Reed, Nick Edgington, and Bruce Futcher for providing strains and/or constructs; Fred Cross and Doug Kellogg for communicating results before publication; Marcus Darrabie, Robin Davis, and Denise Ribar for their able technical assistance; Chris Holley and John York for help with the protein sequence alignments; Sally Kornbluth for critical reading of the manuscript; and Fred Dietrich and Peter Phillippsen for access to the Ashbya SWE1 sequence. We also thank the members of the Lew and Pringle laboratories for stimulating interactions. This work was supported by National Institutes of Health grant GM53050 and a Leukemia and Lymphoma Society Scholar award to D.J.L.
Footnotes
Article published online ahead of print. Mol. Biol. Cell 10.1091/mbc.E02–05–0283. Article and publication date are at www.molbiolcell.org/cgi/doi/10.1091/mbc.E02–05–0283.
REFERENCES
- Bao S, et al. The highly conserved protein methyltransferase, Skb1, is a mediator of hyperosmotic stress response in the fission yeast Schizosaccharomyces pombe. J Biol Chem. 2001;276:14549–14552. doi: 10.1074/jbc.C100096200. [DOI] [PubMed] [Google Scholar]
- Barral Y, Parra M, Bidlingmaier S, Snyder M. Nim1-related kinases coordinate cell cycle progression with the organization of the peripheral cytoskeleton in yeast. Genes Dev. 1999;13:176–187. doi: 10.1101/gad.13.2.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartholomew CR, Woo SH, Chung YS, Jones C, Hardy CF. Cdc5 interacts with the Wee1 kinase in budding yeast. Mol Cell Biol. 2001;21:4949–4959. doi: 10.1128/MCB.21.15.4949-4959.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi E, Pringle JR. ZDS1 and ZDS2, genes whose products may regulate Cdc42p in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:5264–5275. doi: 10.1128/mcb.16.10.5264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop AC, et al. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature. 2000;407:395–401. doi: 10.1038/35030148. [DOI] [PubMed] [Google Scholar]
- Booher RN, Deshaies RJ, Kirschner MW. Properties of Saccharomyces cerevisiae wee1 and its differential regulation of p34CDC28 in response to G1 and G2 cyclins. EMBO J. 1993;12:3417–3426. doi: 10.1002/j.1460-2075.1993.tb06016.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose I, Irazoqui JE, Moskow JJ, Bardes ES, Zyla TR, Lew DJ. Assembly of scaffold-mediated complexes containing Cdc42p, the exchange factor Cdc24p, and the effector Cla4p required for cell cycle-regulated phosphorylation of Cdc24p. J Biol Chem. 2001;276:7176–7186. doi: 10.1074/jbc.M010546200. [DOI] [PubMed] [Google Scholar]
- Chowdhury S, Smith KW, Gustin MC. Osmotic stress and the yeast cytoskeleton: phenotype-specific suppression of an actin mutation. J Cell Biol. 1992;118:561–571. doi: 10.1083/jcb.118.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cid VJ, Shulewitz MJ, McDonald KL, Thorner J. Dynamic localization of the Swe1 regulator Hsl7 during the Saccharomyces cerevisiae cell cycle. Mol Biol Cell. 2001;12:1645–1669. doi: 10.1091/mbc.12.6.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleveland DW, Fischer SG, Kirscner MW, Laemmli UK. Peptide mapping by limited proteolysis in sodium dodecyl sulfate and analysis by gel electrophoresis. J Biol Chem. 1977;252:1102–1110. [PubMed] [Google Scholar]
- Delley PA, Hall MN. Cell wall stress depolarizes cell growth via hyperactivation of RHO1. J Cell Biol. 1999;147:163–174. doi: 10.1083/jcb.147.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drees BL, et al. A. protein interaction map for cell polarity development. J Cell Biol. 2001;154:549–571. doi: 10.1083/jcb.200104057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunphy WG. The decision to enter mitosis. Trends Cell Biol. 1994;4:202–207. doi: 10.1016/0962-8924(94)90142-2. [DOI] [PubMed] [Google Scholar]
- Edgington NP, Futcher B. Relationship between the function and the location of G1 cyclins in S. cerevisiae. J Cell Sci. 2001;114:4599–4611. doi: 10.1242/jcs.114.24.4599. [DOI] [PubMed] [Google Scholar]
- Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbreth M, Yang P, Bartholomeusz G, Pimental RA, Kansra S, Gadiraju R, Marcus S. Negative regulation of mitosis in fission yeast by the shk1 interacting protein skb1 and its human homolog, Skb1Hs. Proc Natl Acad Sci USA. 1998;95:14781–14786. doi: 10.1073/pnas.95.25.14781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlich D, Kutay U. Transport between the cell nucleus and the cytoplasm. Annu Rev Cell Dev Biol. 1999;15:607–660. doi: 10.1146/annurev.cellbio.15.1.607. [DOI] [PubMed] [Google Scholar]
- Harrison JC, Bardes ES, Ohya Y, Lew DJ. A role for the Pkc1p/Mpk1p kinase cascade in the morphogenesis checkpoint. Nat Cell Biol. 2001;3:417–420. doi: 10.1038/35070104. [DOI] [PubMed] [Google Scholar]
- James P, Halladay J, Craig EA. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics. 1996;144:1425–1436. doi: 10.1093/genetics/144.4.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser P, Flick K, Wittenberg C, Reed SI. Regulation of transcription by ubiquitination without proteolysis: Cdc34/SCF(Met30)-mediated inactivation of the transcription factor Met4. Cell. 2000;102:303–314. doi: 10.1016/s0092-8674(00)00036-2. [DOI] [PubMed] [Google Scholar]
- Kaiser P, Sia RAL, Bardes EGS, Lew DJ, Reed SI. Cdc34 and the F-box protein Met30 are required for degradation of the Cdk-inhibitory kinase Swe1. Genes Dev. 1998;12:2587–2597. doi: 10.1101/gad.12.16.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Cook JR, Pollack BP, Kinzy TG, Norris D, Pestka S. Hsl7p, the yeast homologue of human JBP1, is a protein methyltransferase. Biochem Biophys Res Commun. 2000;274:105–111. doi: 10.1006/bbrc.2000.3049. [DOI] [PubMed] [Google Scholar]
- Lew DJ, Reed SI. A cell cycle checkpoint monitors cell morphogenesis in budding yeast. J Cell Biol. 1995;129:739–749. doi: 10.1083/jcb.129.3.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Li MZ, Leibham D, Cortez D, Elledge SJ. The univector plasmid-fusion system, a method for rapid construction of recombinant DNA without restriction enzymes. Curr Biol. 1998;8:1300–1309. doi: 10.1016/s0960-9822(07)00560-x. [DOI] [PubMed] [Google Scholar]
- Longtine MS, Theesfeld CL, McMillan JN, Weaver E, Pringle JR, Lew DJ. Septin-dependent assembly of a cell-cycle-regulatory module in Saccharomyces cerevisiae. Mol Cell Biol. 2000;20:4049–4061. doi: 10.1128/mcb.20.11.4049-4061.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X-J, Lu Q, Grunstein M. A search for proteins that interact genetically with histone H3 and H4 amino termini uncovers novel regulators of the Swe1 kinase in Saccharomyces cerevisiae. Genes Dev. 1996;10:1327–1340. doi: 10.1101/gad.10.11.1327. [DOI] [PubMed] [Google Scholar]
- McMillan JN, Longtine MS, Sia RAL, Theesfeld CL, Bardes ESG, Pringle JR, Lew DJ. The morphogenesis checkpoint in Saccharomyces cerevisiae: cell cycle control of Swe1p degradation by Hsl1p and Hsl7p. Mol Cell Biol. 1999a;19:6929–6939. doi: 10.1128/mcb.19.10.6929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan JN, Sia RAL, Bardes ESG, Lew DJ. Phosphorylation-independent inhibition of Cdc28p by the tyrosine kinase Swe1p in the morphogenesis checkpoint. Mol Cell Biol. 1999b;19:5981–5990. doi: 10.1128/mcb.19.9.5981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan JN, Sia RAL, Lew DJ. A morphogenesis checkpoint monitors the actin cytoskeleton in yeast. J Cell Biol. 1998;142:1487–1499. doi: 10.1083/jcb.142.6.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael WM, Newport J. Coupling of mitosis to the completion of S phase through Cdc34-mediated degradation of Wee1. Science. 1998;282:1886–1889. doi: 10.1126/science.282.5395.1886. [published erratum appears in Science. 1999;128 3, 1835]. [DOI] [PubMed] [Google Scholar]
- Morgan DO. Regulation of the APC and the exit from mitosis. Nat Cell Biol. 1999;1:E47–E53. doi: 10.1038/10039. [DOI] [PubMed] [Google Scholar]
- Patton EE, Peyraud C, Rouillon A, Surdin-Kerjan Y, Tyers M, Thomas D. SCF(Met30)-mediated control of the transcriptional activator Met4 is required for the G(1)-S transition. EMBO J. 2000;19:1613–1624. doi: 10.1093/emboj/19.7.1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton EE, Willems AR, Tyers M. Combinatorial control in ubiquitin-dependent proteolysis: don't Skp the F-box hypothesis. Trends Genet. 1998;14:236–243. doi: 10.1016/s0168-9525(98)01473-5. [DOI] [PubMed] [Google Scholar]
- Raleigh JM, O'Connell MJ. The G(2) DNA damage checkpoint targets both Wee1 and Cdc25. J Cell Sci. 2000;113:1727–1736. doi: 10.1242/jcs.113.10.1727. [DOI] [PubMed] [Google Scholar]
- Richardson HE, Wittenberg C, Cross F, Reed SI. An essential G1 function for cyclin-like proteins in yeast. Cell. 1989;59:1127–1133. doi: 10.1016/0092-8674(89)90768-x. [DOI] [PubMed] [Google Scholar]
- Shulewitz MJ, Inouye CJ, Thorner J. Hsl7 localizes to a septin ring and serves as an adapter in a regulatory pathway that relieves tyrosine phosphorylation of Cdc28 protein kinase in Saccharomyces cerevisiae. Mol Cell Biol. 1999;19:7123–7137. doi: 10.1128/mcb.19.10.7123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sia RAL, Bardes ESG, Lew DJ. Control of Swe1p degradation by the morphogenesis checkpoint. EMBO J. 1998;17:6678–6688. doi: 10.1093/emboj/17.22.6678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sia RAL, Herald HA, Lew DJ. Cdc28 tyrosine phosphorylation and the morphogenesis checkpoint in budding yeast. Mol Biol Cell. 1996;7:1657–1666. doi: 10.1091/mbc.7.11.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S, Grenfell TZ, Garfield S, Erikson RL, Lee KS. Essential function of the polo box of Cdc5 in subcellular localization and induction of cytokinetic structures. Mol Cell Biol. 2000;20:286–298. doi: 10.1128/mcb.20.1.286-298.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreenivasan A, Kellogg D. The Elm1 kinase functions in a mitotic signaling network in budding yeast. Mol Cell Biol. 1999;19:7983–7994. doi: 10.1128/mcb.19.12.7983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stueland CS, Lew DJ, Cismowski MJ, Reed SI. Full activation of p34CDC28 histone H1 kinase activity is unable to promote entry into mitosis in checkpoint-arrested cells of the yeast Saccharomyces cerevisiae. Mol Cell Biol. 1993;13:3744–3755. doi: 10.1128/mcb.13.6.3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wach A. PCR-synthesis of marker cassettes with long flanking homology regions for gene disruptions in S. cerevisiae. Yeast. 1996;12:259–265. doi: 10.1002/(SICI)1097-0061(19960315)12:3%3C259::AID-YEA901%3E3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]