

Abstract
The M2δ peptide self-assembles to form a pentameric bundle of transmembrane α-helices that is a model of the pore-lining region of the nicotinic acetylcholine receptor. Long (>15 ns) molecular dynamics simulations of a model of the M2δ5 bundle in a POPC bilayer have been used to explore the conformational dynamics of the channel assembly. On the timescale of the simulation, the bundle remains relatively stable, with the polar pore-lining side chains remaining exposed to the lumen of the channel. Fluctuations at the helix termini, and in the helix curvature, result in closing/opening transitions at both mouths of the channel, on a timescale of ∼10 ns. On average, water within the pore lumen diffuses ∼4× more slowly than water outside the channel. Examination of pore water trajectories reveals both single-file and path-crossing regimes to occur at different times within the simulation.
INTRODUCTION
The nicotinic acetylcholine receptor (nAChR) is one of most intensively studied ligand gated ion channels, and thus provides a paradigm for the molecular mechanism of fast synaptic neurotransmission. Depolarization of the presynaptic membrane causes release of acetylcholine into the synaptic cleft. Acetylcholine diffuses to the postsynaptic membrane where it binds to the extracellular domains of the nAChR. The overall structure of the nAChR has been studied in some detail by cryoelectron microscopy (Unwin, 1993; Unwin, 1995; Miyazawa et al., 1999; Unwin, 2000), in addition to which the recent x-ray structure of a homologous acetylcholine binding protein (Brejc et al., 2001) provides a model for the extracellular ligand-binding domains. The nAChR is pentameric, the five subunits arranged around a central pore. A number of studies (Imoto et al., 1986; Imoto et al., 1988; Giraudat et al., 1987; Galzi et al., 1992; Changeux et al., 1992; Corringer et al., 2000; Hucho and Hilgenfeld, 1989; Hucho et al., 1996; Leonard et al., 1988; Lester, 1992) have indicated that the transmembrane pore is lined by the second transmembrane helix (M2; see Fig. 1 A) of each subunit. Binding of acetylcholine to the extracellular domains is thought to initiate a wave of conformational change (Grosman et al., 2000) that is propagated through the protein into the transmembrane domain and the M2 helices. This results in a “twist to open” mechanism of channel gating (Unwin, 1995; Unwin, 2000) whereby the central pore is opened to the permeation of cations. A leucine side chain (L11′ in Fig. 1 A) is highly conserved throughout the nAChRs and other members of the superfamily and seems to play a central role in the gating mechanism of these receptors (Revah et al., 1991; Labarca et al., 1995; Corringer et al., 2000; Panicker et al., 2002; Reeves and Lummis, 2002). From the cryoelectron microscopy images, it seems that the pore-lining M2 helices are kinked such that in the closed state of the channel the ring of L11′ side chains form the narrowest region of the pore (Unwin, 1993; Unwin, 2000). However, these side chains do not fully occlude the pore but rather appear to form a hydrophobic barrier to ion permeation (Beckstein et al., 2001). In the open state of the channel the M2 helices change conformation such that the narrowest region of the channel is lined by rings of hydroxyl-containing side chains (e.g., serine or threonine). This assumption is supported by the studies (of e.g., Villarroel et al., 1991; Leonard et al., 1988; Cohen et al., 1992; Lester, 1992) on the effects of mutations of these side chains on ion permeation and block of nAChR. Additional rings of negatively charged residues at the termini of the M2 helices contribute to the permeation and selectivity properties of nAChR (Imoto et al., 1988; Konno et al., 1991).
FIGURE 1.
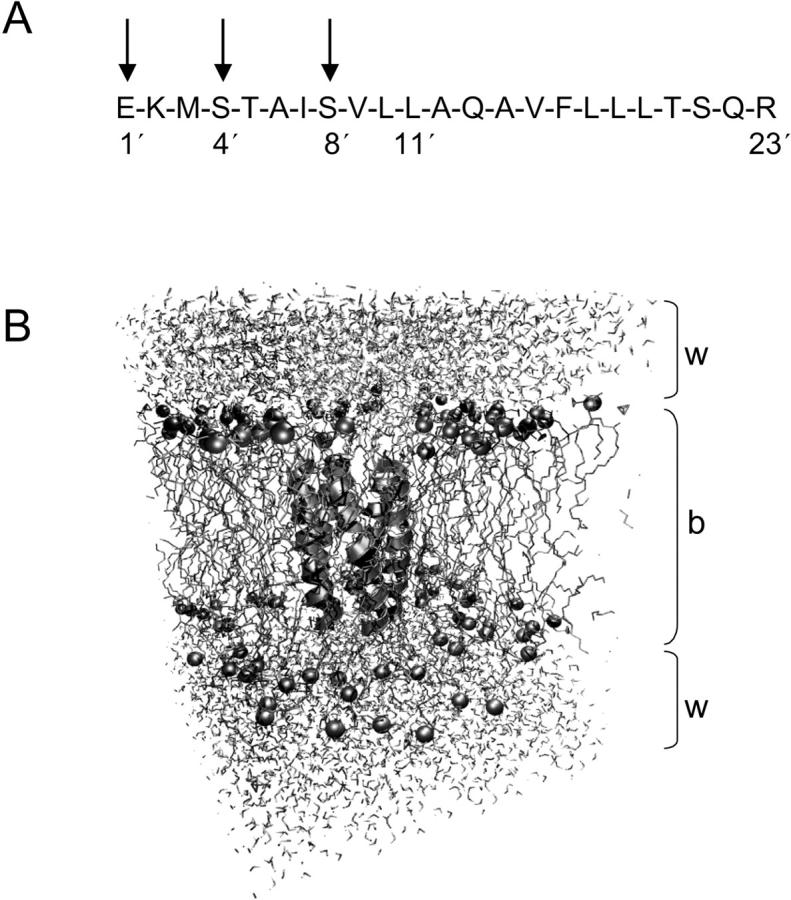
(A) Amino acid sequence of M2δ. Three key pore-lining residues (E1′, S4′, and S8′) are indicated by vertical arrows, as is the highly conserved leucine residue associated with channel gating (L11′). (B) Diagram of the simulation system, with the pentameric helix bundle (in ribbons format) embedded a POPC bilayer (b) with water molecules (w) on either side. The phosphorus atoms of the lipid headgroups are shown as gray spheres.
A number of model ion channels are formed by bundles of transmembrane α-helices. These include the antimicrobial toxin alamethicin (Sansom, 1993; Cafiso, 1994; Tieleman et al., 1999a), the synthetic peptide channel LS3 (Lear et al., 1988; Åkerfeldt et al., 1993; Mitton and Sansom, 1996; Randa et al., 1999), and the simple viral ion channel protein M2 from influenza A (Kovacs and Cross, 1997; Song et al., 2000; Forrest et al., 1998; Sansom et al., 1998; Forrest et al., 2000). A synthetic peptide corresponding to the M2 helix from the δ-subunit of Torpedo nAChR (from now on referred to as M2δ) also belongs to this family. Montal and colleagues have shown that M2δ (Fig. 1 A), a 23-mer peptide, self-assembles in lipid bilayers to form ion channels that share several properties (apart from ligand-gating, of course) with the parent protein (Oiki et al., 1988; Montal, 1995). NMR studies of M2δ have revealed that is α-helical and adopts a transmembrane orientation in lipid bilayers (Bechinger et al., 1991; Opella et al., 1999). Models of pentameric bundles of M2δ helices (i.e., M2δ5 bundles) have been shown to be consistent with the properties of these self-assembled channels (Montal et al., 1993b; Opella et al., 1999). A recent combined NMR and computational study (Tang et al., 2002) suggests a similar model for a stable pentameric bundle of M2 helices from the related glycine receptor. Of course, one must remember that these are models, not definitive structures, but they are consistent with available experimental data and thus may merit evaluation and refinement via more extended simulation studies.
Previous simulations of the M2δ peptide (Law et al., 2000) focused on its conformational dynamics as a function of environment. In water, the M2δ helix partially unfolded to form a molecular hinge in the vicinity of the central leucine (L11′) residue. In a phospholipid bilayer, either as a single transmembrane helix, or as part of a M2δ5 helix bundle, the helix showed less flexibility, but still exhibited a kink about the center of the peptide.
The M2δ5 helix bundle was previously shown to be relatively stable over the duration of a short (2 ns) molecular dynamics (MD) simulation in a POPC bilayer (Law et al., 2000) and more recently related simulations (in a dimyristoylphosphatidylcholine (DMPC) bilayer) have been reported in which formation of a left-handed supercoiled bundle was observed, and in which specific lipid-peptide interactions were analyzed (Saiz and Klein, 2002b; Saiz and Klein, 2002a). In the current study, we explore the conformational dynamics of the M2δ5 bundle for a substantially longer (by an order of magnitude) period of time. This has enabled us to sample possible gating behavior of this helix bundle, and to better characterize the behavior of the water within the channel.
METHODS
M2δ pore model
The M2δ sequence used in these studies is that in Fig. 1 A. The termini were not blocked. The model was generated as described in Law et al., 2000. Briefly, the helices were oriented about the central pore axis so that the pore-lining side chains (E1′, S4′, and S8′) were directed toward the lumen of the pore (as suggested by e.g., Lester, 1992), and such that the L11′ side chains were at the interface between adjacent helices. The helices were tilted by ∼11° so as to form a left-handed bundle. Before simulation, side-chain ionization states were adjusted on the basis of pKA calculations (Adcock et al., 1998) as previous studies had indicated that assumption of default ionization states for all residues resulted in an unstable bundle during short (2 ns) simulations (Law et al., 2000). In particular, of the ring of E1′ side chains, four were deprotonated and one was protonated. All of the C-terminal carboxylates were protonated, and two of the N-terminal amino groups were protonated. Thus the net charge of the M2δ5 bundle used in the simulations was +7.
Simulation system
The setup of the bilayer system was essentially as described by Law et al., 2000, using the approach developed by Tieleman et al., 1999a. The M2δ5 bundle was embedded in a preequilibrated bilayer consisting of 128 1-palmitoyl-2-oleoyl-sn-glycerol-3-phosphatidylcholine (POPC) lipid molecules. A hole was generated in the bilayer by superposition of the lipids and the protein, and subsequent removal of 18 POPC lipids that overlapped with the bundle. The protein/lipid system was solvated with 4663 SPC (Berendsen et al., 1981) waters and seven chloride counterions (added to maintain electroneutrality) and then energy minimized. The resultant system contained 20,937 atoms (Fig. 1 B). After a brief molecular dynamics equilibration stage of 100 ps, during which the protein atoms were restrained, a 17 ns unrestrained production simulation was performed.
MD simulations
MD simulations were carried out as described in earlier papers (Forrest et al., 1999; Tieleman et al., 1999b; Tieleman et al., 1999a; Law et al., 2000), using NPT and periodic boundary conditions. A constant pressure of 1 bar was applied independently in all three directions, using a coupling constant of τP = 1.0 ps (Berendsen et al., 1984). Water, lipid, and peptide were coupled separately to a temperature bath (Berendsen et al., 1984) at 300 K using a coupling constant τT = 0.1 ps. Long-range interactions were dealt with using a twin-range cutoff: 10 Å for van der Waals interactions, and 17 Å for electrostatic interactions. The timestep was 2 fs using LINCS (Hess et al., 1997) to constrain bond lengths, and the force field was based on GROMOS 87 (Hermans et al., 1984). The simple point charge (SPC) water model used (Berendsen et al., 1981) has been shown to behave well in simple lipid/water simulations (Tieleman and Berendsen, 1996). Also, the lipid parameters give reasonable reproduction of experimental properties of a DPPC bilayer, and have been used in previous MD studies of membranes and peptides (Berger et al., 1997; Tieleman et al., 1999b; Tieleman et al., 1999a; Randa et al., 1999).
MD simulations were run on 195 MHz R10000 Origin 2000s and took ∼8 days per processor per 1 ns simulation. Simulations and analysis were carried out using GROMACS v1.6 (Berendsen et al., 1995) (http://www.gromacs.org). Electrostatics calculations for pKA calculations employed a modified UHBD version 5.1 (Davis et al., 1991) (http://chemcca51.ucsd.edu/uhbd.html). Pore radius profiles were calculated using HOLE (Smart et al., 1993; Smart et al., 1996; Smart et al., 1997). The initial bundle model was constructed using X-PLOR (Brünger, 1992) using a simulated annealing protocol detailed in Kerr et al., 1994. Structures were examined using Quanta and RasMol. Molecular graphics images were prepared using VMD (Humphrey et al., 1996), MolScript (Kraulis, 1991), and PovRay (http://www.povray.org).
RESULTS
Dynamic behavior of the bundle
The longer duration of the simulation in this study enables us to examine some of the conformational changes that occur in the initial bundle structure as it relaxes in the lipid bilayer. An overall measure of the drift from the initial structure is provided by the Cα root mean square difference (RMSD) as a function of time relative to the initial structure (Fig. 2 A). This is quite low for a helix bundle model simulation, reaching a value of only ∼2 Å after 15 ns, although it would seem that a much longer simulation would be required to reach an equilibrium. More detailed examination of the time course of the RMSD suggests that most of the movements of the helices within the bundle take place in the first couple of nanoseconds, during which we presume that some degree of optimization of helix conformation and packing occurs. Secondary structure analysis (using DSSP; (Kabsch and Sander, 1983)) confirms that each M2δ peptide remains largely α-helical throughout the duration of the simulation. However, from snapshots of the structure, it is evident that the individual helices within the bundle adopt more curved or kinked conformations than in the initial model. The small increase in RMSD seen in the last 2 ns of the simulation represents the change in structure as the pore opens up (see below).
FIGURE 2.

(A) Cα RMSD versus the initial structure of the M2δ5 bundle as a function of time. Three snapshots of the bundle structure are shown. (B) Cα RMSF (averaged over time and all five helices) as a function of residue number.
The fluctuations of individual residues can be measured as Cα root mean square fluctuations, averaged over time and across all five helices of the bundle (Fig. 2 B). As expected, the fluctuations are larger at the helix termini than at the center. Indeed, between residues 5 and 15, the Cα root mean square fluctuations are <1 Å; again comparable to those seen in simulations based on x-ray structures. The fluctuations are somewhat higher at the C-terminus than at the N-terminus of the helix.
Some of the helix fluctuations are related to kinked and curved conformations of the individual M2δ helices, as analyzed in detail in an earlier paper (Law et al., 2000). In Fig. 3, the distributions of the kink angles are presented for the five helices of the bundle. It can be seen that the range of kink angles is quite broad (from ∼10° to 40°). Thus the initially linear helices are considerably distorted in the bundle. Examination of the structures shows that the helices are kinked in a convex fashion so as to generate a central cavity within the bundle (see below). Comparison of kink angle distributions among the five helices reveals that although there is some considerable degree of overlap, they are not identical. This may be taken as evidence that complete statistical convergence has not occurred during a simulation of ∼20 ns duration.
FIGURE 3.

Helix kink angle distributions for the five helices (H1–H5) of the M2δ5 bundle.
The orientation of the helices relative to the z axis (i.e., the approximate bilayer normal) may also be measured, in term of helix tilt angle. It is known from the analysis of the x-ray structures of membrane proteins that transmembrane helices are generally tilted with respect to the bilayer, with a mean tilt angle of ∼21° (Bowie, 1997). Furthermore, solid-state NMR studies of M2δ peptide in a DMPC bilayer have revealed an overall tilt angle of ∼12° (Opella et al., 1999). The M2δ helices in the bundle at the start of the simulation have a tilt angle of 12°. During the course of the simulation, the individual helices undergo major fluctuations in tilt angles over a range of ∼5°–25° (Fig. 4) on a timescale of ∼2–5 ns. It is also evident from the tilt angle trajectories that there is loss of exact pentameric symmetry in the bundle.
FIGURE 4.

Helix tilt angles (relative to the z axis, i.e., the approximate bilayer normal) of the five M2δ helices as a function of time.
The analyses so far indicate that the M2δ5 is a dynamic assembly, undergoing fluctuations in helix conformation and orientation on a nanosecond timescale, while retaining the overall structure of the bundle. From a more functional perspective, we may examine whether such fluctuations change the nature of the residues forming the pore lining and the helix/helix interfaces. Three key polar residues are thought to play an important role in forming the lining of the N-terminal half of the pore, namely E1′, S4′, and S8′ (Fig. 5 A). Together these form three rings of hydrophilic side chains lining the pore, as is also the case in the parent nAChR (Bertrand et al., 1993). We have analyzed solvent accessible surface areas (data not shown) to demonstrate that these side chains remain exposed to the lumen of the pore throughout the duration of the simulation.
FIGURE 5.
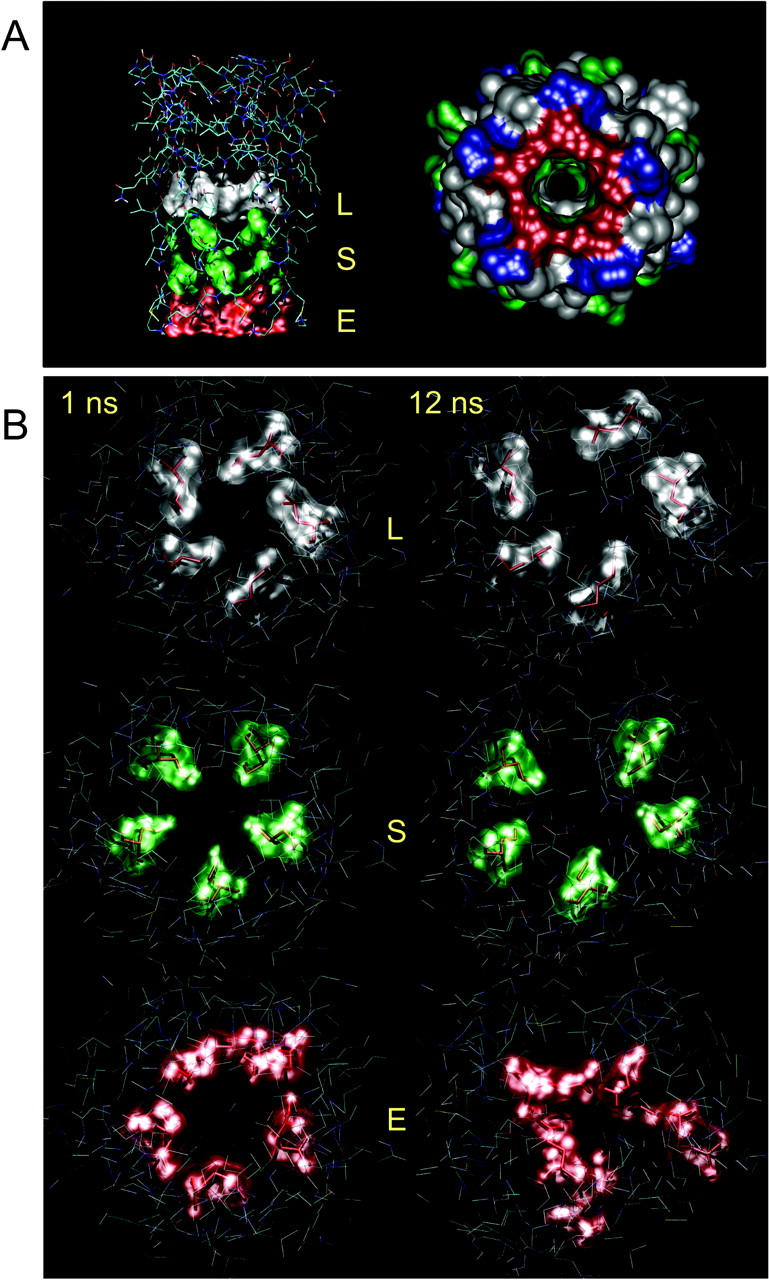
(A) Two views of the M2δ5 bundle. The left-hand image is viewed perpendicular to the pore axis, with the C-termini of the helices at the top of the diagram. The rings of E1′ (red), S4′ and S8′(green), and L11′ residues are shown in space-filling format. The right-hand image is a space-filling model of the bundle, viewed down the pore axis with the N-termini toward the viewer (red, acidic residues; blue, basic residues; green, polar residues; gray, hydrophobic residues). (B) Snapshots at t = 1 and 12 ns of the rings of pore-lining side chains formed by residues L11′ (gray), S8′ (green), and E1′ (red).
In addition to these polar side chains, the L11′ side chain has been examined. In the parent nAChR when in the closed state of the channel this side chain is proposed to be located at the interface between adjacent helices, forming a narrow hydrophobic ring at the center of the transbilayer pore. Interestingly, this is also the case in the M2δ5 model. Snapshots of the channel structure at e.g., 1 ns and 12 ns (Fig. 5 B) suggest that this location of L11′ is maintained throughout the simulation, albeit with some degree of local distortion. Similarly, the two rings of pore-lining serine residues remain pointing into the lumen of the channel, forming a polar environment that is expected to interact favorably with water molecules and with permeant ions. The glutamates (E1′) at the N-termini of the helices show slightly more complex behavior. The additional flexibility of the helix termini enables these side chains to switch between a pore-lining conformation and one in which a glutamate side chain forms an ion pair with a lysine (K2′) from the same helix. K2′ residues are also seen to snorkel so as to form contacts with lipid headgroups and interfacial water molecules, a feature also observed in simulations of a similar system in DMPC (Saiz and Klein, 2002a) and more generally in membrane proteins (Killian and von Heijne, 2000).
Consequences for the structure of the pore
Having examined the dynamic behavior of the M2δ5 bundle, we may analyze the consequences of this in terms of the shape of the transbilayer pore, as this is likely to dominate the channel properties of the assembly. The pore can be characterized in terms of its pore radius profile (Smart et al., 1993), which in turn has an approximate relationship to the conductance of the channel (Smart et al., 1997; Smart et al., 1998). The average pore radius profile over the course of the simulation (Fig. 6 A) reveals a central cavity of radius ∼3 Å, with narrow regions (radius ∼1.3 Å) at both mouths of the pore. The central cavity is reminiscent of that seen in the KcsA channel, although a little smaller. Its average radius is a little less than that of the first solvation shell of a monovalent cation (∼4.5 Å), although it should be remembered that the side chains of the serines could substitute for water molecules in stabilizing an ion in the cavity. The mouth radii are such that a cation would not be able to pass through without being dehydrated. However, it should be remembered that these are average radii, and examination of instantaneous radius profiles shows substantial fluctuations.
FIGURE 6.
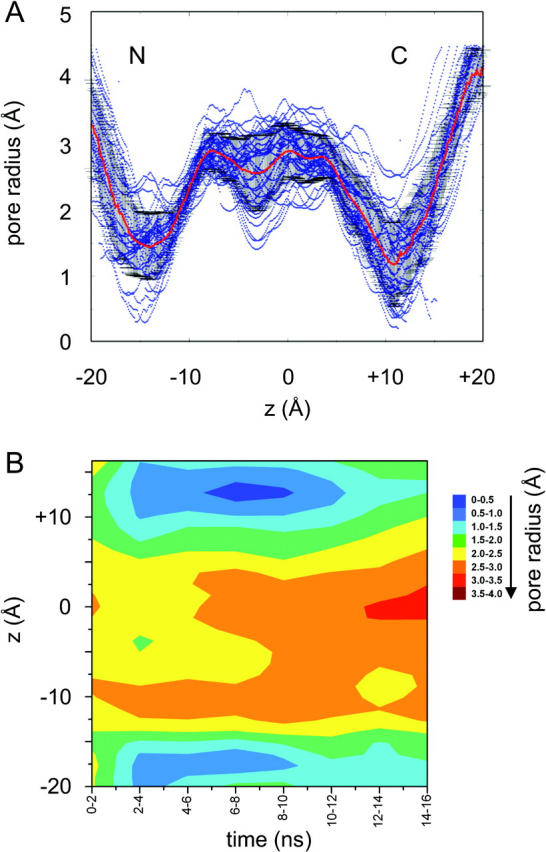
(A) Pore radius profiles over the course of the simulation. The red line is the average pore radius profile over the whole simulation. The black vertical bars represent ±SD. The blue points correspond to individual radius profiles determined every 100 ps during the simulation. N and C indicate the corresponding helix termini. (B) Time evolution of the pore radius profile shown as a contour plot of pore radius (red, wide; blue, narrow) as a function of time (horizontal axis) and position along the pore (vertical axis).
The time dependence of the pore radius profile is shown in Fig. 6 B. The pore seems to go through an overall open-closed-open cycle during the course of the simulation. Thus, for the first nanosecond or so of the simulation, the radius at either mouth is at least 1.5 Å, i.e., large enough to admit a water molecule. The two mouths of the pore then contract, to radii of 1 Å or less from ∼2 to 12 ns, before reopening for the remainder of the simulation.
A more detailed visualization of the changes in pore geometry is presented in Fig. 7, where snapshots of the pore lining are color-coded relative to the radius of a water molecule (Smart et al., 1996). It should be noted that there is complete occlusion of both mouths of the pore from 2.5 to 3 ns, whereas one or other mouth is closed from 7.5 to 8 ns. Both mouths of the pore are fully open at ∼16 ns. Along with the fluctuations in the radius of the central cavity, these changes result in variation of the volume of the pore (with the pore ends defined by the average z coordinate of Cα atoms of the terminal residues of each helix) between 400 Å3 and 700 Å3. Note that this would correspond to a volume sufficient to hold between 13 and 23 water molecules within the channel.
FIGURE 7.

Pore volume as 'a function of time. Snapshots of the helix bundle and pore lining surfaces are shown. The linings are color coded as follows: red, radius <1.4 Å; green, radius between 1.4 and 3 Å; and blue, radius >3 Å.
The pore radius profiles may be used to approximate the conductance of the channel by integrating the electrical resistance of equivalent electrolyte-filled cylinders along the length of the pore and then applying an empirical scale factor to take account of e.g., the reduced mobility of ions within a narrow pore (Smart et al., 1998; Tieleman et al., 2001). The conductance estimated in this fashion is ∼40 pS (in 1M KCl) for the last 2 ns of the simulation. This would correspond to ∼20 pS in 0.5 M KCl and thus is comparable (within the accuracy of the prediction method (Smart et al., 1997) to the experimental conductances of 20 pS for M2δ peptide and 27 pS for M2α4 peptide channels recorded in 0.5 M KCl (Montal et al., 1993a). For reference, the conductance of intact nAChR channels is ∼60 pS in 0.1 M KCl (Imoto et al., 1986), which (assuming linearity) would correspond to ∼300 pS in 0.5 M KCl.
Behavior of water within the pore
It is of some interest to examine the behavior of water within the M2δ5 pore, given ongoing studies of the modification of water's properties within channels (Chiu et al., 1991; Chiu et al., 1999; De Groot and Grubmuller, 2001; Tajkhorshid et al., 2002) and nanopores relative to the bulk state (Hummer et al., 2001; Sansom and Biggin, 2001; Beckstein et al., 2001). Also, analysis of a number of simulations (reviewed in Tieleman et al., 2001) suggests that the overall pattern of reduction in water diffusion coefficients within channels mirrors that of reduction of ionic diffusion coefficients. An overall measure of the water behavior is provided by the diffusion coefficient of water molecules as a function of their average position along the pore axis (Fig. 8). The diffusion coefficients mirror the radius profile of the pore, with a ∼4× reduction in diffusion relative to bulk at the narrow mouths of the pore. This is in general agreement with a meta-analysis of data from a number of simulation studies (reviewed in Tieleman et al., 2001).
FIGURE 8.

Average diffusion coefficients for water molecules as a function of position along the z axis. The broken horizontal line indicates the diffusion coefficient of bulk water in this simulation. Water diffusion coefficients were derived from the mean squared deviation versus time of single water molecules within slices along the z axis of the system.
It is likely that, at least in part, this effect in M2δ5 is due to H-bond interactions with pore-lining side chains. The water molecules within the pore are nonrandomly oriented (data not shown) as a consequence of the interaction of the water dipoles with the parallel helix dipoles. Such behavior has been observed in a number of simulations, of differing degrees of complexity, of water molecules within pores formed by parallel helix bundles (Breed et al., 1996; Mitton and Sansom, 1996; Sansom et al., 1997; Randa et al., 1999; Tieleman et al., 1999a).
A more detailed examination of water trajectories along the pore axis (Fig. 9) reveals complex behavior. Two types of behavior are observed: single filing of waters and path-crossing events within (transiently) wider regions of the pore regimes. In addition to such changes in water trajectories, regions devoid of water molecules can be observed, corresponding to closure events at the mouth of the channel. From examination of a number of such trajectories it can be seen that the timescale for a water molecule to move from one end of the pore to the other is ∼1 ns. This is consistent with e.g., the observed rates of permeation of water through aquaporin pores (Borgnia et al., 1999; Borgnia and Agre, 2001).
FIGURE 9.

Trajectories, projected onto the z axis (which is approximately parallel to the pore axis) of water molecules that were within the pore for at least 90% of the time for the period between 2 and 4 ns. This provides an illustration of the different regimes of water motion within the pore, including single-file (SF) motion and path-crossing (PC) motion.
DISCUSSION
M2δ5 channel properties
Based on the analysis presented above, we may tentatively identify three phases in the behavior of the M2δ5 channel, as summarized in Fig. 10. At the start of the simulation (t = 0–2 ns) the helix bundle remains close to the initial model structure, such that the helices remain unkinked but the channel is open at either mouth. The helices then kink, increasing the volume of the central cavity but also closing one or other mouth of the channel for the central section of the simulation (2–15 ns). Toward the end of the simulation, both mouths of the channel open, whereas the helices remain kinked. Although it is clear from this description that we do not fully sample the conformational dynamics of the helix bundle (which is also demonstrated to be the case by a more formal analysis of the motions of the peptides (Hess et al., unpublished data)), it is also clear that the M2δ5 bundle is capable of undergoing closed ↔ open transitions on a timescale of ∼5 ns and longer. Although this timescale is four orders of magnitude faster than that of the fastest gating events either in the peptide channels (Oiki et al., 1988; Montal, 1995) or in the parent nAChR that has been resolved experimentally (as single channel recording methods have a deadtime of ∼0.1 ms), it is suggestive of an underlying mechanism for channel gating involving helix distortions and/or helix bundle repacking.
FIGURE 10.

Schematic diagram showing the time evolution of the channel. The system begins with straight helices set up with a 5° tilt and the pore open (t = 0–2 ns). The pore then enters a predominantly closed state, in which the helices kink and the mouths of the pore open only transiently (t = 2–15 ns). Toward the end of the simulation (t = 15–17 ns) the pore enters a predominantly open state in which the helices remain kinked.
Biological relevance
These simulations have provided some insights into the conformational dynamics of a model of the M2δ5 bundle on a >10 ns timescale. To what extent are these studies of relevance to more complex, physiologically important ion channels whose gating is on a msec timescale?
The M2δ peptide remains relevant to studies of the intact nAChR. Recent electron microscopy images (Unwin, 2000) continue to support the suggestion that, at its narrowest transmembrane region, the pore running through the center of the nAChR is formed by a bundle of five M2 helices. This also appears to be so in other membranes of the nAChR superfamily, e.g., the 5HT3 receptor (Panicker et al., 2002; Reeves and Lummis, 2002). Thus it seems likely that the behavior of the M2δ5 assembly may cast some light on the conformational dynamics of these more complex channels. In particular, we envisage that fluctuations in helix-helix packing will occur on similar timescales in the simple bundle and in the more complex protein, although the fluctuations in the latter are likely to be damped by interactions of the M2 helices with the rest of the protein.
The behavior of M2δ5 may also be of some relevance to the bacterial mechanosensitive channel MscL. The x-ray structure of MscL in its closed state reveals the pore to be formed by a pentameric bundle, with two α-helices from each subunit (Chang et al., 1998). These helices are able to repack, when the channel-containing membrane is stretched, to form the open conformation of the channel (Sukharev et al., 2001; Biggin and Sansom, 2001). It would be of interest to compare the motions of the pore-lining helices of MscL in MD simulations (Elmore and Dougherty, 2001; Gullingsrud et al., 2001) with those of the M2δ5 bundle.
It is also of interest that the M2δ5 bundle seems to undergo open ↔ closed transitions (albeit on a ∼10 ns timescale) due to fluctuations in helix packing at the mouths of the channel. Both experimental (Perozo et al., 1998; Perozo et al., 1999; Liu et al., 2001) and theoretical (Shen et al., 2002; Shrivastava and Sansom, 2000; Shrivastava and Sansom, 2002; Biggin and Sansom, 2002) approaches indicate that gating of the bacterial potassium channel KcsA occurs via a change in packing of the pore-lining M2 helices at the cytoplasmic mouth of the channel. Recent x-ray studies of a K channel (MthK) captured in an open state indicate that internal (hinge-bending) distortions of these helices is also important (Jiang et al., 2002a; Jiang et al., 2002b). Of course, the timescale of helix repacking in a complex channel protein is likely to be significantly longer (maybe several orders of magnitude) than in a relatively simple self-assembled helix bundle.
The simulations of M2δ5 also add to our knowledge of the complexities of water dynamics in channels and pores. This is of some interest in the context of recent simulation studies of water in aquaporin (De Groot and Grubmuller, 2001) and in the related bacterial protein GlpF (Tajkhorshid et al., 2002), both of which indicate single file motion and a degree of nonrandom orientation of water molecules within their narrow pores. Furthermore, complex behavior of water has been shown in simulations of carbon nanotubes (Hummer et al., 2001) and in simple models of hydrophobic nanopores (Beckstein et al., 2001). By steadily accumulating simulation data from a range of different channel systems, it should be possible to dissect out more general patterns and trends (Tieleman et al., 2001).
Methodological limitations
There are a number of possible methodological limitations that should be considered. The first of these is the use of a cutoff, albeit quite a high cutoff (17 Å), to treat long-range electrostatic interactions. An alternative would be to employ particle mesh Ewald (Darden et al., 1993; Essmann et al., 1995) to treat long-range interactions. This has been shown to yield more realistic simulations of lipid bilayers (Tobias et al., 1997; Tobias, 2001). There have been some concerns that use of particle mesh Ewald may artifactually stabilize peptide structures (Weber et al., 2000), although recent simulations of alamethicin helix bundles in a lipid bilayer (Tieleman et al., 2002) did not indicate this to be a problem. Other possible limitations in the simulation protocol that might merit consideration are the use of NPT rather than NVT, and the forcefield employed. However, comparisons of the results of simulations from different laboratories (using different protocols) on the same peptide channel (Zhong et al., 1998a; Zhong et al., 1998b; Randa et al., 1999) or the same protein channel (Guidoni et al., 1999; Guidoni et al., 2000; Bernèche and Roux, 2000; Shrivastava and Sansom, 2000) do not reveal large differences, suggesting that changes in M2δ5 simulation behavior as a result of changes in simulation protocol might be expected to be relatively minor.
There are two further limitations, which are to some extent coupled. One is the presence of only a very small number of ions (7 Cl−) in the simulation system and the other is the duration of the simulation. It would perhaps be interesting to run an extended simulation with Na+ and Cl− ions present in numbers equivalent to a bulk concentration of 1 M.
It must be remembered that the initial structure of M2δ5 is a model, albeit one that has received some support from e.g., solid-state NMR studies (Montal, 1995; Opella et al., 1999). On the basis of the simulation results presented above we would suggest that this model is relatively stable, at least on a multinanosecond timescale. However, if recent developments in solid-state NMR methods (Marassi and Opella, 1998) could yield a more model-free structure, it would be interesting to see how this behaves in simulations.
Finally we should recall that the simulations are conducted in the absence of a transbilayer voltage difference. Imposition of the latter, which might be possible in future simulations (Roux, 1997), could be expected to stabilize the transbilayer orientation of the M2δ helices, and might therefore affect the dynamics of the helix bundle. Indeed, this has already been seen for a related helix bundle system (Zhong et al., 1998b). Of course, for the intact nAChR (which shows very weak voltage dependence), this is less critical. It is also noted that there is no permeating ion present in the pore and that this may affect the structural dynamics by e.g., interacting with anion side chains at the mouth of the pore.
CONCLUSIONS
What are the more general conclusions we may draw from this and related (Tieleman et al., 2002) studies? The first is that longer (≫1 ns) MD simulations of simple ion channels can start to reveal some aspects of the conformational dynamics of such pores. Although MD simulations clearly cannot reach the timescale of channel gating (∼1 ms) they can already (just) address the timescale of individual permeation events (∼10 ns) and in the near future may be able to extend to the timescales thought to be associated with open channel noise (1 μs = 1000 ns). This in itself will be a useful stage in bridging the gap between (static) structures and (dynamic) physiological processes.
It is significant that meaningful simulation results may be obtained from a relatively simple channel-forming peptide, made up of a fragment of a more complex channel. This helps to reinforce the initial suggestion that such “minimalist” channels (Montal, 1995) may be of biological relevance. It is also conceivable that such assemblies may be of relevance to bionanotechnology, in which case the ability to understand their dynamic properties via simulation is likely to play an important role in the design cycle.
There are two main directions for future studies of M2δ5, already alluded to above. The first is to obtain better structural models, possibly via the application of novel solid-state NMR techniques (Marassi and Opella, 1998; Opella et al., 2001). The second major direction will be to greatly extend the duration of simulations. We estimate that a 1 μs simulation, of a system of this size, would now take ∼24,000 CPU hours. With recent advances in commodity cluster computing, this is not an impossible task.
Acknowledgments
Thanks to our colleagues for their interest and helpful comments.
This work was supported by grants from the Wellcome Trust (to M.S.P.S). D.P.T. is a Scholar of the Alberta Heritage Foundation for Medical Research. R.J.L. is an MRC research student. We are grateful to the Oxford Supercomputer Centre for access to its facilities.
References
- Adcock, C., G. R. Smith, and M. S. P. Sansom. 1998. Electrostatics and the ion selectivity of ligand-gated channels. Biophys. J. 75:1211–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Åkerfeldt, K. S., J. D. Lear, Z. R. Wasserman, L. A. Chung, and W. F. DeGrado. 1993. Synthetic peptides as models for ion channel proteins. Acc. Chem. Res. 26:191–197. [Google Scholar]
- Bechinger, B., Y. Kim, L. E. Chirlian, J. Gesell, J. M. Neumann, M. Montal, J. Tomich, M. Zasloff, and S. J. Opella. 1991. Orientations of amphipathic helical peptides in membrane bilayers determined by solid-state NMR spectroscopy. J. Biomol. NMR. 1:167–173. [DOI] [PubMed] [Google Scholar]
- Beckstein, O., P. C. Biggin, and M. S. P. Sansom. 2001. A hydrophobic gating mechanism for nanopores. J. Phys. Chem. B. 105:12902–12905. [Google Scholar]
- Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, A. DiNola, and J. R. Haak. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81:3684–3690. [Google Scholar]
- Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, and J. Hermans. 1981. Intermolecular Forces. Reidel, Dordrecht.
- Berendsen, H. J. C., D. van der Spoel, and R. van Drunen. 1995. GROMACS: A message-passing parallel molecular dynamics implementation. Comp. Phys. Comm. 95:43–56. [Google Scholar]
- Berger, O., O. Edholm, and F. Jahnig. 1997. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidycholine at full hydration, constant pressure and constant temperature. Biophys. J. 72:2002–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernèche, S., and B. Roux. 2000. Molecular dynamics of the KcsA K+ channel in a bilayer membrane. Biophys. J. 78:2900–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand, D., J. L. Galzi, A. Devillers-Thiéry, S. Bertrand, and J. P. Changeux. 1993. Stratification of the channel domain in neurotransmitter receptors. Curr. Opin. Cell Biol. 5:688–693. [DOI] [PubMed] [Google Scholar]
- Biggin, P. C., and M. S. P. Sansom. 2001. Channel gating: twist to open. Curr. Biol. 11:R364–R366. [DOI] [PubMed] [Google Scholar]
- Biggin, P. C., and M. S. P. Sansom. 2002. Open-state models of a potassium channel. Biophys. J. 83:1867–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgnia, M., S. Nielsen, A. Engel, and P. Agre. 1999. Cellular and molecular biology of the aquaporin water channels. Annu. Rev. Biochem. 68:425–458. [DOI] [PubMed] [Google Scholar]
- Borgnia, M. J., and P. Agre. 2001. Reconstitution and functional comparison of purified GlpF and AqpZ, the glycerol and water channels from Escherichia coli. Proc. Natl. Acad. Sci. USA. 98:2888–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie, J. U. 1997. Helix packing in membrane proteins. J. Mol. Biol. 272:780–789. [DOI] [PubMed] [Google Scholar]
- Breed, J., R. Sankararamakrishnan, I. D. Kerr, and M. S. P. Sansom. 1996. Molecular dynamics simulations of water within models of transbilayer pores. Biophys. J. 70:1643–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brejc, K., W. J. van Dijk, R. V. Klaassen, M. Schuurmans, J. van der Oost, A. B. Smit, and T. K. Sixma. 2001. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature. 411:269–276. [DOI] [PubMed] [Google Scholar]
- Brünger, A. T. 1992. X-PLOR Version 3.1. A System for X-ray Crystallography and NMR. Yale University Press, New Haven, CT.
- Cafiso, D. S. 1994. Alamethicin: a peptide model for voltage gating and protein membrane interactions. Annu. Rev. Biophys. Biomol. Struct. 23:141–165. [DOI] [PubMed] [Google Scholar]
- Chang, G., R. H. Spencer, A. T. Lee, M. T. Barclay, and D. C. Rees. 1998. Structure of the MscL homolog from Mycobacterium tuberculosis: a gated mechanosensitive ion channel. Science. 282:2220–2226. [DOI] [PubMed] [Google Scholar]
- Changeux, J. P., J. I. Galzi, A. Devillers-Thiéry, and D. Bertrand. 1992. The functional architecture of the acetylcholine nicotinic receptor explored by affinity labelling and site-directed mutagenesis. Quart. Rev. Biophys. 25:395–432. [DOI] [PubMed] [Google Scholar]
- Chiu, S. W., E. Jakobsson, S. Subramanian, and J. A. McCammon. 1991. Time-correlation analysis of simulated water motion in flexible and rigid gramicidin channels. Biophys. J. 60:273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu, S. W., S. Subramaniam, and E. Jakobsson. 1999. Simulation study of a gramicidin/lipid bilayer system in excess water and lipid. II. Rates and mechanisms of water transport. Biophys. J. 76:1939–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen, B. N., C. Labarca, L. Czyzk, N. Davidson, and H. Lester. 1992. Tris+/Na+ permeability ratios of nicotinic acetylcholine receptors are reduced by mutations near the intracellular end of the M2 region. J. Gen. Physiol. 99:545–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corringer, P. J., N. Le Novere, and J. P. Changeux. 2000. Nicotinic receptors at the amino acid level. Annu. Rev. Pharmacol. Toxicol. 40:431–458. [DOI] [PubMed] [Google Scholar]
- Darden, T., D. York, and L. Pedersen. 1993. Particle mesh Ewald: an N.log(N) method for Ewald sums in large systems. J. Chem. Phys. 98:10089–10092. [Google Scholar]
- Davis, M. E., J. D. Madura, B. A. Luty, and J. A. McCammon. 1991. Electrostatics and diffusion of molecules in solution: simulations with the University of Houston Brownian dynamics program. Comput. Phys. Comm. 62:187–197. [Google Scholar]
- De Groot, B. L., and H. Grubmuller. 2001. Water permeation across biological membranes: mechanism and dynamics of aquaporin-1 and GlpF. Science. 294:2353–2357. [DOI] [PubMed] [Google Scholar]
- Elmore, D. E., and D. A. Dougherty. 2001. Molecular dynamics simulations of wild-type and mutant forms of the Mycobacterium tuberculosis MscL channel. Biophys. J. 81:1345–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essmann, U., L. Perera, M. L. Berkowitz, T. Darden, H. Lee, and L. G. Pedersen. 1995. A smooth particle mesh Ewald method. J. Chem. Phys. 103:8577–8593. [Google Scholar]
- Forrest, L. R., W. F. DeGrado, G. R. Dieckmann, and M. S. P. Sansom. 1998. Two models of the influenza A M2 channel domain: verification by comparison. Fold. Des. 3:443–448. [DOI] [PubMed] [Google Scholar]
- Forrest, L. R., A. Kukol, I. T. Arkin, D. P. Tieleman, and M. S. P. Sansom. 2000. Exploring models of the influenza A M2 channel: MD Simulations in a lipid bilayer. Biophys. J. 78:55–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest, L. R., D. P. Tieleman, and M. S. P. Sansom. 1999. Defining the transmembrane helix of M2 protein from influenza A by molecular dynamics simulations in a lipid bilayer. Biophys. J. 76:1886–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galzi, J. L., A. Devillers-Thiery, N. Hussy, S. Bertrand, J. P. Changeux, and D. Bertrand. 1992. Mutations in the channel domain of a neuronal nicotinic receptor convert ion selectivity from cationic to anionic. Nature. 359:500–505. [DOI] [PubMed] [Google Scholar]
- Giraudat, J., M. Dennis, T. Heidmann, P. Y. Haumont, F. Lederer, and J. P. Changeux. 1987. Structure of the high-affinity binding site for noncompetitive blockers of the acetylcholine receptor: [3H] chlorpromazine labels homologous residues in the beta and delta chains. Biochemistry. 26:2410–2418. [DOI] [PubMed] [Google Scholar]
- Grosman, C., M. Zhou, and A. Auerbach. 2000. Mapping the conformational wave of acetylcholine receptor channel gating. Nature. 403:773–776. [DOI] [PubMed] [Google Scholar]
- Guidoni, L., V. Torre, and P. Carloni. 1999. Potassium and sodium binding in the outer mouth of the K+ channel. Biochemistry. 38:8599–8604. [DOI] [PubMed] [Google Scholar]
- Guidoni, L., V. Torre, and P. Carloni. 2000. Water and potassium dynamics in the KcsA K+ channel. FEBS Lett. 477:37–42. [DOI] [PubMed] [Google Scholar]
- Gullingsrud, J., D. Kosztin, and K. Schulten. 2001. Structural determinants of MscL gating studied by molecular dynamics simulations. Biophys. J. 80:2074–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermans, J., H. J. C. Berendsen, W. F. van Gunsteren, and J. P. M. Postma. 1984. A consistent empirical potential for water-protein interactions. Biopolymers. 23:1513–1518. [Google Scholar]
- Hess, B., H. Bekker, H. J. C. Berendsen, and J. G. E. M. Fraaije. 1997. LINCS: a linear constraint solver for molecular simulations. J. Comp. Chem. 18:1463–1472. [Google Scholar]
- Hucho, F., and R. Hilgenfeld. 1989. The selectivity filter of a ligand-gated ion channel. FEBS Lett. 257:17–23. [DOI] [PubMed] [Google Scholar]
- Hucho, F., V. I. Tsetlin, and J. Machold. 1996. The emerging three-dimensional structure of a receptor. The nicotinic acetylcholine receptor. Eur. J. Biochem. 239:539–557. [DOI] [PubMed] [Google Scholar]
- Hummer, G., J. C. Rasaiah, and J. P. Noworyta. 2001. Water conduction through the hydrophobic channel of a carbon nanotube. Nature. 414:188–190. [DOI] [PubMed] [Google Scholar]
- Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD: visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]
- Imoto, K., C. Busch, B. Sakmann, M. Mishina, T. Konno, J. Nakai, H. Buyo, Y. Mori, K. Kukuda, and S. Numa. 1988. Rings of negatively charged amino acids determine the acetylcholine receptor channel conductance. Nature. 335:645–648. [DOI] [PubMed] [Google Scholar]
- Imoto, K., C. Methfessel, B. Sakmann, M. Mishina, Y. Mori, T. Konno, K. Fukuda, M. Kurasaki, H. Bujo, Y. Fujita, and S. Numa. 1986. Location of a delta-subunit region determining ion transport through the acetylcholine receptor channel. Nature. 324:670–674. [DOI] [PubMed] [Google Scholar]
- Jiang, Y., A. Lee, J. Chen, M. Cadene, B. T. Chait, and R. MacKinnon. 2002a. Crystal structure and mechanism of a calcium-gated potassium channel. Nature. 417:515–522. [DOI] [PubMed] [Google Scholar]
- Jiang, Y., A. Lee, J. Chen, M. Cadene, B. T. Chait, and R. MacKinnon. 2002b. The open pore conformation of potassium channels. Nature. 417:523–526. [DOI] [PubMed] [Google Scholar]
- Kabsch, W., and C. Sander. 1983. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 22:2577–2637. [DOI] [PubMed] [Google Scholar]
- Kerr, I. D., R. Sankararamakrishnan, O. S. Smart, and M. S. P. Sansom. 1994. Parallel helix bundles and ion channels: molecular modeling via simulated annealing and restrained molecular dynamics. Biophys. J. 67:1501–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killian, J. A., and G. von Heijne. 2000. How proteins adapt to a membrane-water interface. Trends Biochem. Sci. 25:429–434. [DOI] [PubMed] [Google Scholar]
- Konno, T., C. Busch, E. von Kitzing, K. Imoto, F. Wang, J. Nakai, M. Mishina, S. Numa, and B. Sakmann. 1991. Rings of anionic amino acids as structural determinants of ion selectivity in the acetylcholine receptor channel. Proc. R. Soc. Lond. 244:69–79. [DOI] [PubMed] [Google Scholar]
- Kovacs, F. A., and T. A. Cross. 1997. Transmembrane four-helix bundle of influenza A M2 protein channel: structural implications from helix tilt and orientation. Biophys. J. 73:2511–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraulis, P. J. 1991. MolScript: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24:946–950. [Google Scholar]
- Labarca, C., M. W. Nowak, H. Y. Zhang, L. X. Tang, P. Deshpande, and H. A. Lester. 1995. Channel guting governed symmetrically by conserved leucine residues in the M2 domain of nicotinic receptors. Nature. 376:514–516. [DOI] [PubMed] [Google Scholar]
- Law, R. J., L. R. Forrest, K. M. Ranatunga, P. La Rocca, D. P. Tieleman, and M. S. P. Sansom. 2000. Structure and dynamics of the pore-lining helix of the nicotinic receptor: MD simulations in water, lipid bilayers and transbilayer bundles. Proteins: Struct. Func. Genet. 39:47–55. [DOI] [PubMed] [Google Scholar]
- Lear, J. D., Z. R. Wasserman, and W. F. DeGrado. 1988. Synthetic amphiphilic peptide models for protein ion channels. Science. 240:1177–1181. [DOI] [PubMed] [Google Scholar]
- Leonard, R. J., C. G. Labarca, P. Charnet, N. Davidson, and H. A. Lester. 1988. Evidence that the M2 membrane-spanning region lines the ion channel pore of the nicotinic receptor. Science. 242:1578–1581. [DOI] [PubMed] [Google Scholar]
- Lester, H. 1992. The permeation pathway of neurotransmitter-gated ion channels. Annu. Rev. Biophys. Biomol. Struct. 21:267–292. [DOI] [PubMed] [Google Scholar]
- Liu, Y. S., P. Sompornpisut, and E. Perozo. 2001. Structure of the KesA channel intracellular gate in the open state. Nature Struct. Biol. 8:883–887. [DOI] [PubMed] [Google Scholar]
- Marassi, F. M., and S. J. Opella. 1998. NMR structural studies of membrane proteins. Curr. Opin. Struct. Biol. 8:640–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitton, P., and M. S. P. Sansom. 1996. Molecular dynamics simulations of ion channels formed by bundles of amphipathic α-helical peptides. Eur. Biophys. J. 25:139–150. [Google Scholar]
- Miyazawa, A., Y. Fujiyoshi, M. Stowell, and N. Unwin. 1999. Nicotinic acetylcholine receptor at 4.6 angstrom resolution: transverse tunnels in the channel wall. J. Mol. Biol. 288:765–786. [DOI] [PubMed] [Google Scholar]
- Montal, M. 1995. Design of molecular function: channels of communication. Annu. Rev. Biophys. Biomol. Struct. 24:31–57. [DOI] [PubMed] [Google Scholar]
- Montal, M. O., L. K. Buhler, T. Iwamoto, J. M. Tomich, and M. Montal. 1993a. Synthetic peptides and four-helix bundle proteins as model systems for the pore-forming structure of channel proteins. I. Transmembrane segment M2 of nicotinic cholinergic receptor channel is a key pore-lining structure. J. Biol. Chem. 268:14601–14607. [PubMed] [Google Scholar]
- Montal, M. O., T. Iwamoto, J. M. Tomich, and M. Montal. 1993b. Design, synthesis and functional characterization of a pentameric channel protein that mimics the presumed pore structure of the nicotinic cholinergic receptor. FEBS Lett. 320:261–266. [DOI] [PubMed] [Google Scholar]
- Oiki, S., W. Danho, V. Madison, and M. Montal. 1988. M2δ, a candidate for the structure lining the ionic channel of the nicotinic cholinergic receptor. Proc. Natl. Acad. Sci. USA. 85:8703–8707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opella, S. J., F. M. Marassi, J. J. Gesell, A. P. Valente, Y. Kim, M. Oblatt-Montal, and M. Montal. 1999. Structures of the M2 channel-lining segments from nicotinic acetylcholine and NMDA receptors by NMR spectroscopy. Nat. Struct. Biol. 6:374–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opella, S. J., C. Ma, and F. M. Marassi. 2001. Nuclear magnetic resonance of membrane-associated peptides and proteins. Methods Enzymol. 339:285–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panicker, S., H. Cruz, C. Arrabit, and P. A. Slesinger. 2002. Evidence for a centrally located gate in the pore of a serotonin-gated ion channel. J. Neurosci. 22:1629–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perozo, E., D. M. Cortes, and L. G. Cuello. 1998. Three-dimensional architecture and gating mechanism of a K+ channel studied by EPR spectroscopy. Nat. Struct. Biol. 5:459–469. [DOI] [PubMed] [Google Scholar]
- Perozo, E., D. M. Cortes, and L. G. Cuello. 1999. Structural rearrangements underlying K+-channel activation gating. Science. 285:73–78. [DOI] [PubMed] [Google Scholar]
- Randa, H. S., L. R. Forrest, G. A. Voth, and M. S. P. Sansom. 1999. Molecular dynamics of synthetic leucine-serine ion channels in a phospholipid membrane. Biophys. J. 77:2400–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves, D. C., and S. C. R. Lummis. 2002. The molecular basis of the structure and function of the 5-HT3 receptor: a model ligand-gated ion channel. Mol. Membr. Biol. 19:11–26. [DOI] [PubMed] [Google Scholar]
- Revah, F., D. Bertrand, J. L. Galzi, A. Devillers-Thiery, C. Mulle, N. Hussy, S. Bertrand, M. Ballivet, and J. P. Changeux. 1991. Mutations in the channel domain alter desensitization of a neuronal nicotinic receptor. Nature. 353:846–849. [DOI] [PubMed] [Google Scholar]
- Roux, B. 1997. Influence of the membrane potential on the free energy of an intrinsic protein. Biophys. J. 73:2980–2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiz, L., and M. L. Klein. 2002a. Computer simulation studies of model biological membranes. Acc. Chem. Res. 35:482–489. [DOI] [PubMed] [Google Scholar]
- Saiz, L., and M. L. Klein. 2002b. Structure of the pore region of the nicotinic acetylcholine receptor ion channel: a molecular dynamics simulation study. Biophys. J. 82:560a. [Google Scholar]
- Sansom, M. S. P. 1993. Structure and function of channel-forming peptaibols. Quart. Rev. Biophys. 26:365–421. [DOI] [PubMed] [Google Scholar]
- Sansom, M. S. P., and M. S. P. Biggin. 2001. Water at the nanoscale. Nature. 414:156–159. [DOI] [PubMed] [Google Scholar]
- Sansom, M. S. P., L. R. Forrest, and R. Bull. 1998. Viral ion channels: molecular modeling and simulation. Bioessays. 20:992–1000. [DOI] [PubMed] [Google Scholar]
- Sansom, M. S. P., G. R. Smith, C. Adcock, and P. C. Biggin. 1997. The dielectric properties of water within model transbilayer pores. Biophys. J. 73:2404–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, Y. F., Y. F. Kong, and J. P. Ma. 2002. Intrinsic flexibility and gating mechanism of the potassium channel KcsA. Proc. Nat. Acad. Sci. USA. 99:1949–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastava, I. H., and M. S. P. Sansom. 2000. Simulations of ion permeation through a potassium channel: molecular dynamics of KcsA in a phospholipid bilayer. Biophys. J. 78:557–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastava, I. H., and M. S. P. Sansom. 2002. Molecular dynamics simulations and KcsA channel gating. Eur. Biophys. J. 31:207–216. [DOI] [PubMed] [Google Scholar]
- Smart, O. S., J. Breed, G. R. Smith, and M. S. P. Sansom. 1997. A novel method for structure-based prediction of ion channel conductance properties. Biophys. J. 72:1109–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart, O. S., G. M. P. Coates, M. S. P. Sansom, G. M. Alder, and C. L. Bashford. 1998. Structure-based prediction of the conductance properties of ion channels. Faraday Discuss. 111:185–199. [DOI] [PubMed] [Google Scholar]
- Smart, O. S., J. M. Goodfellow, and B. A. Wallace. 1993. The pore dimensions of gramicidin A. Biophys. J. 65:2455–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart, O. S., J. G. Neduvelil, X. Wang, B. A. Wallace, and M. S. P. Sansom. 1996. HOLE: a program for the analysis of the pore dimensions of ion channel structural models. J. Mol. Graph. 14:354–360. [DOI] [PubMed] [Google Scholar]
- Song, Z., F. A. Kovacs, J. Wang, J. K. Denny, S. C. Shekar, J. R. Quine, and T. A. Cross. 2000. Transmembrane domain of M2 protein from influenza A virus studied by solid-state 15N polarization inversion spin exchange at magic angle NMR. Biophys. J. 79:767–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukharev, S., M. Betanzos, C. S. Chiang, and H. R. Guy. 2001. The gating mechanism of the large mechanosensitive channel MscL. Nature. 409:720–724. [DOI] [PubMed] [Google Scholar]
- Tajkhorshid, E., P. Nollert, M. O. Jensen, L. J. W. Miercke, J. O'Connell, R. M. Stroud, and K. Schulten. 2002. Control of the selectivity of the aquaporin water channel family by global orientational tuning. Science. 296:525–530. [DOI] [PubMed] [Google Scholar]
- Tang, P., P. K. Mandal, and Y. Xu. 2002. NMR structures of the second transmembrane domain of the human glycine receptor α1 subunit: model of pore architecture and channel gating. Biophys. J. 83:252–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tieleman, D. P., and H. J. C. Berendsen. 1996. Molecular dynamics simulations of a fully hydrated dipalmitoylphosphatidycholine bilayer with different macroscopic boundary conditions and parameters. J. Chem. Phys. 105:4871–4880. [Google Scholar]
- Tieleman, D. P., H. J. C. Berendsen, and M. S. P. Sansom. 1999a. An alamethicin channel in a lipid bilayer: molecular dynamics simulations. Biophys. J. 76:1757–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tieleman, D. P., P. C. Biggin, G. R. Smith, and M. S. P. Sansom. 2001. Simulation approaches to ion channel structure-function relationships. Quart. Rev. Biophys. 34:473–561. [DOI] [PubMed] [Google Scholar]
- Tieleman, D. P., B. Hess, and M. S. P. Sansom. 2002. Analysis and evaluation of channel models: simulations of alamethicin. Biophys. J. In press.83:2393–2407. [DOI] [PMC free article] [PubMed]
- Tieleman, D. P., M. S. P. Sansom, and H. J. C. Berendsen. 1999b. Alamethicin helices in a bilayer and in solution: molecular dynamics simulations. Biophys. J. 76:40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobias, D. J. 2001. Electrostatics calculations: recent methodological advances and applications to membranes. Curr. Opin. Struct. Biol. 11:253–261. [DOI] [PubMed] [Google Scholar]
- Tobias, D. J., K. C. Tu, and M. L. Klein. 1997. Atomic-scale molecular dynamics simulations of lipid membranes. Curr. Opin. Coll. Interface Sci. 2:15–26. [Google Scholar]
- Unwin, N. 1993. Nicotinic acetylcholine receptor at 9 Å resolution. J. Mol. Biol. 229:1101–1124. [DOI] [PubMed] [Google Scholar]
- Unwin, N. 1995. Acetylcholine receptor channel imaged in the open state. Nature. 373:37–43. [DOI] [PubMed] [Google Scholar]
- Unwin, N. 2000. The Croonian Lecture 2000. Nicotinic acetylcholine receptor and the structural basis of fast synaptic transmission. Phil. Trans. Roy. Soc. Lond. B. 355:1813–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarroel, A., S. Herlitze, M. Koenen, and B. Sakmann. 1991. Location of a threonine residue in the α-subunit M2 transmembrane segment that determines the ion flow through the acetylcholine receptor channel. Proc. R. Soc. Lond. B Biol. Sci. 243:69–74. [DOI] [PubMed] [Google Scholar]
- Weber, W., P. H. Hunenberger, and J. A. McCammon. 2000. Molecular dynamics simulations of a polyalanine octapeptide under Ewald boundary conditions: influence of artificial periodicity on peptide conformation. J. Phys. Chem. B. 104:3668–3675. [Google Scholar]
- Zhong, Q., Q. Jiang, P. B. Moore, D. M. Newns, and M. L. Klein. 1998a. Molecular dynamics simulation of an ion channel. Biophys. J. 74:3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, Q., P. B. Moore, D. M. Newns, and M. L. Klein. 1998b. Molecular dynamics study of the LS3 voltage-gated ion channel. FEBS Lett. 427:267–270. [DOI] [PubMed] [Google Scholar]