

Abstract
Insights into the interacting mode of CXCR4 with SDF-1α are crucial in understanding the structural and functional characteristics of CXCR4 receptor. In this paper a computational pipeline, integrating protein structure prediction, molecular dynamics simulations, automated molecular docking, and Brownian dynamics simulations were employed to investigate the dynamic and energetic aspects of CXCR4 associating with SDF-1α. The entire simulation revealed the surface distribution feature of electrostatic potentials and conformational “open-close” process of the receptor. The possible binding conformation of CXCR4 was identified, and the CXCR4—SDF-1α binding complex was generated. Arg188-Glu277 salt bridge plays an important role for both the extracellular domain conformational change and SDF-1α binding. Two binding sites were mapped at the extracellular domain (Site 1) and inside the transmembrane domain (Site 2), which are composed of conserved residues. Sites 1 and 2 contribute ∼60% and 40% to the binding affinity with SDF-1α, respectively. The binding model is in agreement with most of the experimental data. Transmembrane VI has more significant motion in the harmonious conformational transition of CXCR4 during SDF-1α binding, which may be possibly associated with signal transduction. Based on the modeling and simulation, a binding mechanism hypothesis between CXCR4 and SDF-1α and its relationship to the signal transduction has been proposed.
INTRODUCTION
Chemokines are the largest superfamily of cytokines that regulate the recruitment of various types of leukocytes sites associated with inflammation and many other immune responses (Zlotnik and Yoshie, 2000; Mackay, 2001; Moser and Loetscher, 2001). Stromal cell-derived factor-1α (SDF-1α) is a CXC chemokine with chemoattractant activity for lymphocytes, monocytes, and their progenitor cells (Gerard and Rollins, 2001). It is the only known endogenous ligand for the CXC chemokine receptor 4 (CXCR4) (Schwarz and Wells, 1999). The crystal structure of a variant SDF-1α ([N33A]SDF-1α; see Dealwis et al., 1998; PDB code of 1A15 at 2.2 Å resolution) has shown that SDF-1α adopts a typical chemokine β-β-β-α topology. The NMR studies (Crump et al., 1997; Elisseeva et al., 2000) have demonstrated that SDF-1α binds with the CXCR4 in the form of monomer and the N-terminal eight residues form an important receptor binding patch. Modification of Lys1 and/or Pro2 (Crump et al., 1997) results in loss of activity of CXCR4, but still generates potent SDF-1α antagonist. It was also proposed (Crump et al., 1997; Loetscher et al., 1998) that the RFFESH motif (residues 12–17) is another receptor binding site and launches the initial docking of SDF-1α with its receptor.
CXCR4 belongs to the peptide receptor and rhodopsin-class of G-protein–coupled receptor (GPCR) superfamily (Loetscher et al., 1994). It has been identified as a coreceptor for T-tropic HIV-1 and CD4-independent HIV-2 fusion and infection (Zhao et al., 1999; Moore and Stevenson, 2000; Chabot and Broder, 2000). The coreceptor activity can be impaired by amino acid changes of the conserved elements in CXCR4, especially the conserved residues at the Nter (Brelot et al., 2000). More recent investigations provided molecular support that SDF-1α and CXCR4 have a critical role in determining the metastatic destination of breast cancer cells (Liotta, 2001; Thelen, 2001). Neutralizing the interaction of SDF-1α with CXCR4 can significantly impair metastasis of breast cancer cells to regional lymph nodes and lung (Muller et al., 2001). Therefore, the SDF-1α and its receptor CXCR4 might qualify as targets for “chemoprevention”—the hope for new therapeutics of breast cancer (Schwarz and Wells, 1999; Muller et al., 2001).
Although molecular and cell biological studies have provided some essential insights into the functions of both SDF-1α and CXCR4, the increasingly prominent roles of SDF-1α and CXCR4 in the regulation of HIV-1 infection and metastasis of breast cancer have also been revealed (Gerard and Rollins, 2001; Muller et al., 2001; Babcock et al., 2001; Cheng et al., 2000). These insights have in turn raised new questions. For example, the structural elements of CXCR4 that mediate the interaction with SDF-1α have not been precisely defined due to the difficulty for determining its x-ray structure; in addition, the mode for SDF-1α binding with CXCR4 and the receptor activation has not been clearly demonstrated at molecular level. Therefore, exploring the conformational determinants of the extracellular domain of CXCR4, investigating the dynamic features of CXCR4—SDF-1α binding process, and mapping the receptor activating mechanism become of great interest to us. Structure-function relationship studies (Zhao et al., 1999; Ling et al., 1999; Chabot and Broder, 2000; Cheng et al., 2000; Bennett et al., 2001) demonstrated the importance of CXCR4—SDF-1α in pharmaceutical research, further motivating this theoretical study.
Computational simulation studies can aid in the interpretation of mutagenesis, binding, and other experimental data on GPCRs, and can provide new clues for identifying covered functions of GPCRs and for designing new ligands. Recently, great success has been achieved in the field of structure prediction of GPCRs (Perera et al., 2000; Orry and Wallace, 2000). Combining with experimental information, a lot of matured algorithms (Filizola et al., 1999; Jayasinghe et al., 2001; Baker and Sali, 2001) are capable of predicting the TM structures (Palczewski et al., 2000; Teller et al., 2001). However, constructing appropriate conformational spaces of extracellular or intracellular domains remains a major challenge in the 3D structural modeling of GPCRs. Long-time MD simulations (Duan and Kollman, 1998) take the advantage of iteratively tracking the trajectory of conformational change, and therefore, may capture the ligand binding (or bioactive) conformation of GPCRs. In this study, a robust approach, integrating homology modeling, long-time MD simulations, molecular docking, and Brownian dynamics, has been employed in studying the association process of SDF-1α with CXCR4. The simulation results elucidated the following aspects about CXCR4—SDF-1α interaction: what kind of conformation (bioactive conformation) CXCR4 adopts when it associates with SDF-1α; how SDF-1α binds with CXCR4; and how SDF-1α induces the signal transduction. In addition, these simulations are, to our knowledge, the first of long-time MD study that reveals the bioactive conformation of CXCR4 associating with SDF-1α.
MATERIALS AND METHODS
Experimental studies (Dealwis et al., 1998; Crump et al., 1997; Elisseeva et al., 2000; Loetscher et al., 1998; Zhao et al., 1999; Chabot and Broder, 2000) have indicated that SDF-1α binds with CXCR4 at two binding sites; one lies on the ED and the Nter of CXCR4, and must have a pivotal role in the binding site formation. As mentioned above, there are several methods for modeling the structure of TMs. However, it is difficult for constructing the structure of ED. Furthermore it is more difficult to identify the possible bioactive conformation for CXCR4 that SDF-1α binds with. To solve these problems, we integrated several modeling and simulation methods in this study. The computational pipeline is outlined in Scheme 1. Briefly, the computational flow is as follows. 1) Segmented approach including homology modeling was used to construct the 3D structural model of CXCR4. 2) Long-time MD simulations were carried out on the flexible ED of CXCR4; the lower-energy conformations in the MD trajectory were picked out for following protein-protein docking simulations. 3) Protein-protein docking was divided into two steps: first, the probing ligand extracted from the Nter of SDF-1α was docked to the possible binding site inside the TMs using automated molecular docking method. Next, the BD simulations were performed on the association of the rest part of SDF-1α with all the possible low energy conformations of CXCR4, thus finding the possible binding configuration of SDF-1α—CXCR4 complex according to the binding features and binding free energy. 4) Finally, the entire binding complex was further refined using the molecular mechanics method.
SCHEME 1.

3D structural modeling of CXCR4
The modeling described here utilizes a segmented approach where the N terminus, the transmembranes, and the extracellular loop regions were separately modeled. The intracellular loops and the C terminus were not modeled, as they are not directly involved in the binding of SDF-1α.
Modeling α-helix bundle
Sequence analysis and conserved-residues identification were carried out among chemokine receptors and other rhodopsin-like GPCRs, totally 49 sequences of CC and CXC types were from http://www.expasy.ch/prosite and 115 sequences from http://www.gpcr.org/7tm. Using the crystal structure of bovine rhodopsin (PDB code of 1F88 at 2.80 Å resolution, Palczewski et al., 2000; Teller et al., 2001) as a template, the Homology module of InsightII (version 2000, Accelrys, San Diego, CA) and the ClustalW algorithm (Thompson et al., 1994) were applied in sequence alignment, and the Blosum scoring matrix (Henikoff and Henikoff, 1992) was employed to obtain the best-fit alignment. The best alignment was selected not only according to the value of the alignment score, but also the reciprocal positions of conserved residues. The TMs domain was identified and transformed into α-helices and the nonequivalent amino acids were mutated to produce CXCR4 sequence.
Modeling extracellular loops and Nter
The FASTA program (Pearson, 1990) was used to identify sequence homologs through the in-house database (Huang et al., 2000) containing 700 loops and proteins with medium to high sequence identity. ClustalW (Thompson et al., 1994) was then used to determine the fragments that had higher homology with the loops and the Nter of CXCR4. The reasonable fragment conformation was chosen from the top 10 candidates that had the lowest root mean square (RMS) values and considerable geometrical compatibility. The conserved disulfide bond between residues Cys109 at the beginning of TM III and Cys186 at the middle of extracellular loop 2 (EL-2) was also created and kept as a constraint in the geometric optimization.
Structure optimization
The receptor was optimized using the molecular mechanics method with the following parameters: a distance-dependent dielectric constant of 5.0; nonbonded cutoff 8 Å, Amber force field (Cornell et al., 1995) and Kollman-all-atom charges; and conjugate gradient minimization until the energy gradient RMS <0.05 kcal (mol Å)−1. The whole receptor was minimized to convergence. At the end, the HBPLUS algorithm (McDonald and Thornton, 1994) was used to calculate the interhelical hydrogen bonds, and the modeled structure was validated with PROCHECK (Laskowski et al., 1993) and WHATIF (Vriend and Sander, 1993).
Molecular dynamics simulations
The MD simulation was run on a 64 CPU parallel computer using the program EGO_VIII (Eichinger et al., 2000) including the CHARMM19 force field (Brooks et al., 1983). The TIP3P water model (Jorgensen et al., 1983; Neria et al., 1996) was used to simulate the solvent. The most popular Verlet algorithm (Verlet, 1967) was adopted to integrate the equation of motion and the FAMUSAMM algorithm (Eichinger et al., 1997) was applied to rapidly evaluate electrostatic interactions, while the lengths of bonds involving hydrogen atoms were held fixed with the SHAKE algorithm (Ryckaert et al., 1977).
The initial coordinates of the ED of CXCR4 were extracted from the energy minimized structural model of CXCR4 receptor. The end of each loop together with the end of the Nter connecting helix I was three residues extended to the relative helices. The backbone of these three residues and the peptide bond at the seven ends were kept in torsion angle constraints so that the whole ED could connect to the TMs domain. The ionization states at neutral pH for the acidic, basic residues, and histidines were determined through pKa calculation by using the DelPhi module of molecular modeling software, InsightII (release 2000, Accelrys). The solute was then solvated at the center of a sphere of water molecules, which ensures the whole simulation system to be covered by water molecules at least 10-Å thick. The seven ends connecting the loops and Nter were fixed by adding a stiff harmonic potential so that the loops and Nter had enough moving space during the whole simulation process. To provide a neutral simulation system, the SOLVATE release 1.0 (http://www.mpibpc.gwdg.de.abteilungen/071/solvate/node4.html) was applied to add the counterions in the bulk solution; therefore, the sodium ions were placed in the solvent volume at physiological concentration (0.154 M) obeying the Debye-Hückel distribution. Each charged atom at the surface of the solute was surrounded by a “cloud” of so-called counterions, and the size of this cloud was given by the Debye-Hückel length. The total number of atoms in the simulation system was 26,932, including 1424 atoms of the protein and 8500 water molecules.
After the initial structures were prepared, energy minimization was performed at constant volume for all the water molecules with the criterion of the maximum force of the whole system decreased below 10.00 kcal(mol Å)−1, to optimize poor steric contacts. And then, the whole system was energy minimized with the same criterion. The friction factor τ was set as 0.1 at the end of each integration step during the minimization process, and then changed to 1.0 for the MD equilibrium simulation. As the minimization convergence was reached, the whole system was directly subjected to a slow heating procedure for ∼20 ps in a heat reservoir of 300 K. After that, the system was performed a 2.5-ns (2.5 × 10−9 s) MD simulation. Because the FAMUSAMM algorithm (Eichinger et al., 1997) was adopted, 1 fs (1 × 10−15 s) was used as the time step, and the frequency for analyzing the MD output was set as 1 ps (1 × 10−12 s).
CXCR4—SDF-1α association simulation
Automated molecular docking
For the reason of tackling the interacting mode of SDF-1α with CXCR4, the CAST program (http://cast.engr.uic.edu; see Liang et al., 1998) was used to identify the possible voids and pockets situated in the TMs domain, where were the potential sites for ligand binding (Site 2). Since the Nter of SDF-1α is flexible, the small molecule-protein docking method was used in identifying the possible binding conformation of the Nter. Employing the AutoDock3.0 program (Morris et al., 1998), different probing polypeptides (the Lys-Pro, Lys-Pro-Val, and the Lys-Pro-Val-Ser), extracted from the end of Nter of SDF-1α, were docked into the pocket candidates derived from CAST calculations. The N terminus of these peptides was treated as protonated state to simulate the actual environment of SDF-1α interacting with CXCR4. During the docking process, conformational search was performed using the Solis and Wets local search method (Solis and Wets, 1981), and the Lamarckian genetic algorithm was applied to deal with the ligand-receptor interaction. A series of docking parameters were set on. Not only the atom types but also the generations and the number of runs for the LGA algorithm were edited and properly assigned according to the requirement of the Amber force field. The number of generation, energy evaluation, and docking runs was set to 370,000, 1,500,000, and 10, respectively. The interacting energies of the probing ligands with CXCR4 were assessed by the empirical binding free energy function including the desolvation and the hydrophobic effect (Morris et al., 1998). Thus the score function was sufficient to rank the binding conformations of probing ligands and their orientations associating with Site 2 of CXCR4.
Brownian dynamics simulations
After getting the appropriate binding conformation and orientation of the Nter, the binding feature of the rest part of SDF-1α with CXCR4 can be simulated by the BD method. The flexibility of the ED of CXCR4 was considered by respectively docking SDF-1α into all the possible energy-minima conformations of CXCR4 that were extracted from the above MD simulations. The MacroDox program package (S.H. Northrup, Tennessee Technological University, Cookeville, TN) was used to perform BD simulations.
The detail procedure of BD simulation has been described in several papers (Northrup et al., Tennessee Technological University; Warwicker and Watson, 1982; Smoluchowski, 1917; Ermak and McCammon, 1978; Cui et al., 2001, 2002). Here we only briefly introduce the BD simulation process. The new updated charge file of CHARMM22 (Brooks et al., 1983) was used to assign the charges for SDF-1α and CXCR4. The Tanford-Kirkwood method of Matthew (1985; Matthew and Gurd, 1986) was employed to determine the protonation status of each titratable residue of two proteins at pH of 7.0 and ionic strength of 0.1 M. As the main purpose of BD was to find reasonable recognition conformation of SDF-1α with CXCR4 on the scale of feasible computational facility, the TK recommended partial charges were assigned to CXCR4 and formal charges were assigned to SDF-1α, and the total charge was 9.86 e for ED-CXCR4 and 7.20 e for SDF-1α. Although this test charge model was not as efficient as that of the effective charge method, it has been demonstrated in our previous work that the test charge model produced reasonable results for the interaction of protein-protein binding (Cui et al., 2001, 2002; Fu et al., 2002), therefore, it was applied again in the present study. After charge assignment, the electrostatic potentials about CXCR4 and SDF-1α were calculated by numerically solving the linearized Poisson-Boltzmann equation using the method of Warwicker and Watson (Warwicker and Watson, 1982). The protein interior dielectric constant and solvent dielectric constant were set as 4.0 and 78.3, respectively.
The BD simulation of the two interacting macromolecules in solvent was run stochastically by a series of small displacements chosen from a distribution that is equivalent to the short time solution of the Smoluchowski diffusion equation (Smoluchowski, 1917) derived from different forces. The basic Ermak-McCammon algorithm (Ermak and McCammon, 1978) was employed to simulate the translational and rotational Brownian motion of two interacting proteins. BD simulations of SDF-1α binding to CXCR4 were performed to identify the possible favorable complex, typically by running 3000 trajectories. The mobile SDF-1α was subject to three forces: electrostatic attraction between two proteins, random Brownian force, and frictional force due to solvent viscosity. The closest approaches of SDF-1α complexing with CXCR4 were recorded as trajectories.
Binding model refinement
Synthesizing the results of molecular docking and BD simulations, the bioactive conformation of CXCR4 and the possible SDF-1α—CXCR4 binding model were obtained. During this process, the best binding model was identified from a set of 10 candidates resulting from molecular docking and BD simulations, and the geometrical complementary and the lowest-energy principle were adopted as the criteria. The final structure of the complex was subjected to energy minimization using the same molecular mechanics method just as that for structural optimization of the receptor. The details of the interaction were analyzed using the LIGPLOT program (Wallace et al., 1995).
RESULTS
Structural model of CXCR4 receptor
Structural features
The primary 3D model of CXCR4 (TMs + ED) that resulted from the structural modeling is shown in Fig. 1. The PROCHECK (Laskowski et al., 1993) statistics showed that 90% of the residues in the CXCR4 model are in either the most favored or in the additionally allowed regions of the Ramachandran plot. The overall main chain and side chain parameters, as evaluated by PROCHECK, are all very favorable. The WHATIF (Vriend and Sander, 1993) validation found loose RMS Z-scores, which are typical of modeled structure. The topology of TMs arrangement is in accordance with the helical conformation of GPCRs (Table 1).
FIGURE 1.
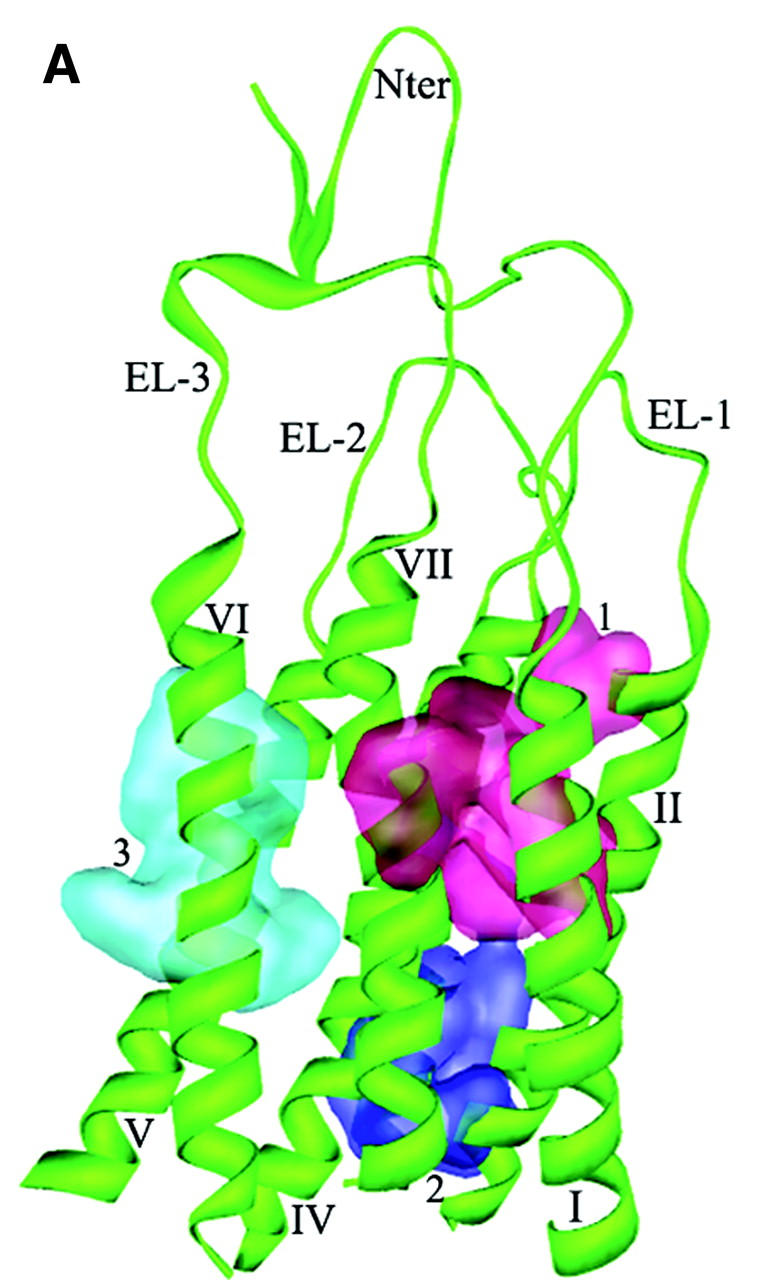

Cartoon representation of 3D model for CXCR4 receptor. Intramolecular hydrophobic interactions represented as three aromatic clusters are also shown: 1) cluster among TMs I, II and VII; 2) cluster among TMs II, III and IV; and 3) cluster among TMs VI and VII. (A) Side view. (B) Top view from the intracellular side (without loops and Nter for clear visual of TMs).
TABLE 1.
Structural data for TMs of CXCR4
| TM | TM center (x, y, z)/Å | Tilt angle of TM (θ)/degree | Bends within TM (χ)/degree |
|---|---|---|---|
| TM I | 12.34, −6.02, −3.28 | 20.96 | Almost straight |
| TM II | 10.16, 2.15, −2.04 | 26.78 | 23.06 (Pro92) |
| TM III | −1.76, 3.75, −0.75 | 27.89 | Almost straight |
| TM IV | −3.29, 13.12, −0.48 | 5.10 | 5.51 (Pro163); 13.48 (Pro170) |
| TM V | −11.87, 1.75, 0.78 | 36.00 | 23.72 (Pro211) |
| TM VI | −7.47, −8.39, 0.65 | 15.96 | 16.12 (Pro254) |
| TM VII | 2.65, −7.10, 2.41 | 5.89 | 6.72 (Pro299) |
| TM packing angles | ||||||
|---|---|---|---|---|---|---|
| TM1-TM2 | TM2-TM3 | TM3-TM4 | TM4-TM5 | TM5-TM6 | TM6-TM7 | TM7-TM1 |
| 156.98 | 159.55 | 157.17 | 149.25 | 160.15 | 161.57 | 164.52 |
Networks of intramolecular interactions
Two kinds of interacting networks are observed: aromatic residue clusters and hydrogen bond (H-bond). In the first network, aromatic residues are observed to assemble three clusters, maintaining the geometry of the TMs by the favorable stacking interactions as in known membrane proteins (Adamian and Liang, 2001; Ulmschneider and Sansom, 2001). One aromatic cluster (Fig. 1, cluster 1) locates at the upside of TMs I, II, and VII, being composed of the conserved residues Tyr45, Phe49, Phe87, Trp94, Phe292, and Phe293; these aromatic interactions make TMs I, II, and VII congregate tightly. Another aromatic cluster (Fig. 1, cluster 2) locates close to the lower part of TMs II, III, and IV formed by the side chains of Tyr76, Tyr121, Phe129, Tyr157, and Trp161. The hydrophobic environment formed by these residues directly affects the conformational changes of the functionally important motif DRY at the C-terminal of TM III, especially the disruption and formation of the salt bridge between Asp133 and Arg134. The third aromatic cluster (Fig. 1, cluster 3), formed by residues Phe248, Phe249, Trp252, Tyr255, and Tyr256, packs TM VI with TMs V and VII through van-der-Waals interactions.
The second network is formed by hydrogen bonds between residues conserved across the chemokine receptor subtypes (Table 2). The presence of hydrogen bond interaction with only TM VII could allow relative movement of TM VI. The specific hydrogen bonds cluster formed among residue Tyr302 and the DRY motif of TM III may be one of the structural constraints keeping the receptor inactive.
TABLE 2.
Hydrogen bonds (beyond the backbone) formed by residues of TMs
| Donor
|
Acceptor
|
||||
|---|---|---|---|---|---|
| Location | Residue | Group | Group | Residue | Distance (Å) |
| TM4-TM2 | Trp161 | NɛH | Nɛ | His79 | 3.28 |
| TM2-TM4 | Tyr76 | OH | O=C< | Glu153 | 3.52 |
| TM2-TM4 | Lys75 | NɛH | OH | Tyr157 | 2.68 |
| TM4-TM2 | Lys154 | NɛH | OH | Ser71 | 2.70 |
| TM2-TM2 | Met72 | NH | OH | Ser71 | 3.00 |
| TM4-TM4 | Thr168 | OH | O=C< | Ala164 | 2.76 |
| TM1-TM2 | Asn56 | NδH | Oδ1− | Asp84 | 2.70 |
| TM1-TM2 | Asn56 | NδH | O=C< | Leu80 | 2.74 |
| TM2-TM2 | Thr90 | OH | O=C< | Leu86 | 2.74 |
| TM5-TM5 | Gln202 | NɛH | NɛH | His203 | 3.18 |
| TM5-TM5 | Ser217 | OH | O=C< | Ile213 | 2.74 |
| TM5-TM5 | Ser224 | OH | O=C< | Cys220 | 2.74 |
| TM6-TM6 | Thr240 | OH | O=C< | Ala237 | 2.71 |
| TM7-TM6 | Thr287 | OH | Oδ1 | Asp262 | 2.66 |
| TM1-TM7 | Tyr45 | OH | Oɛ1 | Glu288 | 2.60 |
| TM7-TM3 | Tyr302 | OH | Oδ1 | Asp133 | 2.61 |
| TM3-TM3 | Arg134 | Nη1H | Oδ2 | Asp133 | 2.59 |
| TM3-TM3 | Arg134 | Nη2H | Oδ2 | Asp133 | 2.60 |
| TM3-TM7 | Arg134 | Nη1H | OH | Tyr302 | 2.61 |
| TM2-TM7 | Arg77 | NɛH | O=C< | Ala303 | 3.26 |
| TM2-TM7 | Arg77 | Nη1H | O=C< | Ala303 | 2.73 |
| TM2-TM2 | Arg77 | Nη2H | O=C< | Thr73 | 2.73 |
| TM2-TM7 | Thr73 | OH | O=C< | Gly306 | 2.84 |
| TM2-TM2 | Ser81 | OH | O=C< | Leu78 | 2.71 |
| TM3-TM3 | His113 | NδH | O=C< | His113 | 2.87 |
| TM3-TM3 | Ser122 | OH | O=C< | Asn119 | 2.71 |
| TM3-TM3 | Ser123 | OH | O=C< | Leu120 | 2.75 |
| TM3-TM3 | Ser131 | OH | O=C< | Ala128 | 2.73 |
| TM7-TM7 | His294 | Nɛ | O=C< | Leu290 | 2.78 |
MD simulations
Structural dynamics
As indicated in Fig. 2, the total energy (potential energy plus kinetic energy) of the system decreases rapidly to the average value of −6.59 × 105 kcal/mol. After ∼200 ps, the whole system is becoming stable. Using the total energy fluctuation as the criteria for monitoring conformational changes, the local energy-minima conformations were identified and the corresponding points on the energy curve are also indicated in Fig. 2 (dotted lines).
FIGURE 2.

Total energy changes of the solvated CXCR4 system as the function of time. Dotted lines indicate the local energy-minima representative of typical conformations.
The time-evolution RMS deviation (RMSD) and relative RMS fluctuation (RMSF) of individual residues of the Nter are graphically shown in Fig. 3. Due to its flexible nature, the conformation of the Nter changes rapidly and dramatically as reflected in the RMSD curve (Fig. 3 A). Compared with the great fluctuations of several residues at the tail of Nter and residues of Tyr7 and Met16, the fluctuation scale of residues 9–14 is relatively small (Fig. 3 B). Conformational analysis demonstrates that there are two conserved motifs in the secondary structure of Nter. One motif (motif 1), formed by Ile4 to Tyr12, has the secondary structure of β-sheets. The superimposed conformations, taken from the snapshots with an interval of 50 ps plus those at the energy-minima, are graphically represented in Fig. 3 C. Residues Ile4 to Ile6 and Ser9 to Tyr12 have formed paralleling β-sheets, and residues Tyr7 to Thr8 are at the turn of the β-sheets. Another motif (motif 2) is composed of Thr13 to Asp22, which contains several negative-charged residues such as Glu14, Glu15, and Asp20. As shown in Fig. 3 D, residues Thr13 to Glu15 and Asp20 to Asp22 in motif 2 form a β-sheet–like secondary structure. The whole structures of these two motifs are conserved over a long time period as indicated by the curves of RMSD changes in Fig. 3 A.
FIGURE 3.

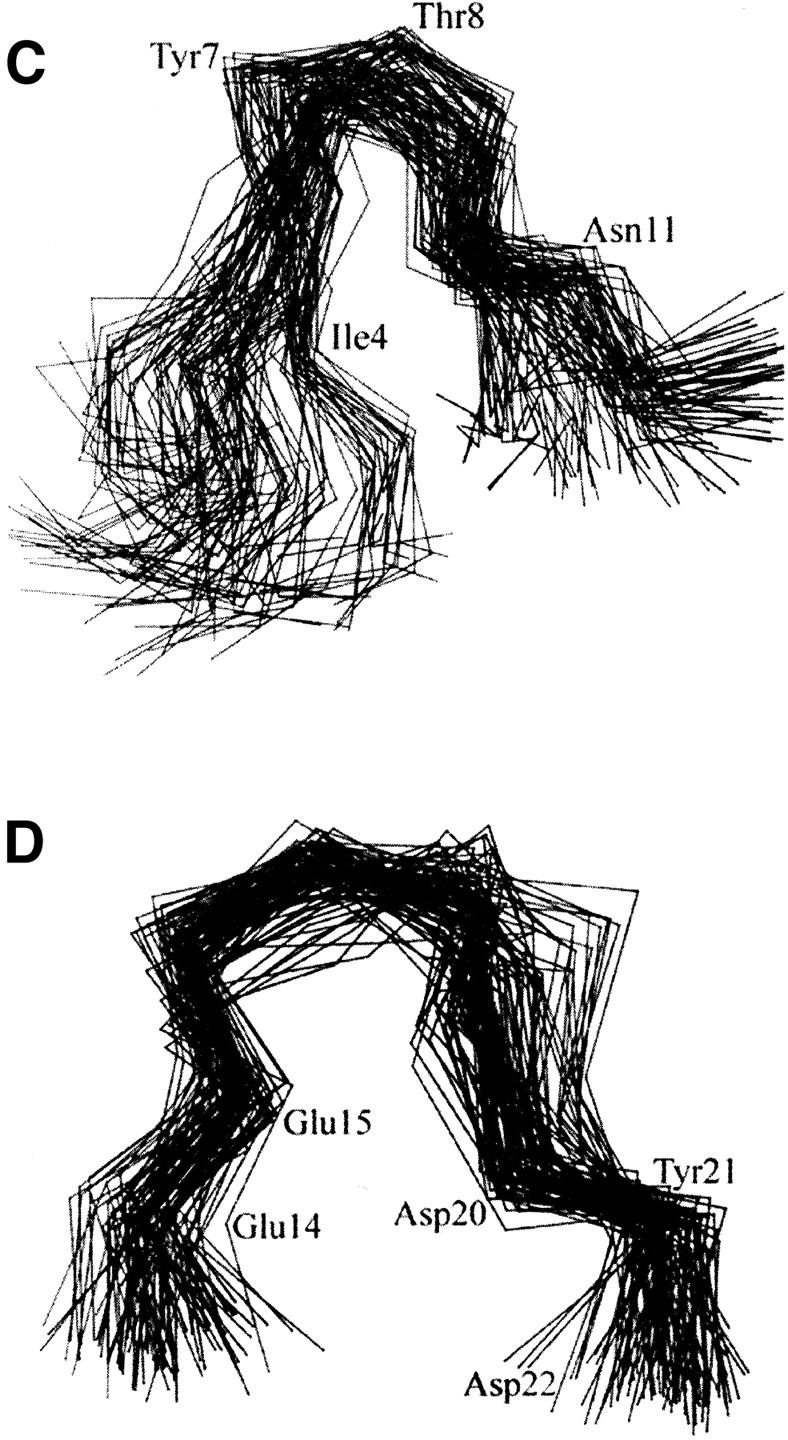
(A) Time evolution of RMSD from initial structure of MD simulations on Nter and two motifs: motif 1, from residue Ile4 to Tyr12 and motif 2, from residue Thr13 to Asp22. (B) The RMS fluctuations of the Nter and three loops (EL-1, EL-2, and EL-3) calculated from trajectories at 300 K. All the values were averaged over individual amino acids. (C) and (D) Stereo view of Cα superposition of conformations from MD snapshots every 50 ps for motif 1 (C) and motif 2 (D) at Nter.
Extracellular loops have small scale of structural movements except for EL-2. From the RMSF values in Fig. 3 B we can see that EL-1 and EL-3 have little structural perturbation on the whole ED-CXCR4. Conformational fluctuation of EL-3 comes from the flexibility of itself and the structural mediation by the Nter. Only several residues in EL-1 and EL-3 have great contribution to the RMS fluctuations of the loops. These residues are Val99, Trp102, and Phe104 from EL-1, and Ile269, Ile270, and Cys274 from EL-3. Most residues of EL-1 have been involved in hydrophobic interactions with the extracellular end of TMs I and II. Residues 188–195 of EL-2 contribute the most part to the conformational fluctuations.
Electrostatic properties of ED-CXCR4
To explore surface properties, electrostatic potentials on the surface of CXCR4 were calculated using the method as described above and representatively shown by local energy-minima conformations in Fig. 4. The surface of ED-CXCR4, which is in direct contact with the bulky solution at the outside of the cell membrane, bears a large part of negative electrostatic potential. The negative electrostatic potential results mainly from negative charged side chains of residues Glu2, Asp10, Glu14, Glu15, Asp20, Asp22, Glu26, Glu31, and Glu32 of the Nter; Asp97 of EL-1; Glu179, Asp181, Asp182, Asp187, and Asp193 of EL-2; and Glu269, Glu276, and Glu278 of EL-3. Although the electrostatic potential distribution was heavily modulated by the tertiary structure and the exterior shape of ED-CXCR4, the negative electrostatic potential around residues Asp10, Glu14, Glu15, and Asp20 kept highly conservative throughout all the conformational fluctuations (Fig. 4). From the viewpoint of Coulombic interactions, the negative electrostatic potential around these surface-exposed residues may act as the important attracting force in the initial step of association process for SDF-1α—CXCR4 interaction. Meanwhile, negative electrostatic potential area could also be found around the conserved residues of Asp262 and Glu288 inside the TMs domain, and this site is located ∼15 Å far away from the negative electrostatic potential field of the conserved cluster at Nter. The features of electrostatic potential distribution of one energy-minimum conformation at 1.228-ns trajectory as shown in Fig. 4 indicate that these two negative electrostatic potential sites would cooperatively interact with the positive-charged groups of a ligand.
FIGURE 4.
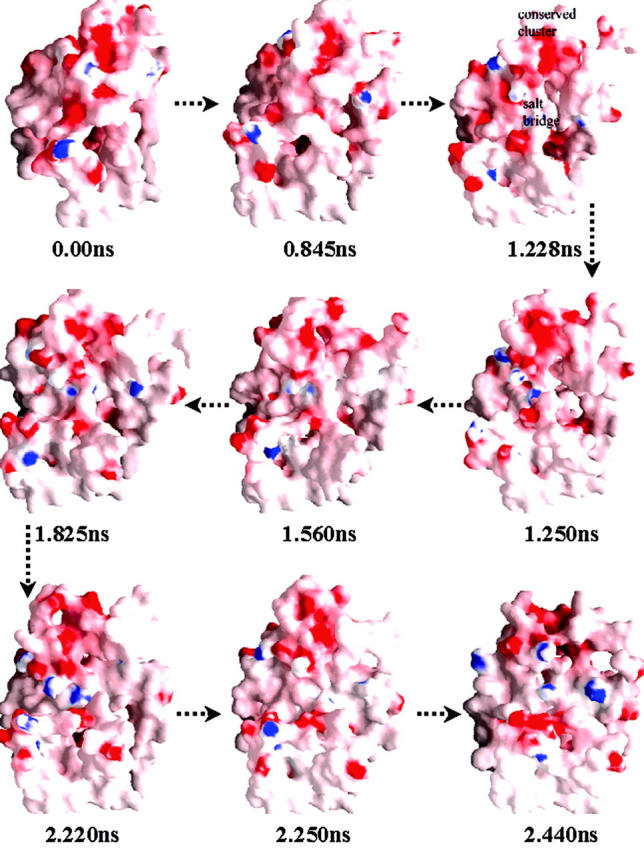
The energy-minima conformations (corresponding to dotted lines in Fig. 2) representing “opening” and “closing” of the binding site at ED-CXCR4. All conformations are shown in the style of molecular surface colored by its electrostatic potential (the color from red to blue on the color panel, which shows the electrostatic potential from negative to positive, numerically represents the range from −10.5 to +10.5). Most part of the TMs is not shown for visual clearance of ED-CXCR4. This figure was generated by using the GRASP (Nicholls et al., 1991) program.
Conformational “open-close” process of ED-CXCR4
Another interesting finding from the MD simulations is the opening and closing process of the “mouth” shaped void situated at the top part of ED-CXCR4 (Fig. 4). During the first several hundred ps, the “mouth” gradually opens up as the conformation of ED-CXCR4 becomes loosely packed. This change could be found from the comparison of the trajectory at 0.845 ns with the initial compact structure. The “mouth” grows up to be the largest ∼1.228-ns trajectory as the Nter and the three loops are at the most relaxed state. The conformation of ED-CXCR4 opens most extensively and readily to adopt complementary molecules at that period. The conserved electrostatic potential area around motif 2 is fully naked to the exterior solvents. This state of conformation lasts ∼100 ps as shown in Fig. 2. Gradually, the “mouth” comes into its closing period as the simulations go on. It becomes smaller and smaller after a time period of about 1.0 ns, and almost closes up at 2.44 ns (Fig. 4).
Arg188-Glu277 salt bridge
The charged residues from loops have a crucial function in the process of the active site formation at ED-CXCR4. Two representatives of them, Arg188 from EL-2 and Glu277 from EL-3 (which have close contacts and formed the “tooth” protruding into the “mouth” as obviously shown in Fig. 4), were selected to analyze the possible electrostatic interactions. Several interatomic distances between the charged groups of their side chains were computed and their interacting mode was examined (Fig. 5). Distances from Cζ (Arg188) to Cδ (Glu277), Nη1 (Arg188) to Cδ (Glu277), and Nη2 (Arg188) to Cδ (Glu277) are ∼5 Å at the first 400-ps simulation. Accordingly, the side chains of Arg188 and Glu277 interact with each other through direct electrostatic interactions and hydrogen bonding (Fig. 5 B). As the simulations move on, at ∼450 ps, the distances between these atoms increase to ∼8 Å (Fig. 5 A). At this time, the side chain of Glu277 turns away from the side chain of Arg188, breaking the direct H-bonds formed between these two side chains, and a network of H-bonds forms, linking the side chains of Arg188 and Glu277 by several water molecules. This dynamic state (Fig. 5 C) lasts ∼1.10 ns. This indicates that the salt bridge acts like a “bolt,” so that when it unlocks, the ED-CXCR4 may open its mouth. As will be seen later, when SDF-1α binds with CXCR4, this salt bridge adopts its unlocked state.
FIGURE 5.


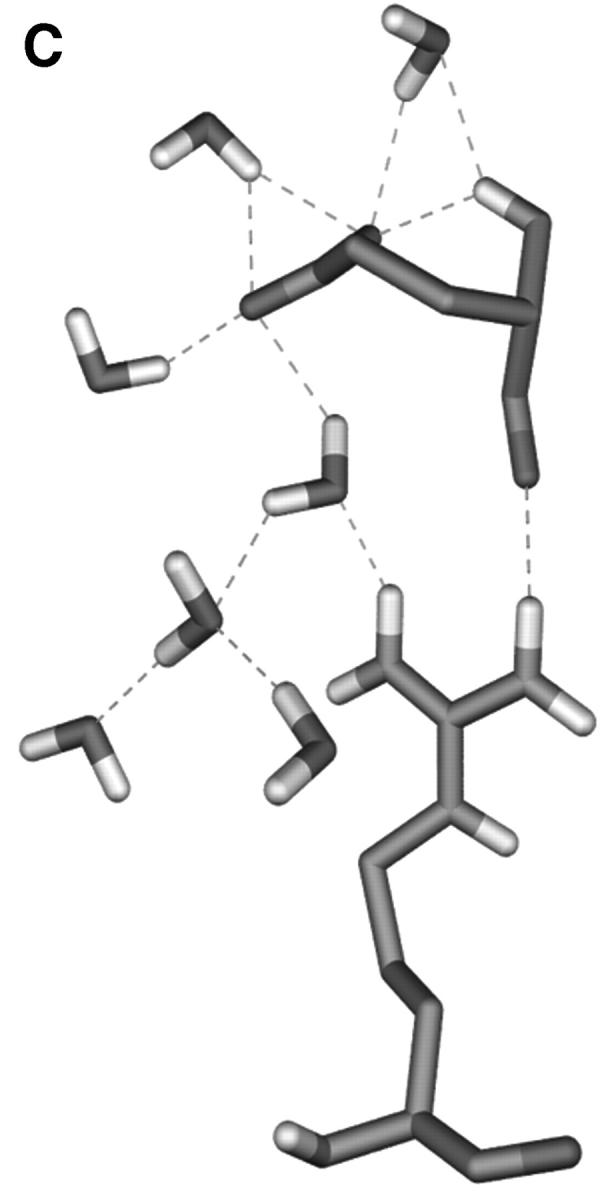
(A) Distance fluctuations for atoms (Cζ, Nη1, and Nη2 of Arg188; Cδ of Glu277) of the Arg188-Glu277 salt bridge. -▪-: Distance from CζArg188 to CδGlu277; -•-: distance from Nη1Arg188 to CδGlu277; and -▴-: distance from Nη2Arg188 to CδGlu277. (B) and (C) Typical interaction modes (B, Mode I; C, Mode II) of Arg188-Glu277 pair including relative water molecules.
Interactions between SDF-1α and CXCR4
Contacts between SDF-1α and CXCR4
CAST (Liang et al., 1998) calculations indicate that there is a ligand-binding pocket situated near the extracellular side of TMs domain among TMs III, V, VI, and VII (Site 2). This binding site is partly covered by ED-CXCR4, especially by the side chains of Arg188 and Phe189 in EL-2. Molecular docking identified the binding orientation and conformation for residues Lys1 to Leu5 at Nter of SDF-1α. BD simulations and binding free energy estimations, considering all the energy-minima conformations (Fig. 4) of ED-CXCR4 as the possible interactive conformations, demonstrate that conformation at 1.228 ns in the MD trajectory CXCR4 (Fig. 2) is the most favorable active conformation for SDF-1α binding. Glu2, Asp10, Glu14, Glu15, Asp20, and Asp22 at the Nter of CXCR4 are the major components for Site 1. Arg8, Arg12, Arg41, and Arg47 of SDF-1α may interact with Site 1 through electrostatic attraction.
Integrating results of molecular docking and BD simulations, we obtained the 3D model of SDF-1α—CXCR4 complex. This model was further optimized by molecular mechanics. The optimized structure of SDF-1α—CXCR4 complex, and the principal hydrophobic and hydrogen bonding interactions are represented in Fig. 6 and Table 3. In general, SDF-1α interacts with CXCR4 via its Nter by electrostatic and hydrophobic interactions, and hydrogen bonding. The atomic contacts between side chains of Arg188 and Glu277 were replaced by reasonable interactions between the ligand and the receptor, indicating the salt bridge is at its unlocked state in the ligand-receptor complex. The whole SDF-1α stays above the ED of CXCR4 with its cationic head penetrating into the binding pocket inside TMs domain (Site 2). As shown in Fig. 6 and Table 3, two major electrostatic interaction networks including hydrogen bonding are formed around the charged residue pairs: one is formed between Arg8 and Arg12 of SDF-1α and Glu15 and Asp20 of CXCR4, and another is formed between Lys1 of SDF-1α and Asp262 of CXCR4, which are in correspondence with the binding site in ED-CXCR4 (Site 1) and the binding site inside TMs domain (Site 2), respectively.
FIGURE 6.
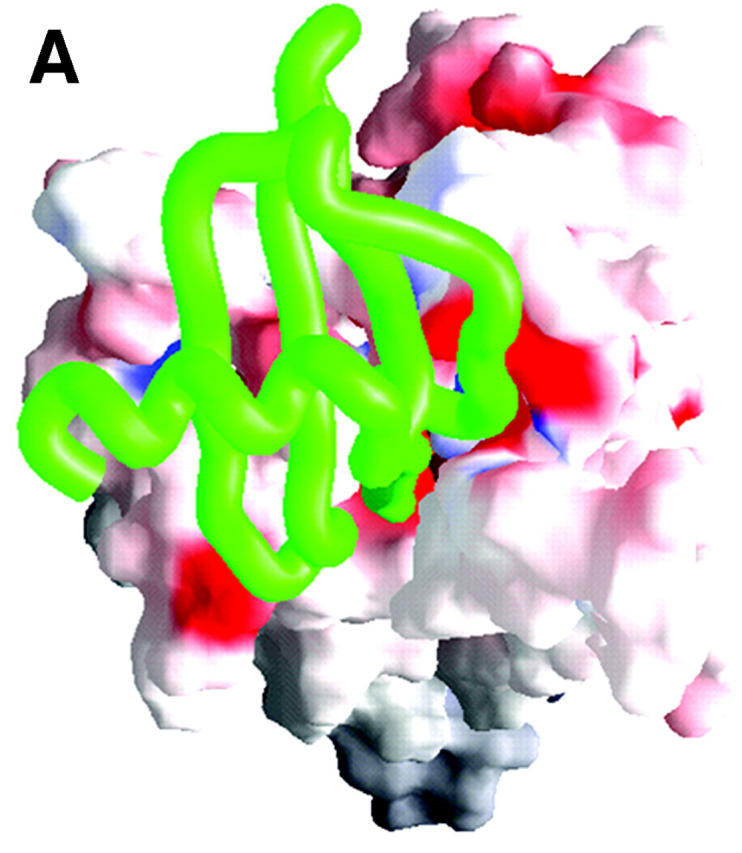
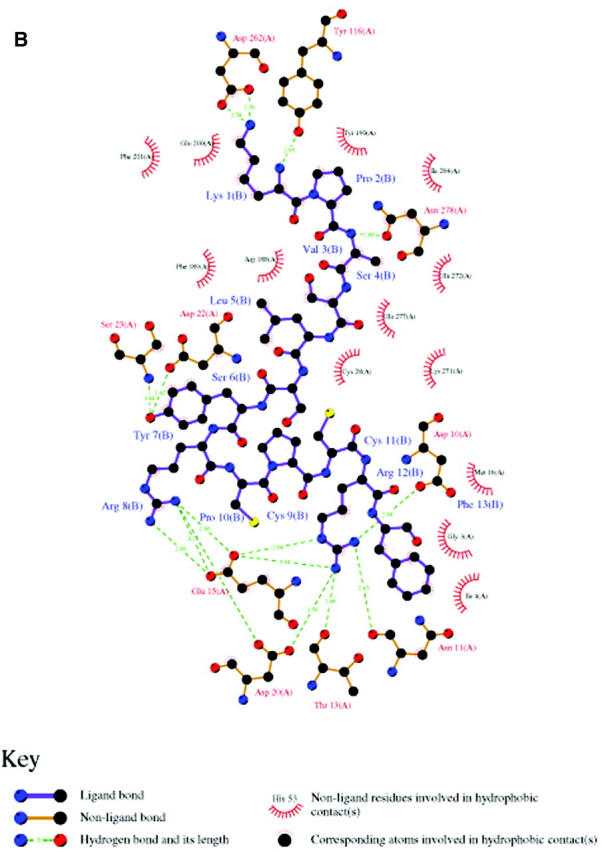
(A) A typical final complex of SDF-1α—CXCR4. CXCR4 is represented as a molecular surface colored by electrostatic potential (the range of the color panel is the same as that in Fig. 4), and SDF-1α as a green worm-like structure. (B) Schematic depiction (generated by using LIGPLOT program; see Wallace et al., 1995) of main interactions between SDF-1α (only the first 13 residues were included) and CXCR4.
TABLE 3.
Hydrophobic contacts and hydrogen bonds between side chains of SDF-1α and CXCR4
| Hydrophobic contacts
| ||||
|---|---|---|---|---|
| SDF-1α
|
CXCR4
|
|||
| Residue | Atom | Atom | Residue | Distance (Å) |
| Lys1 | C | Cδ1 | Ile284 | 3.81 |
| Lys1 | Cα | Cδ1 | Ile284 | 4.57 |
| Lys1 | Cγ | Cδ | Gln200 | 4.80 |
| Lys1 | Cβ | Cδ | Gln200 | 3.96 |
| Lys1 | Cγ | Cɛ1 | Phe201 | 4.77 |
| Lys1 | Cδ | Cɛ1 | Phe201 | 4.66 |
| Lys1 | Cγ | Cζ | Phe201 | 4.66 |
| Lys1 | Cδ | Cζ | Phe201 | 4.10 |
| Lys1 | Cɛ | Cγ | Asp262 | 3.39 |
| Pro2 | Cδ | Cɛ1 | Tyr190 | 3.94 |
| Pro2 | Cδ | Cζ | Tyr190 | 4.10 |
| Pro2 | Cα | Cδ1 | Ile284 | 4.56 |
| Val3 | Cα | Cα | Asn278 | 4.66 |
| Val3 | C | Cγ | Asn278 | 4.30 |
| Val3 | C | Cα | Asn278 | 4.26 |
| Val3 | C | Cβ | Asn278 | 4.32 |
| Val3 | Cβ | Cγ | Gln272 | 4.76 |
| Val3 | Cα | Cβ | Gln272 | 4.76 |
| Val3 | Cβ | Cβ | Gln272 | 3.55 |
| Ser4 | Cβ | Cδ2 | Phe189 | 4.76 |
| Ser4 | Cβ | Cɛ2 | Phe189 | 4.62 |
| Ser4 | C | Cβ | Glu277 | 4.88 |
| Ser4 | Cβ | Cγ | Arg188 | 3.92 |
| Ser4 | Cα | Cγ | Arg188 | 3.91 |
| Leu5 | Cδ1 | Cβ | Cys28 | 4.34 |
| Leu5 | Cδ2 | Cβ | Cys28 | 4.31 |
| Leu5 | Cδ2 | Sγ | Cys28 | 3.84 |
| Leu5 | Cδ2 | Cɛ | Lys271 | 3.53 |
| Leu5 | Cδ1 | Cɛ | Lys271 | 3.78 |
| Leu5 | Cδ2 | Cδ | Lys271 | 3.76 |
| Tyr7 | Cζ | Cβ | Ser23 | 3.57 |
| Tyr7 | Cɛ2 | Cβ | Ser23 | 3.36 |
| Tyr7 | Cδ2 | Cβ | Ser23 | 3.84 |
| Tyr7 | Cɛ1 | Cα | Asp22 | 3.68 |
| Arg8 | Cζ | C | Asp20 | 3.79 |
| Arg8 | Cζ | Cδ | Glu15 | 3.75 |
| Arg8 | Cδ | Cβ | Asn11 | 3.56 |
| Arg12 | Cγ | Cδ1 | Ile4 | 3.67 |
| Arg12 | Cζ | Cα | Gly3 | 3.85 |
| Phe13 | Cζ | Cγ1 | Ile4 | 3.56 |
| Phe13 | Cζ | C | Gly3 | 3.85 |
| Phe13 | Cδ2 | Cγ | Glu15 | 3.83 |
| Phe13 | Cδ2 | Cβ | Glu15 | 3.80 |
| Phe13 | Cβ | C | Met16 | 3.68 |
| Hydrogen bonds
| ||||
|---|---|---|---|---|
| Donor
|
Acceptor
|
|||
| Residue | Group | Group | Residue | Distance (Å) |
| Lys1 | NζH | Oδ2 | Asp262 | 2.58 |
| Lys1 | NζH | Oδ1 | Asp262 | 2.59 |
| Lys1 | NH | OH | Tyr116 | 2.95 |
| Val3 | NH | Oδ1 | Asn278 | 2.83 |
| Tyr7 | OH | NH | Ser23 | 3.41 |
| Tyr7 | OH | Oδ2 | Asp22 | 2.62 |
| Arg8 | NH1 | Oδ2 | Asp20 | 2.61 |
| Arg8 | NH2 | Oɛ2 | Glu15 | 2.69 |
| Arg8 | NH1 | Oɛ1 | Glu15 | 2.60 |
| Arg8 | NH1 | Oɛ2 | Glu15 | 3.25 |
| Arg12 | NɛH | Oɛ1 | Glu15 | 2.66 |
| Arg12 | NH2 | Oδ1 | Asp20 | 2.59 |
| Arg12 | NH2 | Oɛ1 | Glu15 | 3.04 |
| Arg12 | NH2 | O | Thr13 | 2.69 |
| Arg12 | NH1 | O | Asn11 | 2.65 |
| Arg12 | NH1 | Oδ2 | Asp10 | 2.64 |
Relative contributions of two binding sites
Using the free energy calculation method encoded in the AutoDock program (Morris et al., 1998), we calculated the binding free energies of SDF-1α and its Sites 1 and 2 binding fragments with CXCR4. The calculated binding free energy between Lys-Pro-Val-Ser (residues 1–4) at the Nter of SDF-1α and CXCR4 (ΔGbind(1−4)) is −6.59 kcal/mol, and that between residues 5–13 and CXCR4 (ΔGbind(5−13)) is −9.59 kcal/mol. The calculated total binding free energy ΔGbind of SDF-1α with CXCR4 is −16.62 kcal/mol, indicating that the interactions between Sites 1 and 2 of CXCR4 with SDF-1a contribute to most of the binding energy, and Site 1 binding is ∼1.5 times stronger than Site 2.
DISCUSSION
We have built a 3D structural model of CXCR4, and performed long-time MD simulations and protein-protein interaction modeling on the SDF-1α—CXCR4 system. The entire modeling and simulation provide a lot of new insights into the interaction between SDF-1α and CXCR4. Although the model of CXCR4 is tentative and requires further comparison with the x-ray structure coming up in the future, some basic subjects could be demonstrated according to the simulation results.
Structure-function relationships
Interpretation of experimental data
The proposed binding model of SDF-1α with CXCR4 (Fig. 6) could be used in explaining the structure-function relationship of SDF-1a-CXCR4 binding and other related mutagenesis experiments (Crump et al., 1997; Chabot and Broder, 2000; Brelot et al., 2000; Gerlach et al., 2001; Hatse et al., 2001; Gupta et al., 2001; Zhou et al., 2001). The binding model indicates that the cationic end of Lys1 of SDF-1α forms two strong H-bonds with the side chain of Asp262 in Site 2 of CXCR4, and it is apparent that strong electrostatic interaction should exist between these two residues (Fig. 6 B). These interactions are well in agreement with the Asp262Asn (D262N) mutation experiment, which significantly reduces the binding affinity of SDF-1a and the potency of SDF-1α—induced intracellular calcium signaling (Hatse et al., 2001). The importance of the conserved residue Asp262 was also highlighted by the experimental phenomena that the positive-charged molecules, such as AMD3100 and its analogs, could inhibit the bindings of SDF-1α mediated chemotaxis and [35S]-GTPγS (Gerlach et al., 2001). Moreover, the 3D model of SDF-1α—CXCR4 is supported by several other experimental results (Crump et al., 1997; Brelot et al., 2000): Site 1 at ED-CXCR4 involves in SDF-1α binding but not signaling; residues Glu14, Glu15, and Tyr21 at the Nter of CXCR4 have particular importance in the binding with SDF-1α; and potent antagonism by mutations of Lys1 and/or Pro2 of SDF-1α (Crump et al., 1997) surely resulted in decrease of the binding affinity of SDF-1α with CXCR4. In addition, it was already suggested that there was a similar binding pocket in the TMs domain of CXCR4 to that in CCR5 (Moore and Stevenson, 2000; Chabot and Broder, 2000; Dragic et al., 2000), but it has not been demonstrated at atomic level. Our simulations have mapped this binding pocket position and features inside the TMs (Site 2, Fig. 6, and Table 3), indicating that most of the residues situated in the binding pocket are considerably conserved. The consistency between the 3D model of the ligand-receptor complex and the experimental results indicates the reasonability of the modeled structure, and the binding pocket may act as a starting point for structure-based ligand design.
Most of the site-directed mutagenesis both on Nter of SDF-1α and two binding sites of CXCR4 have a direct effect on SDF-1α binding, and then resulting in decrease or even loss of activation of the receptor (Gerlach et al., 2001; Hatse et al., 2001; Gupta et al., 2001; Zhou et al., 2001). However, several mutageneses demonstrated that some residues not involved in the direct binding contacts between SDF-1α and CXCR4 may also affect the receptor's bindings and activation, such as the mutation studies on Asp97, Asp171, and Glu288 in the TMs domain; Asp187, Tyr190, Asp193, and Glu268 in ED-CXCR4; and Glu2, Tyr7, Tyr12, and Tyr21 at the Nter of CXCR4 (Loetscher et al., 1998; Chabot and Broder, 2000; Brelot et al., 2000; Gerlach et al., 2001; Hatse et al., 2001; Gupta et al., 2001; Zhou et al., 2001). These mutations on negative-charged or aromatic residues appear to modulate the packing and folding of the receptor itself, or affect the interaction between the TMs and the membrane.
Importance of Arg188-Glu277 salt bridge
As indicated above, electrostatic interactions that originated from charged amino acids play a major role in determining how SDF-1α recognizes and interacts with CXCR4. However, compared with other charged residues, the Arg188-Glu277 pair has not yet been appreciated by mutagenesis and binding or functional studies (Gerlach et al., 2001; Hatse et al., 2001; Gupta et al., 2001; Zhou et al., 2001). As shown in Fig. 5, intramolecular interactions between Arg188 and Glu277 directly influence the final complex formation of SDF-1α—CXCR4. In the inactive conformation of CXCR4, the Nter and ELs pack tightly, the salt bridge is locked by H-bonds and electrostatic interactions; as the MD simulations go on, the salt bridge is unlocked by several water molecules (Fig. 5 C), and the mouth of ED-CXCR4 opens for SDF-1α binding.
Binding mechanism of SDF-1α with CXCR4
As represented in the dynamic and energetic aspects (Figs. 2–6), the binding mechanism of SDF-1α with CXCR4 could be clarified at molecular level, and the binding process is summarized in Fig. 7. Typically, CXCR4 could adopt its lowest-energy conformation and keep itself in the inactive state through intramolecular interactions (R state as shown in Fig. 7 A). The tightly packed ED-CXCR4 becomes gradually relaxed, and transits between several energy-minima conformations by molecular thermodynamic motion and redistribution of its electrostatic potentials on its surface (Fig. 4). As the chemokine factor SDF-1α approaches the receptor, a kind of electrostatic signal is transmitted to the molecular surface of CXCR4. In responding to this specific stimulus, CXCR4 changes its conformation and turns into its Rt state (Fig. 7 A). Initialized by electrostatic attraction, SDF-1α binds with CXCR4 at Site 1. As a result of steric complementary and further electrostatic attraction, binding at Site 1 promotes the disruption of the Arg188-Glu277 salt bridge. Site 2 inside the TMs domain is totally exposed and therefore could bind with the Nter of SDF-1α. This two-site binding action drives CXCR4 receptor to its bioactive conformation (R* state in Fig. 7 A). During the whole binding process, electrostatic stimulus is transferred to the intracellular end through the motion of TMs domain. Some positively charged residues near the intracellular end of TMs domain act as the final processor for the stimulant, and the active receptor is ready to couple with G-proteins; thus, the active CXCR4 transfers signals from outside to inside of the cell. Similar to the transmembrane signaling of aspartate receptor (Ottemann et al., 1999), the harmonious conformational changes during SDF-1α—CXCR4 binding just like a piston movement. This is in agreement with the generally recognized conclusion that significant conformational rearrangements must be accompanied in the process of agonist-induced activation and signal transduction of GPCRs (Palczewski et al., 2000; Teller et al., 2001; Gether and Kobilka, 1998; Shapiro et al., 2000; Grobner et al., 2000).
FIGURE 7.

Overall conformational transitions in the process of CXCR4 binding with SDF-1α. (A) CXCR4 changes from its lowest-energy conformation (R) to transition state (Rt). Interaction between the Arg188-Glu277 salt bridge changes from Mode I to Mode II at same time. After recognition of conserved negative-charged cluster (− symbol, in red, Site1 represented as molecular surface; the range of the color panel is the same as that in Fig. 4) at Nter of CXCR4 with positive-charged cluster (+ symbol, molecular surface indicated in blue) at Nter of SDF-1α (shown as black arrow); conformational changes of ED-CXCR4 results in exposure of Site 2 in TMs domain. Thereafter, when SDF-1α—CXCR4 complex is formed, the receptor comes to its active state (R*). (B) Top view of conformation comparison for R (in green) with R* (in blue). TM VI has more significant conformational change while the whole RMSD of Cα of TMs is 0.95.
Using TM truncation and reconstruction approach, it has been tested (Ling et al., 1999) that deletion of TMs I and II has no obvious impairment on CCR5 and CXCR4 for their function as normal chemokine receptors in mediating chemokine-stimulated chemotaxis, Ca2+ influx, and activation of pertussis toxin-sensitive G-proteins. These experimental results combined with other studies (Chabot and Broder, 2000; Brelot et al., 2000; Ling et al., 1999; Gerlach et al., 2001; Hatse et al., 2001; Gupta et al., 2001; Zhou et al., 2001) directly emphasized the relative importance of different TMs in the process of signal transduction. Comparing the conformation R state of CXCR4 with its R* state, the radius of gyration (RG) changes from 2.217 nm to 2.246 nm, and the RMSD of the Cα atoms of the seven TMs has the value of 0.95. These geometrical parameters indicate that in binding with SDF-1α, conformation of ED-CXCR4 changes dramatically (Figs. 4 and 5), and the conformations of the TMs also change in some degree and become more relaxed. Structural alignment indicates that small changes occur for TMs I, II, III, IV, V, and VII; however, great motion happens for TM VI due to the pressure of ED-CXCR4 (Fig. 7 B). This indicates that TM VI plays an important role in the process of SDF-1α binding, conformational change, electrostatic stimulus transferring, and G-protein coupling, which is in agreement with the weak interaction of TM VI with other TMs (Fig. 1). Therefore we suggest much attention should be paid to TM VI in the functional studies of CXCR4.
Acknowledgments
We extend great thanks to Professor Helmut Grubmüller for his kindly offering the EGO program, to Professor Arthur J. Olson for his provision of the AutoDock 3.0 program, and to Professor S. H. Northrup for his kindness in offering us the MacroDox 3.2.2 program. The authors also wish to thank Dr. Wenbo Zhang of Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, for his helpful discussion. The MD calculations were performed on the ShenWei-I supercomputer at the Shanghai Supercomputer Center.
This work was supported by the Ministry of Science and Technology (Grants 1999053907 and 1999054003), the 863 High-Tech Project (Grant 2001AA235131), the National Natural Science Foundation of China (Grants 29725203 and 39825110), Chinese Academy of Sciences (Grant KSCX2-2-02), and the State Key Program of Basic Research of China (Grant 1998051115).
Abbreviations used: TM, transmembrane; MD, molecular dynamics; BD, Brownian dynamics; ED, extracellular domain; RMSD (F), root mean square deviation (fluctuation); Nter, N-terminus.
References
- Adamian, L., and J. Liang. 2001. Helix-helix packing and interfacial pairwise interactions of residues in membrane proteins. J. Mol. Biol. 311:891–907. [DOI] [PubMed] [Google Scholar]
- Babcock, G. J., T. Mirzabekov, W. Wojtowicz, and J. Sodroski. 2001. Ligand-binding characteristics of CXCR4 incorporated into paramagnetic proteoliposomes. J. Biol. Chem. 276:38433–38440. [DOI] [PubMed] [Google Scholar]
- Baker, D., and A. Sali. 2001. Protein structure prediction and structural genomics. Science. 294:93–96. [DOI] [PubMed] [Google Scholar]
- Bennett, T. A., T. A. Key, V. V. Gurevic, G. R. Neubig, E. R. Prossnitz, and L. Sklar. 2001. Real-time analysis of G protein-coupled receptor reconstitution in a solubilized system. J. Biol. Chem. 276:22453–22460. [DOI] [PubMed] [Google Scholar]
- Brelot, A., N. Heveker, M. Montes, and M. Alizon. 2000. Identification of residues of CXCR4 critical for human immunodeficiency virus coreceptor and chemokine receptor activities. J. Biol. Chem. 275:23736–23744. [DOI] [PubMed] [Google Scholar]
- Brooks, B. R., R. E. Bruccoleri, B. D. Olasfon, D. J. States, S. Swaminathan, and M. Karplus. 1983. CHARMM: a program for macromolecular energy, minimization and dynamics calculations. J. Comput. Chem. 4:187–217. [Google Scholar]
- Chabot, D., and C. C. Broder. 2000. Substitutions in a homologous region of extracellular loop 2 of CXCR4 and CCR5 alter coreceptor activities for HIV-1 membrane fusion and virus entry. J. Biol. Chem. 275:23774–23782. [DOI] [PubMed] [Google Scholar]
- Cheng, Z. J., J. Zhao, Y. Sun, W. Hu, Y. L. Wu, B. Cen, G. X. Wu, and G. Pei. 2000. β-arrestin differentially regulates the chemokine receptor CXCR4-mediated signaling and receptor internalization, and this implicates multiple interaction sites between β-arrestin and CXCR4. J. Biol. Chem. 275:2479–2485. [DOI] [PubMed] [Google Scholar]
- Cornell, W. D., D. P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell, and P. A. Kollman. 1995. A new force field for molecular mechanical simulation on nucleic acids and proteins. J. Am. Chem. Soc. 117:5179–5197. [Google Scholar]
- Crump, M. P., J. H. Gong, P. Loetscher, K. Rajarathnam, A. Amara, F. Arenzana-Seisdedos, J. L. Virelizier, M. Baggiolini, B. D. Sykes, and I. Clark-Lewis. 1997. Solution structure and basis for functional activity of stromal cell-derived factor-1; dissociation of CXCR4 activation from binding and inhibition of HIV-1. EMBO J. 16:6996–7007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui, M., J. H. Shen, J. M. Biggs, X. M. Luo, X. J. Tan, H. L. Jiang, K. X. Chen, and R. Y. Ji. 2001. Brownian dynamics with hydrodynamic interactions. Biophys. J. 80:1659–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui, M., J. H. Shen, J. M. Biggs, W. Fu, J. J. Wu, Y. M. Zhang, X. M. Luo, Z. W. Chi, R. Y. Ji, H. L. Jiang, and K. X. Chen. 2002. Brownian dynamics simulations of the recognition of the scorpion toxin P05 with the small-conductance calcium activated potassium channels. J. Mol. Biol. 318:417–428. [DOI] [PubMed] [Google Scholar]
- Dealwis, C., E. J. Fernandez, D. A. Thompson, R. J. Simon, M. A. Siani, and E. Lolis. 1998. Crystal structure of chemically synthesized [N33A] stromal cell-derived factor 1α, a potent ligand for the HIV-1 “fusion” coreceptor. Proc. Natl. Acad. Sci. USA. 95:6941–6946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragic, T., A. Trkola, D. A. D. Thompson, E. G. Cormier, F. A. Kajumo, E. Maxwell, S. W. Lin, W. W. Ying, S. O. Smith, T. P. Sakmar, and J. P. Moore. 2000. A binding pocket for a small molecule inhibitor of HIV-1 entry within the transmembrane helics of CCR5. Proc. Natl. Acad. Sci. USA. 97:5639–5644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan, Y., and P. A. Kollman. 1998. Pathways to a protein folding intermediate observed in a 1-microsecond simulation in aqueous solution. Science. 282:740–744. [DOI] [PubMed] [Google Scholar]
- Eichinger, M., H. Grubmüller, H. Heller, and P. Tavan. 1997. FAMUSAMM: an algorithm for rapid evaluation of electrostatic interactions in molecular dynamics simulations. J. Comput. Chem. 18:1729–1749. [Google Scholar]
- Eichinger, M., H. Heller, and H. Grubmüller. 2000. EGO—An efficient molecular dynamics program and its application to protein dynamics simulations. In Workshop on Molecular Dynamics on Parallel Computers. R. Esser, P. Grassberger, J. Grotendorst, and M. Lewerenz, editors. World Scientific, Singapore, Germany. 397–412.
- Elisseeva, E. L., C. M. Slupsky, M. P. Crump, I. Clark-Lewis, and B. D. Sykes. 2000. NMR studies of active N-terminal peptides of stromal cell-derived factor-1. Structural basis for receptor binding. J. Biol. Chem. 275:26799–26805. [DOI] [PubMed] [Google Scholar]
- Ermak, L., and J. A. McCammon. 1978. Brownian dynamics with hydrodynamic interactions. J. Chem. Phys. 69:1352–1360. [Google Scholar]
- Filizola, M., M. Carteni-Farina, and J. J. Perez. 1999. Modeling the 3D structure of rhodopsin using a de-novo approach to build G-protein-coupled receptors. J. Phys. Chem. 103:2520–2527. [Google Scholar]
- Fu, W., M. Cui, J. M. Briggs, X. Q. Huang, B. Xiong, Y. M. Zhang, X. M. Luo, J. H. Shen, R. Y. Ji, H. L. Jiang, and K. X. Chen. 2002. Brownian dynamics simulations of the recognition of the scorpion toxin maurotoxin with the voltage-gated potassium ion channels. Biophys. J. 83:2370–2385. [DOI] [PMC free article] [PubMed]
- Gerard, C., and B. J. Rollins. 2001. Chemokines and disease. Nat. Immunol. 2:108–115. [DOI] [PubMed] [Google Scholar]
- Jayasinghe, S., K. Hristova, and S. H. White. 2001. Energetics, stability, and prediction of transmembrane helices. J. Mol. Biol. 312:927–934. [DOI] [PubMed] [Google Scholar]
- Gerlach, L. O., R. T. Skerlj, G. J. Bridger, and T. W. Schwartz. 2001. Molecular interactions of cyclam and bicyclam non-peptide antagonists with the CXCR4 chemokine receptor. J. Biol. Chem. 276:14153–14160. [DOI] [PubMed] [Google Scholar]
- Gether, U., and B. K. Kobilka. 1998. G protein-coupled receptors. II. Mechanism of agonist activation. J. Biol. Chem. 273:17979–17982. [DOI] [PubMed] [Google Scholar]
- Grobner, G., U. Burnett, C. Glaubitz, G. Cho, A. J. Bason, and A. Watts. 2000. Observations of light-induced structural changes of retinal within rhodopsin. Nature. 405:810–813. [DOI] [PubMed] [Google Scholar]
- Gupta, S. K., K. Pillarisetti, R. A. Thomas, and N. Aiyar. 2001. Pharmacological evidence for complex and multiple site interaction of CXCR4 with SDF-1alpha: implications for development of selective CXCR4 antagonists. Immunol. Lett. 78:29–34. [DOI] [PubMed] [Google Scholar]
- Hatse, S., K. Princen, L. O. Gerlach, G. Bridger, G. Henson, E. Clercq, T. W. Schwartz, and D. Schols. 2001. Mutation of Asp (171) and Asp (262) of the chemokine receptor CXCR4 impairs its coreceptor function for human immunodeficiency virus-1 entry and abrogates the antagonistic activity of AMD3100. Mol. Pharm. 60:164–173. [DOI] [PubMed] [Google Scholar]
- Henikoff, S., and J. G. Henikoff. 1992. Amino-acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA. 89:10915–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, X. Q., H. L. Jiang, X. M. Luo, K. X. Chen, R. Y. Ji, Y. Cao, and G. Pei. 2000. Building three-dimensional structures of HIV-1 coreceptor CCR5 and its complex with antagonist TAK779 by comparative molecular modeling. Acta Pharmacol. Sin. 21:521–528. [PubMed] [Google Scholar]
- Jorgensen, W. L., J. Chandrasekhar, J. D. Madura, R. W. Impey, and M. L. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- Laskowski, R. A., M. W. MacArthur, D. S. Moss, and J. M. Thornton. 1993. PROCHECK—a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26:283–291. [Google Scholar]
- Liang, J., H. Edelsbrunner, and C. Woodward. 1998. Anatomy of protein pockets and cavities: measurement of binding site geometry and implications for ligand design. Protein Sci. 7:1884–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling, K., P. Wang, J. Zhao, Y. L. Wu, Z. J. Chen, G. X. Wu, W. Hu, L. Ma, and G. Pei. 1999. Five-transmembrane domains appear sufficient for a G protein-coupled receptor: functional five-transmembrane domain chemokine receptors. Proc. Natl. Acad. Sci. USA. 96:7922–7926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liotta, L. A. 2001. An attractive force in metastasis. Nature. 410:24–25. [DOI] [PubMed] [Google Scholar]
- Loetscher, M., T. Geiser, T. O'Reilly, R. Zwahlen, M. Baggiolini, and B. Moser. 1994. Cloning of a human seven-transmembrane domain receptor, LESTR, that is highly expressed in leukocytes. J. Biol. Chem. 269:232–237. [PubMed] [Google Scholar]
- Loetscher, P., J. H. Gong, B. Dewald, M. Baggiolini, and I. Clark-Lewis. 1998. N-terminal peptides of stromal cell-derived factor-1 with CXC chemokine receptor 4 agonist and antagonist activities. J. Biol. Chem. 273:22279–22283. [DOI] [PubMed] [Google Scholar]
- Mackay, C. 2001. Chemokines: immunology's high impact factors. Nat. Immunol. 2:95–101. [DOI] [PubMed] [Google Scholar]
- Matthew, J. B. 1985. Electrostatic effects in proteins. Annu. Rev. Biophys. Biophys. Chem. 14:387–417. [DOI] [PubMed] [Google Scholar]
- Matthew, J. B., and F. R. Gurd. 1986. Calculation of electrostatic interactions in proteins. Methods Enzymol. 130:413–436. [DOI] [PubMed] [Google Scholar]
- McDonald, I. K., and J. M. Thornton. 1994. Satisfying hydrogen bonding potential in proteins. J. Mol. Biol. 238:777–793. [DOI] [PubMed] [Google Scholar]
- Moore, J. P., and M. Stevenson. 2000. New targets for inhibitors of HIV-1 replication. Nat. Rev. Mol. Cell Biol. 1:40–49. [DOI] [PubMed] [Google Scholar]
- Morris, G. M., D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew, and A. J. Olson. 1998. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 19:1639–1662. [Google Scholar]
- Moser, B., and P. Loetscher. 2001. Lymphocyte traffic controlled by chemokines. Nat. Immunol. 2:123–128. [DOI] [PubMed] [Google Scholar]
- Muller, A., B. Homey, H. Soto, N. F. Ge, D. Catron, M. E. Buchanan, T. McClanahan, E. Murphy, W. Yuan, S. N. Wagner, J. L. Barrera, A. Mohar, E. Verastegui, and A. Zlotnik. 2001. Involvement of chemokine receptors in breast cancer metastasis. Nature. 410:50–56. [DOI] [PubMed] [Google Scholar]
- Neria, E., S. Fisher, and M. Karplus. 1996. Simulation of activation energies in molecular systems. J. Chem. Phys. 105:1902–1921. [Google Scholar]
- Nicholls, A., K. A. Sharp, and B. Honig. 1991. Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins: Struct. Funct. Genet. 11:281–296. [DOI] [PubMed] [Google Scholar]
- Orry, A. J. W., and B. A. Wallace. 2000. Modeling and docking the endothelin G-protein-coupled receptor. Biophys. J. 79:3083–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottemann, K. M., W. Z. Xiao, Y. K. Shin, and D. E. Koshland, Jr. 1999. A pistil model for transmembrane signaling of the aspartate receptor. Science. 285:1751–1754. [DOI] [PubMed] [Google Scholar]
- Palczewski, K., T. Kumasaka, T. Hori, C. A. Behnke, H. Motoshima, B. A. Fox, I. Trong, D. C. Teller, T. Okada, R. E. Stenkamp, M. Yamamoto, and M. Miyano. 2000. Crystal structure of rhodopsin: a G protein-coupled receptor. Science. 289:730–734. [DOI] [PubMed] [Google Scholar]
- Pearson, W. R. 1990. Rapid and sensitive sequence comparison with FASTAP and FASTA. Methods Enzymol. 183:63–98. [DOI] [PubMed] [Google Scholar]
- Perera, L., C. Foley, T. A. Darden, D. Stafford, T. Mather, C. T. Esmon, and L. G. Pedersen. 2000. Modeling zymogen protein C. Biophys. J. 79:2925–2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckaert, J. P., G. Ciccotti, and H. J. C. Berendsen. 1977. Numerical integration of the Cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 23:327–341. [Google Scholar]
- Schwarz, M. K., and T. N. C. Wells. 1999. Interfering with chemokine networks—the hope for new therapeutics. Curr. Opin. Chem. Biol. 3:407–417. [DOI] [PubMed] [Google Scholar]
- Shapiro, D. A., K. Kristiansen, W. K. Kroeze, and B. L. Roth. 2000. Differential modes of agonist binding to 5-hydroxytryptamine2A serotonin receptors revealed by mutation and molecular modeling of conserved in transmembrane residues region 5. Mol. Pharm. 58:877–886. [DOI] [PubMed] [Google Scholar]
- Smoluchowski, M. V. 1917. Versuch einer mathematischen theorie der koagulationskinetik kolloider loeschungen. Z. Phys. Chem. 92:129–168. [Google Scholar]
- Solis, F. J., and R. J. B. Wets. 1981. Minimization by random search techniques. Maths. Opera. Res. 6:19–30. [Google Scholar]
- Teller, D. C., T. Okada, C. A. Behnke, K. Palczewski, and R. E. Stenkamp. 2001. Advances in determination of a high-resolution three-dimensional structure of rhodopsin, a model of G-protein-coupled receptors (GPCRs). Biochemistry. 46:7761–7772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thelen, M. 2001. Dancing to the tune of chemokines. Nat. Immunol. 2:129–134. [DOI] [PubMed] [Google Scholar]
- Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties, and weight matrix choice. Nucleic Acids Res. 22:4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulmschneider, M. B., and M. S. P. Sansom. 2001. Amino acid distributions in integral membrane protein structures. Biochim. Biophys. Acta. 1512:1–14. [DOI] [PubMed] [Google Scholar]
- Verlet, L. 1967. Computer experiments on classical fluids. I. Thermodynamical properties of Lennard-Jones molecules. Phys. Rev. 159:98–103. [Google Scholar]
- Vriend, G., and C. Sander. 1993. Quality control of protein models—directional atomic contact analysis. J. Appl. Crystallogr. 26:47–60. [Google Scholar]
- Wallace, A. C., R. A. Laskowski, and J. M. Thornton. 1995. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 8:127–134. [DOI] [PubMed] [Google Scholar]
- Warwicker, J., and H. C. Watson. 1982. Calculation of the electric potential in the active site cleft due to alpha-helix dipoles. J. Mol. Biol. 157:671–679. [DOI] [PubMed] [Google Scholar]
- Zhao, J., L. H. Ben, Y. L. Wu, W. Hu, K. Ling, S. M. Xin, L. N. Nie, L. Ma, and G. Pei. 1999. Anti-HIV agent trichosanthin enhances the capabilities of chemokines to stimulate chemotaxis and G protein activation, and this is mediated through interaction of trichosanthin and chemokine receptors. J. Exp. Med. 190:101–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, N. M., Z. W. Luo, J. S. Luo, D. X. Liu, J. W. Hall, R. J. Pomerantz, and Z. W. Huang. 2001. Structural and functional characterization of human CXCR4 as a chemokine receptor and HIV-1 coreceptor by mutagenesis and molecular modeling studies. J. Biol. Chem. 276:42826–42833. [DOI] [PubMed] [Google Scholar]
- Zlotnik, A., and O. Yoshie. 2000. Chemokines: a new classification system and their role in immunity. Immunity. 12:121–127. [DOI] [PubMed] [Google Scholar]