

Abstract
There is growing evidence that activated synovial fibroblasts, as part of a complex cellular network, play an important role in the pathogenesis of rheumatoid arthritis. In recent years, significant progress has been made in elucidating the specific features of these fibroblasts. It has been understood that although macrophage and lymphocyte secreted factors contribute to their activation, rheumatoid arthritis synovial fibroblasts (RA-SFs) do not merely respond to stimulation by pro-inflammatory cytokines, but show a complex pattern of molecular changes also maintained in the absence of external stimulation. This pattern of activation is characterized by alterations in the expression of regulatory genes and signaling cascades, as well as changes in pathways leading to apoptosis. These together result in the upregulation of adhesion molecules that mediate the attachment of RA-SFs to the extracellular matrix and in the overexpression of matrix degrading enzymes that mediate the progressive destruction of the joints. In addition, activated RA-SFs exert specific effects on other cell types such as macrophages and lymphocytes. While the initiating step in the activation of RA-SFs remains elusive, several key pathways of RA-SF activation have been identified. However, there is so far no single, specific marker for this phenotype of RA-SF. It appears that activated RA-SFs are characterized by a set of specific properties which together lead to their aggressive behavior.
Keywords: fibroblasts, rheumatoid arthritis
Introduction
Rheumatoid arthritis (RA) is a chronic disorder primarily affecting the joints and leading to their progressive destruction. This joint destruction is a unique and most prominent feature of RA that not only distinguishes the disease from other arthritic conditions, but also determines the outcome of RA in most affected individuals [1]. Research in recent years has produced exciting new information about the pathogenesis of RA. Apart from the multitude of specific data that have been published by many investigators, two fundamental concepts have been of particular importance. First, it has been understood that the pathogenesis of RA is characterized by the concerted action of different cell types that, through highly specific communication, create the great variety of signs and symptoms characteristic of RA. Based on this awareness, the initial view of active players and passive responders has been replaced largely by that of several cell types being involved actively in the pathogenesis of disease [2,3]. Second, it has become clear that inflammation and auto-immunity are key features of RA, but do not constitute by far the only relevant mechanisms of disease. Instead, growing evidence suggests that, in addition to macrophages and T cells, activated synovial fibroblasts (SFs) play a major role in both initiating and driving RA [4]. Therefore, these cells have been studied intensively over recent years, and it has been understood that the specific properties of activated RA-SFs not only cause their aggressive behavior, but may also explain some other aspects of RA. By focusing on recent data, this review will outline the current knowledge about the molecular changes in activated RA-SFs and point to the consequences with respect to the pathogenesis of disease.
Morphology and altered behavior of activated RA-SFs
A key observation concerning SFs in RA was the description of different subpopulations within the synovial membrane [5]. It is now well established that RA-SFs in the most superficial layer of the synovium--the lining--differ substantially not only from SFs in normal synovial tissue, but also from SFs in deeper layers of the rheumatoid synovium. These differences appear characteristic for RA and can be seen both in the morphology and in biological properties. Specifically, RA-SFs in the lining layer display numerous features of cellular activation that ultimately result in an aggressive, invasive behavior. At a morphological level, a more rounded shape, a large pale nucleus and prominent nucleoli characterize the activated phenotype of RA-SFs. In culture, these cells grow in an anchorage-independent manner [6] over multiple passages and can escape contact inhibition. The most characteristic feature is the ability of activated RA-SFs to attach to the articular cartilage and deeply invade the extracellular matrix. Apart from the question of what initiates cellular activation, the problem of whether the activation of RA-SFs is an intrinsic property of these cells or is purely a response to inflammatory cytokines has been of crucial importance. Growing evidence interestingly suggests that the changes resulting in the activation (sometimes also called partial transformation) of RA-SFs constitute stable long-term alterations that are maintained even in the absence of pro-inflammatory cytokines. The most convincing piece of evidence supporting this hypothesis has come from the severe combined immunodeficient mouse co-implantation model of RA [7]. In this model, isolated RA-SFs are implanted with normal human cartilage under the renal capsule of severe combined immunodeficient mice. Because of the lack of a functional immune system, these mice do not reject the implants, allowing one to study the interaction of the RA-SFs with the normal articular cartilage. As shown in several studies, RA-SFs attach to and deeply invade the co-implanted cartilage in this in vivo model of RA [7,8,9,10]. In contrast, normal SFs, osteoarthritis SFs or dermal fibroblasts do not show such an invasive behavior. This experimental setting investigates the invasive behavior of RA-SFs in the absence of human inflammatory cells and cytokines; therefore, the differences in the behavior of RA-SFs, osteoarthritis SFs and normal SFs have been taken as evidence for the stable, intrinsic activation of RA-SFs.
Alterations in signaling pathways and apoptosis
Several lines of evidence indicate that both the altered morphology and the aggressive behavior of RA-SFs result from specific changes in the transcription of disease-relevant genes and in intracellular signaling cascades. Among the factors involved in the activation of RA-related genes, the AP-1 transcription factor is of special interest. It consists of the c-Fos and c-Jun elements, and is key in signaling pathways that ultimately lead to the degradation of cartilage and bone. Several studies have shown that c-fos is overexpressed in the RA synovium [11,12,13,14]. Asahara et al analyzed nuclear extracts from synovial cells and found a high DNA-binding activity of AP-1 in the tissues of RA patients, but negligible activity in osteoarthritis tissue specimens [15]. This is important because the promoters of several disease-relevant genes such as those encoding matrix metalloproteinases (MMPs) contain binding sites for AP-1. In addition, the AP-1 transcription factor is involved in the tissue-specific expression of MMPs [16]. Interestingly, AP-1 activity is found predominantly in fibroblast-like cells, and correlates with both the in situ expression of c-fos and c-jun mRNA and disease activity [15].
The transcription factor NF-κ B is also activated in RA-SFs [17], and appears to be important in perpetuating disease as well as in mediating synovial inflammation [17,18,19,20]. Not only is NF-κ B induced by several pro-inflammatory cytokines, but also its activation conversely stimulates the transcription of disease-relevant genes such as those encoding adhesion molecules and cytokines. Consequently, overexpression of Iκ Ba, the natural inhibitor of NF-κ B, leads to the downregulation of pro-inflammatory cytokines. In addition, recent data have linked NF-κ B with reduced apoptosis of synovial cells [17]. Like AP-1, NF-κ B induces the expression of MMPs [21], but it is unclear which regulatory pathway contributes most significantly to the altered expression of MMPs in RA.
Tumor suppressor genes have recently become of interest in explaining important features of cellular activation of RA-SFs. Thus, alterations in the expression and function of p53 have been associated with some important features of RA-SFs because Firestein et al described somatic mutations of p53 in RA-SFs [22]. The predicted amino acid substitutions, interestingly, are similar to those commonly observed in a variety of tumors. It was concluded from these data that, by resulting in a nonfunctional `cell death suppressor', such mutations could contribute to an extended life span of activated cells aggressively destroying cartilage and bone. Further evidence supporting this hypothesis has emerged from recent observations demonstrating that the inhibition of p53 causes the transformation of normal SFs into cells with aggressive behavior characteristic of that seen in RA [23]. As shown in a study by Kullmann et al, RA patients from another part of the world did not reveal mutations of p53 [24]; therefore, it may be hypothesized that inhibition of p53 constitutes only part of the mechanism of cellular activation in RA, and that the pattern of cellular changes differs within the affected patients.
Our laboratory recently demonstrated that aggressive RA-SFs lack the expression of mRNA for the novel tumor suppressor PTEN [10], the protein product of which exhibits tyrosine phosphatase activity as well as extensive homology to the cytoskeletal proteins tensin and auxillin [25]. In situ hybridization on a RA synovium revealed a distinct expression pattern of PTEN, with negligible staining in the lining layer but abundant expression in the sublining. In contrast, normal synovial tissue exhibited homogeneous staining for PTEN [10]. Only 40% of cultured RA-SFs expressed PTEN, and severe combined immunodeficient mouse experiments showed no staining for PTEN in those cells aggressively invading the cartilage. A recent study of interest by Di Cristofano et al demonstrated that PTEN is involved critically not only in the aggressive behavior of cells, but also as an essential mediator of Fas response [26], providing a further link between invasiveness and resistance to apoptosis in activated cells.
The question of whether the massive synovial hyperplasia characteristic for RA is mainly due to the proliferation of RA-SFs or to defective apoptosis is still a matter for discussion. Over recent years, evidence has been accumulated for both hypotheses. On the basis of the well-known phenomenon that RA-SFs can grow very fast over multiple passages in vitro, cell proliferation has been considered the major mechanism for the hyperplastic growth of the RA synovium. Support for this notion has come from the demonstration that key regulators of cellular proliferation, such as the proto-oncogene c-myc, are overexpressed in RA [27]. In addition, recent data from Hashiramoto et al and from our own laboratory indicate that inhibition of c-myc through gene transfer reduces the growth of RA-SFs [9,28]. In the context of synovial cell proliferation, interesting data have arisen from a study by Taniguchi et al [29], which show that gene transfer with the p16INK4asenescence gene only inhibited the growth of SFs in vitro, but ameliorated the course of rat adjuvant arthritis as well when applied in vivo [29].
The hypothesis that proliferation of activated RA-SFs constitutes the major driving force of synovial hyperplasia has been challenged by observations indicating SFs do not proliferate faster than cells from osteoarthritis patients [30]. Studies using 3H-thymidine incorporation revealed a proliferation rate of only 1-5% [31], and mitosis is also rarely found in RA synovial tissue [32]. There is accumulating evidence that an alteration in apoptosis contributes significantly to the synovial hyperplasia in RA. Despite earlier reports, based on results from experiments using DNA labeling techniques, suggesting an increased rate of apoptosis among RA-SFs [33], recent data have demonstrated that apoptosis is a rare event in the rheumatoid synovium [34]. The lack of apoptosis in RA-SFs appears to have two aspects: the missing ability of RA-SFs to complete pathways initiated by pro-apoptotic stimuli, and the partial resistance to such stimuli. The former is documented by the aforementioned discrepancy between the high number of DNA strand breaks and the low frequency of completed apoptosis. The resistance of RA-SFs to apoptosis is caused by the prominent upregulation of anti-apoptotic molecules in RA-SFs. In this context, the induction of apoptosis through the Fas/Apo-1/CD95 pathway is of particular interest [35]. Several reports have demonstrated that RA-SFs are susceptible to Fas-induced apoptosis [35]. This appears to apply only to about 20% of RA-SFs however; the majority are not only unable to complete apoptosis, but are also resistant to the induction of apoptosis through the Fas pathway. Recent data indicate that this specific feature of RA-SFs is linked to the expression of several anti-apoptotic molecules that counteract key pathways in the apoptosis cascade. In this context, we have demonstrated most recently the overexpression of sentrin in RA-SFs [36]. Sentrin (also called SUMO-1) is a ubiquitin-like protein that has been shown to interact with the signal-competent forms of Fas/Apo-1 and tumor necrosis factor (TNF) receptor-1, and thereby to protect cells against apoptosis mediated by Fas and TNF [37]. Sentrin is, interestingly, found predominantly in RA-SFs of the lining layer. In contrast, normal synovial cells express very little sentrin [36]. Quantitative analysis of sentrin expression in cultured SFs revealed an increase of sentrin mRNA expression of greater than 30-fold as compared with osteoarthritis SFs and normal fibroblasts [36]. Taken together, alteration in the ability to undergo apoptosis may not only play a role in the hyperplasia of the rheumatoid synovium, but also, even more importantly, extend the life span of activated RA-SFs at sites of matrix degradation, thus contributing indirectly to the destruction of articular structures.
Adhesion molecules
One crucial consequence of the activation of RA-SFs is the expression and upregulation of different adhesion molecules that mediate the attachment of these cells to the cartilage. Among the different adhesion molecules that contribute to this process, β 1 integrins appear to be involved most significantly. The expression of β 1 integrins such as VLA-3, VLA-4 and VLA-5 on SFs has been demonstrated in several studies [38,39]. Rinaldi et al also demonstrated that antibodies against β 1 integrin inhibited, at least in part, the binding of RA-SFs to the extracellular matrix [38]. Several integrins function as fibronectin receptors, and therefore it has been suggested that the fibronectin-rich environment of the RA cartilage surface facilitates the adhesion of RA-SFs to the articular cartilage. This notion was supported by data from Müller-Ladner et al showing that CS-1, a spliced isoform of fibronectin, is expressed highly in the RA synovium [40]. However, integrins not only function as receptor molecules, but also interact with several signaling pathways involved in the pathogenesis of RA [41]. The expression of early cell cycle genes such as c-fos and c-myc is specifically stimulated by integrin-mediated cell adhesion. Gene expression driven by the fos promoter shows strongly synergistic activation by integrin-mediated cell adhesion [42,43]. These data contribute to the understanding that the role of adhesion molecules in RA is not restricted to the attachment of the synovium to cartilage and bone, but involves complex regulatory functions including the recruitment of inflammatory cells as well as the induction of MMP expression.
Role of RA-SFs in matrix degradation
It is now well understood that RA-SFs contribute significantly to matrix degradation in RA through the expression of matrix degrading enzymes such as MMPs [44] and cathepsins [45] (Fig. 1). Interest has so far focused mainly on the MMPs, and numerous reports have implicated MMPs in rheumatoid joint destruction [46,47,48,49,50]. MMPs have not only been found at elevated levels in synovial fluid of RA patients [51], but in situ studies have revealed strong expression of MMPs in the rheumatoid synovium [50,52]. RA-SFs in the lining layer or at the sites of cartilage invasion have been identified as the major source of MMPs. This pattern of distribution is similar to that for proto-oncogenes and consistent with the notion that proto-oncogenes are involved in the activation of MMP genes. It was also shown that the expression levels of some MMPs, such as MMP-13, correlate with elevated levels of systemic inflammation markers [53]. The recently described membrane type (MT)-MMPs have also been found in activated RA-SFs of the lining layer [54]. This is important because some of the known MT-MMPs not only degrade extracellular matrix components, but also activate other disease-relevant MMPs, such as MMP-2 and MMP-13 [55]. In a recent study that compared the expression of MT-MMPs in RA, we found that MT1-MMP was expressed most intensely in fibroblasts and macrophages, and also in osteoclast-like cells at sites of bone resorption [54]. From this study, we concluded that MT1-MMP and, potentially, MT3-MMP contribute significantly to matrix degradation in RA.
Figure 1.
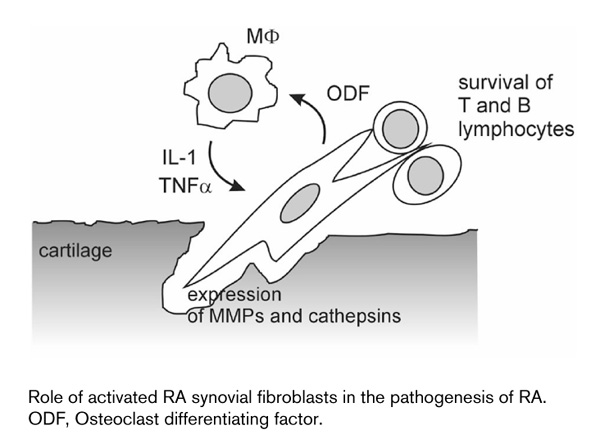
Role of activated RA synovial fibroblasts in the pathogenesis of RA. ODF, Osteoclast differentiating factor.
Cathepsins are another major group of proteases involved in joint destruction [45]. The cysteine proteases cathepsin B and cathepsin L, particularly, have been shown to be activated by proto-oncogenes and upregulated in RA synovium at sites of cartilage invasion [56]. The finding that transfection of fibroblasts with the ras proto-oncogene leads to cellular transformation accompanied by the induction of cathepsin L [57] agrees with data demonstrating the combined expression of ras and cathepsin L in RA-SFs [58]. Several studies have also shown that pro-inflammatory cytokines such as interleukin-1 (IL-1) and TNFα can stimulate the production of cathepsins B and L by SF-like cells [59,60]. Recent data have also implicated cathepsin K in the matrix degradation by RA-SFs through demonstrating the expression of cathepsin K in RA-SFs and macrophages [61].
Interaction of RA-SFs with other cell types
Although it is now believed that the activation of RA-SFs is caused by a set of events and can be maintained in the absence of human inflammatory cytokines, pro-inflammatory cytokines play a key role in the stimulation of RA-SFs towards an aggressive behavior (Fig. 1). Among the variety of cytokines and factors that have been shown to enhance the activation and destructive potential of RA-SFs, TNFα and IL-1 are of particular importance and have been investigated in detail. This has led to the development of novel therapeutic strategies targeting these cytokines. The delivery of TNFα receptor fusion proteins as well as novel monoclonal antibodies against TNFα has provided the most promising results [62,63,64]. It was demonstrated that administration of anti-TNFα antibodies results not only in an improvement of inflammation, but also in a reduction in the levels of MMPs in the sera of RA patients. In this context, data from clinical trials indicate that, in addition to reducing inflammation, such treatment in combination with methotrexate may impact on the progression of radiological changes (RN Maini, personal communication). However, the complexity of RA-SF activation as already outlined raises the question of to what extent inhibition of TNFα can influence the long-term outcome of disease. This is because recent data indicate that TNFα may exert different effects on different subtypes of RA-SF through a variation in the distribution of TNF receptor variants in the lining and sublining [65]. Data from animal models of RA are more intriguing, suggesting that, despite a certain overlap in the effect of TNFα and IL-1, there are distinct differences with respect to inflammation and matrix degradation [66]. While TNFα appears to have a more pronounced effect on the extent of synovitis, IL-1 may have a greater impact on joint destruction. As demonstrated by Bendele et al, the combination of methotrexate and anti-IL-1 treatment resulted in a 97% reduction of bone resorption [67].
The notion that RA-SFs not only react passively to pro-inflammatory stimuli but also contribute actively to the pathogenesis of RA is supported by recent findings demonstrating a significant effect of RA-SFs on macrophages and lymphocytes in the RA synovium. It appears that RA-SFs play a significant role in the activation of macrophages in the lining and their differentiation into multinucleated, osteoclast-like bone resorbing cells. As cited in a review by Goldring and Gravallese published in Arthritis Research, recent studies have identified a potent regulator of osteoclastogenesis, the osteoclast differentiating factor in the RA synovium [68]. Shigeyama et al interestingly demonstrated that RA-SFs produce large amounts of osteoclast differentiating factor in vivo, which correlates with the ability of these cells to induce osteoclastogenesis in peripheral blood monocytes in vitro [69]. Thus, RA-SFs contribute to the degradation of the extra-cellular matrix not only directly through the release of matrix degrading enzymes, but also indirectly through effecting the differentiation and activation of osteoclasts (Fig. 1). This close cooperation of macrophages and fibroblasts in the RA synovium is documented further by Hamann et al [70], who demonstrated that the cooperation of synovial macrophages and fibroblasts is also mediated through direct cell-cell interactions via ligation of CD55 on RA-SFs with CD97 on macrophages. These interactions appear to take place predominantly in the synovial lining that mediates the progressive destruction of cartilage and bone.
Other data indicate that RA-SFs also influence the accumulation and survival of lymphocytes in the RA synovium. Franz et al demonstrated that RA-SFs constitute an important source of IL-16, which has been demonstrated to be chemoattractive for CD4-positive T cells [71]. IL-16 also appears responsible--at least in part--for the anergic state of T cells in the synovial tissue [72]. Interesting data concerning the effects of RA-SFs have also emerged from a study by Salmon et al demonstrating that isolated synovial T cells rapidly undergo programmed cell death, whereas a co-culture with SFs prevented apoptosis [73]. SFs derived from patients with RA not only prevent T cells from apoptosis: elegant studies by de Groot and co-workers have shown that RA-SFs also prevent B lymphocytes from programmed cell death [74].
Conclusion
On the basis of these data, it can be concluded that RA-SFs contribute significantly to the survival of both B and T lymphocytes in the diseased synovium, and thereby maintain inflammation (Fig. 1). Based on the fact that RA-SFs are pivotal players in the pathogenesis of joint destruction, we have transferred specific genes to these cells [8,9,75] to explore the distinct pathways of synovial cell activation [76,77].
References
- Gay S, Gay RE, Koopman WJ. Molecular and cellular mechanisms of joint destruction in rheumatoid arthritis: two cellular mechanisms explain joint destruction? Ann Rheum Dis. 1993;52 (suppl 1):S39–S47. doi: 10.1136/ard.52.suppl_1.s39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firestein GS. Invasive fibroblast-like synoviocytes in rheumatoid arthritis. Passive responders or transformed aggressors? Arthritis Rheum. 1996;39:1781–1790. doi: 10.1002/art.1780391103. [DOI] [PubMed] [Google Scholar]
- Pap T, Franz JK, Gay RE, Gay S. Has research on lymphocytes hindered progress in rheumatoid arthritis? In Challenges in Rheumatoid Arthritis Edited by Bird H, Snaith M Oxford: Blackwell Science; 1999.
- Franz JK, Pap T, Müller-Ladner U, Gay RE, Burmester GR, Gay S. T-cell independent joint destruction. . In T Cells in Arthritis Edited by Miossec P, van-den BW, Firestein GS Basel: Birkenhäuser, 1998. pp. 55–74.
- Fassbender HG. Histomorphological basis of articular cartilage destruction in rheumatoid arthritis. . Coll Relat Res. 1983;3:141–155. doi: 10.1016/s0174-173x(83)80040-5. [DOI] [PubMed] [Google Scholar]
- Lafyatis R, Remmers EF, Roberts AB, Yocum DE, Sporn MB, Wilder RL. Anchorage-independent growth of synoviocytes from arthritic and normal joints. Stimulation by exogenous platelet-derived growth factor and inhibition by transforming growth factor-beta and retinoids. . J Clin Invest. 1989;83:1267–1276. doi: 10.1172/JCI114011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller-Ladner U, Kriegsmann J, Franklin BN, Matsumoto S, Geiler T, Gay RE, Gay S. Synovial fibroblasts of patients with rheumatoid arthritis attach to and invade normal human cartilage when engrafted into SCID mice. . Am J Pathol. 1996;149:1607–1615. [PMC free article] [PubMed] [Google Scholar]
- Müller-Ladner U, Roberts CR, Franklin BN, Gay RE, Robbins PD, Evans CH, Gay S. Human IL-1Ra gene transfer into human synovial fibroblasts is chondroprotective. . J Immunol. 1997;158:3492–3498. [PubMed] [Google Scholar]
- Nawrath M, Hummel KM, Pap T, Müller-Ladner U, Gay RE, Moelling K, Gay S. Effect of dominant negative mutants of raf -1 and c-myc on rheumatoid arthritis synovial fibroblasts in the SCID mouse model [abstract]. Arthritis Rheum. 1998;41:S95–S95. [Google Scholar]
- Pap T, Franz JK, Hummel KM, Jeisy E, Gay RE, Gay S. Activation of synovial fibroblasts in rheumatoid arthritis: lack of expression of the tumour suppressor PTEN at sites of invasive growth and destruction. . Arthritis Res. 2000;2:59–65. doi: 10.1186/ar69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooley S, Herlitzka I, Hanselmann R, Ermis A, Henn W, Remberger K, Hopf T, Welter C. Constitutive expression of c-fos and c-jun, over-expression of ets-2, and reduced expression of metastasis suppressor gene nm23-H1 in rheumatoid arthritis. Ann Rheum Dis. 1996;55:298–304. doi: 10.1136/ard.55.5.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimbacher B, Aicher WK, Peter HH, Eibel H. Measurement of transcription factor c-fos and EGR-1 mRNA transcription levels in synovial tissue by quantitative RT-PCR. Rheumatol Int. 1997;17:109–112. doi: 10.1007/s002960050018. [DOI] [PubMed] [Google Scholar]
- Knotny E, Ziolkowska M, Dudzinka E, Filipowicz-Sosnowska A, Ryzewska A. Modified expression of c-Fos and c-Jun proteins and production of interleukin-1 beta in patients with rheumatoid arthritis. . Clin Exp Rheumatol. 1995;13:51–57. [PubMed] [Google Scholar]
- Trabandt A, Aicher WK, Gay RE, Sukhatme VP, Fassbender HG, Gay S. Spontaneous expression of immediately-early response genes c-fos and egr-1 in collagenase-producing rheumatoid synovial fibroblasts. Rheumatol Int. 1992;12:53–59. doi: 10.1007/BF00300977. [DOI] [PubMed] [Google Scholar]
- Asahara H, Fujisawa K, Kobata T, Hasunuma T, Maeda T, Asanuma M, Ogawa N, Inoue H, Sumida T, Nishioka K. Direct evidence of high DNA binding activity of transcription factor AP-1 in rheumatoid arthritis synovium. Arthritis Rheum. 1997;40:912–918. doi: 10.1002/art.1780400520. [DOI] [PubMed] [Google Scholar]
- Benbow U, Brinckerhoff CE. The AP-1 site and MMP gene regulation: what is all the fuss about? Matrix Biol. 1997;15:519–526. doi: 10.1016/s0945-053x(97)90026-3. [DOI] [PubMed] [Google Scholar]
- Miagkov AV, Kovalenko DV, Brown CE, Didsbury JR, Cogswell JP, Stimpson SA, Baldwin AS, Makarov SS. NF-kappaB activation provides the potential link between inflammation and hyperplasia in the arthritic joint. . Proc Natl Acad Sci USA. 1998;95:13859–13864. doi: 10.1073/pnas.95.23.13859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Z, Boyle DL, Manning AM, Firestein GS. AP-1 and NF-kappaB regulation in rheumatoid arthritis and murine collagen-induced arthritis. . Autoimmunity. 1998;28:197–208. doi: 10.3109/08916939808995367. [DOI] [PubMed] [Google Scholar]
- Marok R, Winyard PG, Coumbe A, Kus ML, Gaffney K, Blades S, Mapp PI, Morris CJ, Blake DR, Kaltschmidt C, Baeuerle PA. Activation of the transcription factor nuclear factor-kappaB in human inflamed synovial tissue. Arthritis Rheum. 1996;39:583–591. doi: 10.1002/art.1780390407. [DOI] [PubMed] [Google Scholar]
- Palombella VJ, Conner EM, Fuseler JW, Destree A, Davis JM, Laroux FS, Wolf RE, Huang J, Brand S, Elliott PJ, Lazarus D, McCormack T, Parent L, Stein R, Adams J, Grisham MB. Role of the proteasome and NF-kappaB in streptococcal cell wall-induced polyarthritis. Proc Natl Acad Sci USA. 1998;95:15671–15676. doi: 10.1073/pnas.95.26.15671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincenti MP, Coon CI, Brinckerhoff CE. Nuclear factor kappaB/p50 activates an element in the distal matrix metalloproteinase 1 promoter in interleukin-1beta-stimulated synovial fibroblasts. Arthritis Rheum . 1998;41:1987–1994. doi: 10.1002/1529-0131(199811)41:11<1987::AID-ART14>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Firestein GS, Echeverri F, Yeo M, Zvaifler NJ, Green DR. Somatic mutations in the p53 tumor suppressor gene in rheumatoid arthritis synovium. Proc Natl Acad Sci USA. 1997;94:10895–10900. doi: 10.1073/pnas.94.20.10895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pap T, Aupperle KR, Jeisy E, Gay RE, Firestein GS, Gay S. Inhibition of endogenous p53 by gene transfer increases the invasiveness of fibroblast-like synoviocytes in the SCID mouse model of rheumatoid arthritis (RA) [abstract]. Arthritis Rheum. 1999;42:S163. [Google Scholar]
- Kullmann F, Judex M, Neudecker I, Lechner S, Jüsten HP, Green DR, Wessinghage D, Firestein GS, Gay S, Schöllmerich J, Müller-Ladner U. Analysis of the p53 tumor suppressor gene in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 1999;42:1594–1600. doi: 10.1002/1529-0131(199908)42:8<1594::AID-ANR5>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- Di Cristofano A, Kotsi P, Peng YF, Cordon-Cardo C, Elkon KB, Pan-dolfi PP. Impaired Fas response and autoimmunity in Pten+/-mice. Science. 1999;285:2122–2125. doi: 10.1126/science.285.5436.2122. [DOI] [PubMed] [Google Scholar]
- Qu Z, Garcia CH, O'Rourke LM, Planck SR, Kohli M, Rosenbaum JT. Local proliferation of fibroblast-like synoviocytes contributes to synovial hyperplasia. Results of proliferating cell nuclear antigen/cyclin, c-myc, and nucleolar organizer region staining. Arthritis Rheum. 1994;37:212–220. doi: 10.1002/art.1780370210. [DOI] [PubMed] [Google Scholar]
- Hashiramoto A, Sano H, Maekawa T, Kawahito Y, Kimura S, Kusaka Y, Wilder RK, Kato H, Kondo M, Nakajima H. C-myc antisense oligodeoxynucleotides can induce apoptosis and down-regulate Fas expression in rheumatoid synoviocytes. Arthritis Rheum. 1999;42:954–962. doi: 10.1002/1529-0131(199905)42:5<954::AID-ANR14>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Taniguchi K, Kohsaka H, Inoue N, Terada Y, Ito H, Hirokawa K, Miyasaka N. Induction of the p16INK4a senescence gene as a new therapeutic strategy for the treatment of rheumatoid arthritis. Nature Med. 1999;5:760–767. doi: 10.1038/10480. [DOI] [PubMed] [Google Scholar]
- Aicher WK, Heer AH, Trabandt A, Bridges SL, Jr, Schroeder HW, Jr, Stransky G, Gay RE, Eibel H, Peter HH, Slebenlist U, Gay S. Overexpression of zinc-finger transcription factor Z-225/Egr-1 in synoviocytes from rheumatoid arthritis patients. J Immunol. 1994;152:5940–5948. [PubMed] [Google Scholar]
- Nykanen P, Bergroth V, Raunio P, Nordstrom D, Konttinen YT. Phenotypic characterization of 3H-thymidine incorporating cells in rheumatoid arthritis synovial membrane. Rheumatol Int. 1986;6:269–271. doi: 10.1007/BF00541318. [DOI] [PubMed] [Google Scholar]
- Mohr W, Hummler N, Peister B, Wessinghage D. Proliferation of pannus tissue cells in rheumatoid arthritis. Rheumatol Int. 1986;6:127–132. doi: 10.1007/BF00270349. [DOI] [PubMed] [Google Scholar]
- Firestein GS, Yeo M, Zvaifler NJ. Apoptosis in rheumatoid arthritis synovium. J Clin Invest. 1995;96:1631–1638. doi: 10.1172/JCI118202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima T, Aono H, Hasunuma T, Yamamoto K, Shirai T, Hirohata K, Nishioka K. Apoptosis and functional Fas antigen in rheumatoid arthritis synoviocytes. . Arthritis Rheum. 1995;38:485–491. doi: 10.1002/art.1780380405. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Okamoto K, Kobata T, Hasumuna T, Nishioka K. Apomodulation as a novel therapeutic concept for the regulation of apoptosis in rheumatoid synoviocytes. Curr Opin Rheumatol. 1999;11:188–193. doi: 10.1097/00002281-199905000-00007. [DOI] [PubMed] [Google Scholar]
- Franz JK, Pap T, Hummel KM, Nawrath M, Aicher WK, Shigeyama Y, Müller-Ladner U, Gay RE, Gay S. Expression of sentrin, a novel anti-apoptotic molecule at sites of synovial invasion in rheumatoid arthritis. Arthritis Rheum. 2000;43:599–607. doi: 10.1002/1529-0131(200003)43:3<599::AID-ANR17>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Okura T, Gong L, Kamitani T, Wada T, Okura I, Wei CF, Chang HM, Yeh ET. Protection against Fas/APO-1- and tumor necrosis factor-mediated cell death by a novel protein, sentrin. J Immunol. 1996;157:4277–4281. [PubMed] [Google Scholar]
- Rinaldi N, Schwarz EM, Weis D, Leppelmann JP, Lukoschek M, Keilholz U, Barth TF. Increased expression of integrins on fibroblast-like synoviocytes from rheumatoid arthritis in vitro correlates with enhanced binding to extracellular matrix proteins. Ann Rheum Dis. 1997;56:45–51. doi: 10.1136/ard.56.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Hirata S, Andoh Y, Kubo H, Nakagawa N, Nishibayashi Y, Mizuno K. An immunohistochemical and immunoelectron microscopic study of adhesion molecules in synovial pannus formation in rheumatoid arthritis. Rheumatol Int. 1996;16:53–60. doi: 10.1007/BF01816436. [DOI] [PubMed] [Google Scholar]
- Müller-Ladner U, Elices MJ, Kriegsmann J, Strahl D, Gay RE, Firestein GS, Gay S. Alternatively spliced CS-1 fibronectin isoform and its receptor VLA-4 in rheumatoid synovium demonstrated by in situ hybridization and immunohistochemistry. J Rheumatol . 1997;24:1873–1880. [PubMed] [Google Scholar]
- Schwartz MA. Integrins, oncogenes, and anchorage independence. J Cell Biol. 1997;139:575–578. doi: 10.1083/jcb.139.3.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dike LE, Farmer SR. Cell adhesion induces expression of growth-associated genes in suspension-arrested fibroblasts. Proc Natl Acad Sci USA. 1988;85:6792–6796. doi: 10.1073/pnas.85.18.6792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dike LE, Ingber DE. Integrin-dependent induction of early growth response genes in capillary endothelial cells. J Cell Sci. 1996;109:2855–2863. doi: 10.1242/jcs.109.12.2855. [DOI] [PubMed] [Google Scholar]
- Parks WC, Mecham RP. Matrix Metalloproteinases San Diego: Academic Press. 1998.
- Müller-Ladner U, Gay RE, Gay S. Cysteine proteinases in arthritis and inflammation. Perspect Drug Discovery Design. 1996;6:87–98. [Google Scholar]
- Gravallese EM, Darling JM, Ladd AL, Katz JN, Glimcher L. In situ hybridization studies on stromelysin and collagenase mRNA expression in rheumatoid synovium. Arthritis Rheum . 1991;34:1071–1084. doi: 10.1002/art.1780340903. [DOI] [PubMed] [Google Scholar]
- Firestein GS, Paine M. Expression of stromelysin and TIMP in rheumatoid arthritis synovium. Am J Pathol. 1992;140:1309–1314. [PMC free article] [PubMed] [Google Scholar]
- Konttinen YT, Ceponis A, Takagi M, Ainola M, Sorsa T, Sutinen M, Salo T, Ma J, Santavirta S, Seiki M. New collagenolytic enzymes/cascade identified at the pannus-hard tissue junction in rheumatoid arthritis: destruction from above. Matrix Biol. 1998;17:585–601. doi: 10.1016/s0945-053x(98)90110-x. [DOI] [PubMed] [Google Scholar]
- Kikuchi H, Shimada W, Nonaka T, Ueshima S, Tanaka S. Significance of serine proteinase and matrix metalloproteinase systems in the destruction of human articular cartilage. Clin Exp Pharmacol Physiol. 1996;23:885–889. doi: 10.1111/j.1440-1681.1996.tb01138.x. [DOI] [PubMed] [Google Scholar]
- Lindy O, Konttinen YT, Sorsa T, Ding Y, Santavirta S, Ceponis A, López-Otín C. Matrix-metalloproteinase 13 (collagenase 3) in human rheumatoid synovium. Arthritis Rheum. 1997;40:1391–1399. doi: 10.1002/art.1780400806. [DOI] [PubMed] [Google Scholar]
- Clark IM, Powell LK, Ramsey S, Hazelman BL, Cawston TE. The measurement of collagenase, TIMP and collagenase-TIMP complex in synovial fluids from patients with osteoarthritis and rheumatoid arthritis. Arthritis Rheum. 1993;36:372–380. doi: 10.1002/art.1780360313. [DOI] [PubMed] [Google Scholar]
- Okada Y, Takeuchi N, Tomita K, Nakanishi I, Nagase H. Immunolocalization of matrix metalloproteinase 3 (stromelysin) in rheumatoid synovioblasts (B cells): correlation with rheumatoid arthritis. Ann Rheum Dis. 1989;48:645–653. doi: 10.1136/ard.48.8.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westhoff CS, Freudiger D, Petrow P, Seyfert C, Zacher J, Kriegsmann J, Pap T, gay S, Stiehl P, Gromnica-Ihle E, Wernicke D. Characterization of collagenase 3 (matrix metalloproteinase 13) messenger RNA expression in the synovial membrane and synovial fibroblasts of patients with rheumatoid arthritis. Arthritis Rheum. 1999;42:1517–1527. doi: 10.1002/1529-0131(199907)42:7<1517::AID-ANR27>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Pap T, Shigeyama Y, Kuchen S, Fernihough JK, Simmen B, Gay RE, Billingham M, Gay S. Differential expression of membrane-type matrix metalloproteinases (MT-MMPs) in rheumatoid arthritis (RA). Arthritis Rheum. 2000;43:1226–1232. doi: 10.1002/1529-0131(200006)43:6<1226::AID-ANR5>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Sato H, Takino T, Okada Y, Cao J, Shinagawa A, Yamamoto E, Seiki M. A matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature. 1994;370:61–65. doi: 10.1038/370061a0. [DOI] [PubMed] [Google Scholar]
- Cunnane G, Fitzgerald O, Hummel KM, Gay RE, Gay S, Bresnihan B. Collagenase, cathepsin B and cathepsin L gene expression in the synovial membrane of patients with early inflammatory arthritis. Rheumatology (Oxford) 1999;38:34–42. doi: 10.1093/rheumatology/38.1.34. [DOI] [PubMed] [Google Scholar]
- Lemaire R, Flipo RM, Monte D, Dupressoir T, Duquesnoy B, Cesbron JY, Janin A, Capron A, Lafyatis R. Synovial fibroblast-like cell transfection with the SV40 large T antigen induces a transformed phenotype and permits transient tumor formation in immunodeficient mice. J Rheumatol. 1994;21:1409–1419. [PubMed] [Google Scholar]
- Trabandt A, Aicher WK, Gay RE, Sukhatme VP, Nilson HM, Hamilton RT, McGhee JR, Fassbender HG, Gay S. Expression of the collagenolytic and Ras-induced cysteine proteinase cathepsin L and proliferation-associated oncogenes in synovial cells of MRL/I mice and patients with rheumatoid arthritis. Matrix. 1990;10:349–361. doi: 10.1016/s0934-8832(11)80142-3. [DOI] [PubMed] [Google Scholar]
- Lemaire R, Huet G, Zerimech F, Grard G, Fontaine C, Duquesnoy B, Flipo RM. Selective induction of the secretion of cathepsins B and L by cytokines in synovial fibroblast-like cells. Br J Rheumatol. 1997;36:735–743. doi: 10.1093/rheumatology/36.7.735. [DOI] [PubMed] [Google Scholar]
- Huet G, Flipo RM, Colin C, Janin A, Hemon B, Collyn dM, Lafyatis R, Duquesnoy B, Degand P. Stimulation of the secretion of latent cysteine proteinase activity by tumor necrosis factor alpha and interleukin-1. Arthritis Rheum. 1993;36:772–780. doi: 10.1002/art.1780360606. [DOI] [PubMed] [Google Scholar]
- Hummel KM, Petrow PK, Franz JK, Müller-Ladner U, Aicher WK, Gay RE, Brömme D, Gay S. Cysteine proteinase cathepsin K mRNA is expressed in synovium of patients with rheumatoid arthritis and is detected at sites of synovial bone destruction. J Rheumatol. 1998;25:1887–1894. [PubMed] [Google Scholar]
- Brennan FM, Browne KA, Green PA, Jaspar JM, Maini RN, Feldmann M. Reduction of serum matrix metalloproteinase 1 and matrix metalloproteinase 3 in rheumatoid arthritis patients following anti-tumour necrosis factor-alpha (cA2) therapy. Br J Rheumatol. 1997;36:643–650. doi: 10.1093/rheumatology/36.6.643. [DOI] [PubMed] [Google Scholar]
- Maini RN, Elliott M, Brennan FM, Williams RO, Feldmann M. TNF blockade in rheumatoid arthritis: implications for therapy and pathogenesis. APMIS. 1997;105:257–263. doi: 10.1111/j.1699-0463.1997.tb00567.x. [DOI] [PubMed] [Google Scholar]
- Moreland LW, Baumgartner SW, Schiff MH, Tindall EA, Fleischmann RM, Weaver AL, Ettlinger RE, Cohen S, Koopman WJ, Mohler K, Widmer MB, Blosch CM. Treatment of rheumatoid arthritis with a recombinant human tumor necrosis factor receptor (p75)-Fc fusion protein. N Engl J Med . 1997;337:141–147. doi: 10.1056/NEJM199707173370301. [DOI] [PubMed] [Google Scholar]
- Alsalameh S, Winter K, A-Ward R, Wendler J, Kalden JR, Kinne RW. Distribution of TNF-α, TNF-R55 and TNF-R75 in the rheumatoid synovial membrane: TNF receptors are localized preferentially in the lining layer; TNF-α is distributed mainly in the vicinity of TNF receptors in the deeper layers. Scand J Immunol . 1999;49:278–285. doi: 10.1046/j.1365-3083.1999.00458.x. [DOI] [PubMed] [Google Scholar]
- Kuiper S, Joosten LA, Bendele AM, Edwards CK, Arntz O, Helsen M, van de Loo FA, van den Berg BW. Different roles of TNFα and IL-1 in murine streptococcal wall arthritis. Cytokine . 1998;10:690–702. doi: 10.1006/cyto.1998.0372. [DOI] [PubMed] [Google Scholar]
- Bendele A, Sennello G, McAbee T, Frazier J, Chlipala E, Rich B. Effects of interleukin 1 receptor antagonist alone and in combination with methotrexate in adjuvant arthritic rats. J Rheumatol. 1999;26:1225–1229. [PubMed] [Google Scholar]
- Goldring SR, Gravallese EM. Mechanisms of bone loss in inflammatory arthritis: diagnosis and therapeutic implications. Arthritis Res. 2000;2:33–37. doi: 10.1186/ar67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigeyama Y, Pap T, Kunzler P, Rethage J, Simmen B, Gay RE, Gay S. Rheumatoid arthritis (RA) synovial fibroblasts express osteoclast differentiating factor (ODF) mRNA at sites of joint destruction [abstract]. Arthritis Rheum. 1999;42:S283. [Google Scholar]
- Hamann J, Wishaupt JO, van Lier RA, Smeets TJ, Breedveld FC, Tak PP. Expression of the activation antigen CD97 and its ligand CD55 in rheumatoid synovial tissue. Arthritis Rheum. 1999;42:650–658. doi: 10.1002/1529-0131(199904)42:4<650::AID-ANR7>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Franz JK, Kolb S, Hummel KM, Lahrtz F, Neidhart M, Aicher WK, Pap T, Gay RE, Fontana A, Gay S. Interleukin-16 produced by synovial fibroblasts mediates chemoattraction to CD4+ T-cells in rheumatoid arthritis. Eur J Immunol. 1998;28:2661–2671. doi: 10.1002/(SICI)1521-4141(199809)28:09<2661::AID-IMMU2661>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Ogasawara H, Takeda-Hirokawa N, Sekigawa I, Hashimoto H, Kaneko Y, Hirose S. Inhibitory effect of interleukin-16 on interleukin-2 production by CD4+ T cells. Immunology. 1999;96:215–219. doi: 10.1046/j.1365-2567.1999.00693.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon M, Scheel Toellner D, Huissoon AP, Pilling D, Shamsadeen N, Hyde H, D'Angeac AD, Bacon PA, Emery P, Akbar AN. Inhibition of T cell apoptosis in the rheumatoid synovium. J Clin Invest. 1997;99:439–446. doi: 10.1172/JCI119178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindhout E, van Eijk M, van Pel M, Lindeman J, Dinant HJ, de Groot C. Fibroblast-like synoviocytes from rheumatoid arthritis patients have intrinsic properties of follicular dendritic cells. J Immunol. 1999;162:5949–5956. [PubMed] [Google Scholar]
- Müller-Ladner U, Evans CH, Franklin BN, Roberts CR, Gay RE, Robbins PD, Gay S. Gene transfer of cytokine inhibitors into human synovial fibroblasts in the SCID mouse model. Arthritis Rheum. 1999;42:490–497. doi: 10.1002/1529-0131(199904)42:3<490::AID-ANR14>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Jorgensen C, Gay S. Gene therapy in osteoarticular diseases: where are we? Immunol Today. 1998;19:387–391. doi: 10.1016/s0167-5699(98)01296-1. [DOI] [PubMed] [Google Scholar]
- Pap T, Müller-Ladner U, Gay RE, Gay S. Gene therapy in rheumatoid arthritis: how to target joint destruction? Arthritis Res. 1999;1:5–9. doi: 10.1186/ar3. [DOI] [PMC free article] [PubMed] [Google Scholar]