

Abstract
Gramicidin is a hydrophobic peptide that assembles as a head-to-head dimer in lipid membranes to form water-filled channels selective to small monovalent cations. Two diastereoisomeric forms, respectively SS and RR, of chemically modified channels in which a dioxolane ring links the formylated N-termini of two gramicidin monomers, were shown to form ion channels. To investigate the structural basis underlying experimentally measured differences in proton conductance in the RR and SS channels, we construct atomic-resolution models of dioxolane-linked gramicidin dimers by analogy with the native dimer. A parametric description of the linker compatible with the CHARMM force field used for the peptide is derived by fitting geometry, vibrational frequencies, and energy to the results of ab initio calculations. The linker region of the modified gramicidin dimers is subjected to an extensive conformational search using high-temperature simulated annealing, and free-energy surfaces underlying the structural fluctuations of the channel backbone at 298K are computed from molecular dynamics simulations. The overall secondary structure of the β-helical gramicidin pore is retained in both linked channels. The SS channel is found in a single conformation resembling that of the native dimer, with its peptide bonds undergoing rapid librations with respect to the channel axis. By contrast, its RR counterpart is characterized by local backbone distortions in which the two peptide bonds flanking the linker are markedly tilted in order to satisfy the pitch of the helix. In these distorted structures, each of the two carbonyl groups points either in or out of the lumen. Flipping these two peptides in and out involves thermally activated transitions, which results in four distinct conformational states at equilibrium with one another on a nanosecond time scale. This work opens the way to detailed comparative studies of structure–function relationships in biological proton ducts.
INTRODUCTION
Ion channels are transmembrane proteins that are directly implicated in numerous vital cell functions. When an ion channel is in the open state, it allows the passage of specific ions against their transmembrane electrochemical gradients. Diffusion of ions across membranes determines the electrical excitability of nerve and muscle, the contraction of muscle fibers, and the secretion of hormones and neurotransmitters by many different cells. In general, ion channels are extremely complex structures consisting of thousands of amino acids that can usually be found associated with other protein complexes (Hille, 2001; Andersen and Koeppe, 1992). In addition, ion channels can be modified by phosphorylation and/or glycosylation. This organizational complexity makes the understanding of the fine physico-chemical mechanisms involved in ion permeation a major and important challenge and underlines the need for fundamental studies of ion transport in comparatively simpler ion channels whose structure and function are well characterized.
Such a system is provided by gramicidin A (gA), a hydrophobic pentadecapeptide secreted by Bacillus brevis. The primary structure of gA consists mostly of an alternating sequence of L and D amino acids, formyl-Val1-Gly2-Ala3-Dleu4-Ala5-Dval6-Val7-Dval8-Trp9-Dleu10-Trp11-DLeu12-Trp13-Dleu14-Trp15-ethanolamine. The peptide adopts a right-sided β6.3 helix structure in various molecular environments (Arseniev et al., 1985; Ketchem et al., 1993, 1997). In lipid bilayers, the association of the amino termini of two gA molecules via hydrogen bonds results in the formation of an ion channel that is selective for small monovalent cations (Tian and Cross, 1999). In this active dimeric form, the helix defines a cylindrical pore 4 Å in diameter that accommodates a single file of water molecules and provides a conduit for ions. In the single-file region or lumen of the channel, the partial desolvation of cations is partly compensated by the polar peptide bonds lining the walls of the pore (and defining the helix conformation). Disruption of intermolecular hydrogen bonds between gA monomers results in loss of ion channel activity. The lifetime of gA dimers in reconstituted lipid planar bilayers is of the order of 100 ms.
Dioxolane-linked gA dimers were originally designed and synthesized by Stankovic et al. (1989) to stabilize the head-to-head assembly of the pore through covalent linkage. To this end, two gA monomers were covalently linked via a dioxolane group inserted between their formylated N-termini. The linked molecules form ion channels in lipid bilayers. These channels, like native gA, are selective for monovalent cations but their average lifetime in the open state is considerably longer than that of native gA dimers. Because of the presence of two chiral carbon atoms in the dioxolane linker, the chemical synthesis route leads to two distinct diastereoisomers, in which the linkers are respectively SS and RR (Stankovic et al., 1989, 1990; Cukierman et al., 1997). Significant differences have been characterized between the properties of channels formed respectively by SS and RR dioxolane-linked gA dimers in planar lipid bilayers (Cukierman et al., 1997; Quigley et al., 1999, 2000; Cukierman, 2000; de Godoy and Cukierman, 2001). In particular, under experimental conditions in which protons are the permeating cations, it has been shown that the single-channel conductance (gH) is two- to fourfold larger in the SS than in the RR dimer. In addition, these channels differ in the shape of current–voltage relationships. Furthermore, the SS dimer is extremely stable in various types of lipid bilayers, where it remains essentially in the open state (with an open probability greater than 95%) for several hours or longer. By contrast, in some experimental conditions (see Quigley et al., 1999; Armstrong et al., 2001; Armstrong and Cukierman, 2002) the open state of the RR channel lasts only a few minutes, after which it dwells into a conformational state that does not conduct protons (inactivated state). Finally, the relationship between gH and proton concentration in bulk solution, [H+], is qualitatively different in the SS and RR channels. The log–log plot of gH versus [H+] is linear in a wide range of proton concentrations in the SS channel but not in its RR counterpart (Cukierman, 2000).
Given the chemical similarity of these two channels, the relative structural simplicity of native gA and of SS- and RR-dioxolane-linked gA channels, and the meaningful differences in their properties, it is of interest to explore the potential relationships between structure and function of these ion channels. Such an undertaking is made possible by the wealth of experimental and theoretical data on the gA dimer. The structure of the native gA dimer has been characterized at high resolution (Arseniev et al., 1985; Ketchem et al., 1993, 1997) and the atomic basis for ionic permeation has been the object of numerous computational studies (for reviews, see Roux, 2002; Roux and Karplus, 1994; and references therein). In particular, theoretical studies of proton translocation via a Grotthuss relay mechanism along the chain of water molecules embedded in the channel lumen have provided insight into the role of the peptidic matrix in assisting proton transport (Pomès and Roux, 1996, 2002). The structural basis of phenomenological differences between SS- and RR-dioxolane-linked channels has been addressed in the past using computational models (Stankovic et al., 1989; Crouzy et al., 1994; Quigley et al., 1999). These studies considered rotameric states of the linkers differing by the topology of the dioxolane ring with respect to the channel lumen. Based on partially rigid models built by analogy with the native dimer, Stankovic et al. (1989) concluded that contrary to the SS linker, conformations of the RR linker with dioxolane extending outside of the pore were incompatible with the backbone topology of a β-helix, and proposed a closure mechanism involving the intercalation of the dioxolane ring into the channel lumen via conformational isomerization of the linker's backbone. Crouzy et al. (1994) used molecular dynamics simulations to study the pathway and the rate of this proposed closure mechanism. Other molecular models suggested that the RR linker may induce backbone distortions in the open state which could have direct implications to proton permeation (Quigley et al., 1999); however, these models were based on rigid descriptions of the dioxolane ring and did not include the effect of thermal fluctuations.
In the present study, we seek to model in greater detail the structure and fluctuations of linked channels in their open state, over time scales comparable to that of ionic permeation. The hypothesis underlying the present approach is that the structural origin of measurable perturbations of ionic currents in dioxolane-linked channels lies in moderate structural differences. The basis for this assumption is threefold: (i) the chemical structures of RR and SS channels are almost identical, (ii) both molecules form channels, and (iii) the single-channel proton conductance of the SS channel is highly similar to that of native gA (Quigley et al., 1999), suggesting that functional differences are a consequence of structural perturbations in the middle of the channel.
The work described below combines a dedicated force field with full treatment of the internal flexibility of the linker and extensive search of the conformational space accessible to the linked channels. To this end, we first develop accurate molecular mechanics force field parameters for the dioxolane linker. We then use molecular dynamics simulations to construct atomic-resolution models of the SS and RR dioxolane linked gA channels. Finally, we analyze the equilibrium structure, conformational distribution, and dynamic fluctuations of native and linked channels at 298 K. Significant differences in the structural properties and in the nature of dynamic fluctuations of native gA and of the two linked channels are identified and analyzed. While this study confirms that the RR linker induces structural distortions to the channel backbone, it also reveals that the stress imposed by the RR dioxolane linker to the β-helix leads to four distinct conformational states that differ essentially in the topology of the backbone in the immediate vicinity of the linker. These four rotamers are in equilibrium with each other in the nanosecond time range. Because the permeation of ions in biological channels occurs on a similar time scale, these results may bear direct implications for molecular mechanisms of gating and functional dynamics in ion channels.
METHODS
A set of empirical potential energy functions was developed to reproduce the distribution of states obtained with quantum mechanical (QM) ab initio calculations in the full conformational space accessible to the linker. These parameters were integrated into an existing molecular mechanics (MM) force field (MacKerell et al., 1998) widely used in computational studies of biopolymers. Molecular structures of dioxolane-linked channels were obtained from molecular dynamics (MD) simulations. Models of SS and RR channels analogous to the β-helical pore formed by the native gA dimer were derived using high-temperature simulated annealing with conformational restraints on the backbone. This initial conformational search of linked channels was confined to the vicinity of the linker (middle turn of the five-turn helix), with gradual relaxation of backbone restraints in the subsequent refinement of the channel structures. Finally, relaxed conformations of the channels were obtained from MD simulations at room temperature; the structure and fluctuations of the native and linked gA channels were characterized with potential of mean-force free-energy surfaces.
Force field parametrization
As a linker of two head-to-head pentadecapeptide monomers, the dioxolane ring is inserted between two formylated N-termini, adding two carbon atoms to the polypeptidic main chain of gramicidin (Fig. 1). A meaningful description of conformational space of the linked channels requires parametrization not only of the ring itself, but also of the main chain, which in the linked channels is augmented by three CC bonds relative to the native gA dimer. Empirical potential energy functions are essential for MD simulations. As a nonstandard biomolecular fragment, the dioxolane linker is not adequately parametrized in existing force fields. We constructed new MM parameters compatible with a conventional biomolecular force field, the CHARMM22 parameter set (MacKerell et al., 1998). We describe below the development of such a set of parameters for a molecule containing the dioxolane ring and the two adjacent peptide bonds. To ensure transferability to (and compatibility with) the rest of the force field, all the CHARMM22 parameters pertaining to the peptide bond (MacKerell et al., 1998) were retained in the process of fitting the novel parameters to the geometry, vibrational frequencies, and energy of the linker fragment obtained from QM calculations.
FIGURE 1.

(Top) Dioxolane ring and (bottom) RR-dioxolane flanked by two peptide bonds, RR-[1,3] dioxolane-4,5-dicarboxylic acid bis-methylamide. Heavy atom and torsion definitions are shown. This and subsequent representations of molecular structures were prepared with the program VMD (Humphey et al., 1996).
High-level ab initio calculations are the alternative to experimental data for force field development, particularly when a small molecular fragment is used for parameterization. The two small fragments chosen for ab initio calculations were [1,3]dioxolane and (R,R) [1,3]dioxolane-4,5-dicarboxylic acid bis-methylamide (Fig. 1). The latter molecule will henceforth be referred to as dioxolane dipeptide. The program Gaussian 98, Revision A.9 (Frisch et al., 1998) was used in all ab initio calculations, together with the RHF/6-311G** level of theory. Hartree-Fock (HF) energy optimizations followed by CHELPG analysis (Breneman and Wiberg, 1990) were performed on the two fragments. The resulting equilibrium bond distances and bond angles were used in CHARMM potential energy functions (Brooks et al., 1983) along with the CHELPG-derived partial atomic charges. In order to fit the force constants of the potential energy terms, HF frequencies were calculated. A scale factor of 0.9 (Florian and Johnson, 1994) was applied to the HF frequencies for comparison to MM force field frequencies.
To compare QM and MM results over the range of conformational space accessible to the dipeptidic fragment, potential energy surfaces were constructed from QM and MM calculations. The QM energy maps were obtained by full gradient optimization. The first energy map was computed at fixed values of the dihedral angle θ (O11-C10-C1-O5, see Fig. 1) from 0 to 330° in increments of 30° (5° near energy minima). The second surface was computed by covering the full range of dihedral angle θ by increments of 30° and by varying δ (O5-C1-C2-O3) by increments of 10° between −31° and 29°. Finally, another two-dimensional potential energy surface was constructed by optimizing 144 structures of the RR-dioxolane dipeptide at grid points separated by 30° in the θ, θ′ (O13-C12-C2-O3) dihedral space.
The program CHARMM (Brooks et al., 1983), version 27, was used in all MM and subsequent MD calculations. The following sequence presided over the refinement of MM parameters: (i) fit of the equilibrium geometry and vibrational frequencies of the dioxolane ring; (ii) iterative refinement of the parameters of the dipeptide fragment based on θ and (θ,δ) maps and on the structure and relative energy of the two stable dipeptide conformers. The accuracy of geometries and energies was checked from a comparison of the two (θ,θ′) maps.
Molecular dynamics simulations
The force field developed for the linker was used to model the structure of dioxolane-linked gramicidin channels with MD simulations. The initial structures of the linker region of RR and SS dioxolane-linked gA channels were adopted from a previous study (Quigley et al., 1999), while the experimentally determined structure of Arseniev et al. (1985) was used as a reference from which symmetric conformational restraints were determined. The simulations consisted of two stages. First, high-temperature simulated annealing was used to search conformational space and to determine the stable conformational states of linked SS and RR channels. Some of the resulting structures were then subjected to further simulations in order to characterize the equilibrium distribution and the dynamic fluctuations of these conformational states at room temperature (298 K). All simulations used Langevin dynamics with a friction coefficient of 5 ps−1 on nonhydrogen atoms.
High-temperature simulated annealing
Two independent high-temperature MD simulations employing different distributions of initial velocities were generated for both RR- and SS-linked channels. Each system was heated to 5000 K. The basic assumption of β-helical structure led to imposing backbone restraints in the high-temperature runs; to prevent the system from unfolding, all the heavy atoms beyond valine residues 1 and 1′ were subjected to strong harmonic restraints of 10.0 kcal/mol/Å2. The dioxolane ring and its two adjacent peptide bonds were not constrained, while softer restraints of 3.0 kcal/mol/Å2 were applied to the remaining atoms of the two Val1 residues. 20-ps trajectories were generated with a time step of 0.5 fs. Twenty structures were recorded at intervals of 2000 MD steps in each of the high-temperature runs. Each of the 80 resulting “hot” structures was then gradually cooled to around 200 K with a succession of 10-ps MD runs at a rate of 80% starting at 4000 K. The cooling process was followed by energy minimization, during which harmonic restraints applied to the backbone atoms beyond the two valine residues were reduced to 5.0 kcal/mol/Å2. The analysis of the resulting “cool” structures led to the identification of clusters in conformational space corresponding to distinct conformational states (see Results). One structure from each of these clusters was selected for subsequent MD simulations at 298 K, during which backbone restraints imposed on the outer turns of the β-helix were gradually relaxed so as to let the strain in the linker region (middle half-turn of the helix) diffuse via structural adjustments in neighboring residues.
Room-temperature simulations
Eight energy-minimized structures, seven RR and one SS, were initially selected for long MD simulations at room temperature. All side-chain restraints on non-Trp residues were removed, and the restraints imposed on the backbone and on Trp side chains were reduced in two stages. First, the central two turns of the gA channel, i.e., six residues beyond the dioxolane on each side, were fully relaxed without any restraints. The rest of the heavy backbone atoms was subjected to soft restraints of 1.0 kcal/mol/Å2. These weak restraints allow backbone atoms of the channel to move more freely than before so as to adjust to the presence of the linker. Following thermalization and equilibration at 298 K over 1 ns, these restraints were further reduced to a minimal set described below.
Side-chain constraints on the eight Trp residues are necessary in order to palliate the absence of the lipid bilayer, which tethers the indole rings via hydrophobic and hydrogen-bonding interactions (Woolf and Roux, 1994). Likewise, restraints on the backbone of each of the outer turns of the helix (i.e., turns #1 and 5) are necessary to prevent unfolding of the channel in vacuo. A “minimal set” consisted of: (1) harmonic restraining terms with a force constant of 10 kcal/mol/rad2 on χ1 and χ2 torsions of each Trp side chain so as to retain the experimentally determined conformations of Arseniev et al. (1985); and (2) harmonic restraints imposed to the N, Cα, C, and O atoms of residues 11–15 as well as the ethanolamine Cα in each monomer to force them to remain within a cylindrical shell 4 Å from the channel axis, with a restoring force constant of 5 kcal/mol/Å2 acting outside the range 3.5 < r < 4.5 Å. By design, these restraints prevent unfolding of the outer turns while allowing for axial stretch and radial twist adjustments of the helix in response to the insertion of the linker, without overly constraining the tilt and librations of peptide bonds with respect to the channel axis. The choice of the restraining force constants was justified a posteriori by the agreement in the magnitude of peptide tilts and the amplitude of their librations with high-resolution experimental data obtained for the native gA dimer (see Results and Discussion).
A total of 80,000 conformations were generated from 16-ns MD simulations for each of the three channels. The SHAKE algorithm (Ryckaert et al., 1977) was applied to fix the length of all the bonds involving hydrogen atoms to their equilibrium value, allowing a relative large time step of 2 fs. Potential of mean force (PMF) surfaces were computed from the equilibrium distribution of dihedral angles θ and θ′ and peptide tilt angles α and α′ (see Results).
RESULTS
Dioxolane linker
Dioxolane is a five-member ring containing two oxygen atoms and three sp3 carbon atoms (Fig. 1). Full geometry optimization of dioxolane with quantum mechanical ab initio calculations at the HF/6-311G** level reveals two stable states corresponding to half-chair conformations, with the dihedral angle δ (O-C-C-O) = ±27.3°. These two half-chair rotameric states are separated by a small energy barrier of 0.24 kcal/mol. The envelope conformation, at δ = 0°, is not stable energetically but instead lies at the top of the barrier.
The minimum-energy conformations of [1,3]dioxolane-4,5-dicarboxylic acid bis-methylamide (the RR-dioxolane dipeptide) can be described in terms of three torsional angles, θ (O5-C1-C10-O11), θ′ (O3-C2-C12-O13), and δ (O5-C1-C2-O3) (see Fig. 1). One energy minimum finds the dioxolane ring in the half-chair conformation (δ = −31.0°), with both θ and θ′ antiperiplanar or trans. The other half-chair conformation, at δ ≅ 30°, is not an energy minimum due to repulsion between the two carbonyl O atoms, O11 and O13. Two envelope energy minima (δ = 3.3°) are identified. Each of them has one of the torsions θ or θ′ in trans-conformation at 173.9°, with the other in synperiplanar or cis-conformation (14.9°). These envelope conformations are stabilized by an internal hydrogen bond between the two peptide bonds. The energy difference between the half-chair and envelope conformers is small (0.036 kcal/mol), and the energy barrier between the two is ∼2.3 kcal/mol. Because θ, δ, and θ′ define the local backbone conformation of the linked channels, the structure and energy of the linker in the space described by these three torsions are the focus of the parameterization of the dioxolane linker described below.
Table 1 lists the MM potential energy functions and associated parameters retained for the dioxolane linker. In order to define a unique set of parameters for both RR and SS linkers, all the energy terms used must be symmetric with respect to the two diastereoisomers. Thus, of all possible torsional energy functions for the dioxolane ring, only that for δ was retained. Urey-Bradley terms were introduced to remedy the missing torsion terms and to improve the frequency fitting.
TABLE 1.
MM potential energy functions and parameters for [1,3]dioxolane-4,5-dicarboxylic acid bis-methylamide
| Kb (b−b0)2
|
||
|---|---|---|
| Bond length | Kb | b0 |
| C1-C2 | 285.0 | 1.525 |
| C1-O5 C2-O3 | 303.0 | 1.404 |
| C4-O3 C4-O5 | 328.0 | 1.391 |
| C1-H6 C2-H7 | 304.0 | 1.084 |
| C4-H8 C4-H9 | 304.0 | 1.085 |
| C1-C10 C2-C12 | 250.0 | 1.524 |
| Kθ (θ−θ0)2
|
KUB (S−S0)2
|
|||
|---|---|---|---|---|
| Bond angle & Urey-Bradley | Kθ | θ0 | KUB | S0 |
| O3-C4-O5 | 78.0 | 107.04 | 25.00 | 2.240 |
| H8-C4-H9 | 36.5 | 110.20 | 5.40 | 1.780 |
| O5-C4-H8 O5-C4-H9 | 38.5 | 109.89 | 22.53 | 2.040 |
| O5-C1-H6 O3-C2-H7 | 37.3 | 109.74 | 22.53 | 2.043 |
| C1-C2-H7 C2-C1-H6 | 41.5 | 112.63 | 22.53 | 2.180 |
| O5-C1-C2 O3-C2-C1 | 85.0 | 102.69 | ||
| C1-O5-C4 C2-O3-C4 | 85.0 | 107.49 | ||
| C1-C10-N14 C2-C12-N16 | 85.0 | 114.90 | ||
| C1-C10-O11 C2-C12-O13 | 80.0 | 120.23 | ||
| O5-C1-C10 O3-C2-C12 | 32.0 | 113.13 | ||
| C2-C1-C10 C1-C2-C12 | 33.0 | 111.64 | ||
| C10-C1-H6 C12-C2-H7 | 22.0 | 107.95 | ||
| Kχ (1 + cos(n χ−δ))
|
|||
|---|---|---|---|
| Torsional angle | Kχ | n | δ |
| O5-C1-C10-O11 | 1.10 | 2 | 180.0 |
| O3-C2-C12-O13 | 1.10 | 2 | 180.0 |
| O5-C1-C2-O3 | 3.00 | 1 | 180.0 |
| 0.48 | 5 | 0.0 | |
|
ɛij[(Rminij/rij)12−(Rminij/rij)6]
|
qi qj/ɛ1 rij
|
|||
|---|---|---|---|---|
| Nonbonded interaction | ɛi | Rmini/2 | qi | |
| C1 C2 | −0.020 | 2.27 | 0.240 | |
| O3 O5 | −0.160 | 1.77 | −0.512 | |
| C4 | −0.055 | 2.17 | 0.540 | |
| H6 H7 | −0.022 | 1.32 | 0.022 | |
| H8 H9 | −0.022 | 1.32 | −0.020 | |
The parameters involving the dioxolane ring are shown. Atom numbering is defined in Fig. 1. The standard CHARMM22 parameters (MacKerell et al., 1998) are used for the peptide bonds.
The normal mode frequencies of dioxolane obtained successively with quantum ab initio HF/6-311G** (QM) and with the empirical (MM) force field are listed in Table 2. Overall, the QM and MM frequencies are in good agreement. Discrepancies in the ordering and/or in the assignment of normal mode frequencies persist for modes 6, 7, and 9–12. These differences reflect the sensitivity of normal mode frequencies to the choice of MM parameters, especially for C–O bond stretching and methylene rocking motions. The iterative optimization of normal mode frequencies was stopped once it became difficult to improve the match of low frequencies without compromising the overall fit of frequencies and energies.
TABLE 2.
Vibrational frequencies of dioxolane
| Mode | QM frequency | Assignment | MM frequency | Assignment | |||
|---|---|---|---|---|---|---|---|
| 1 | 73.1 | τ′(60) | τ (40) | 72.2 | τ′ (101) | ||
| 2 | 251.9 | τ (56) | τ′ (38) | 207.2 | τ (99) | ||
| 3 | 686.7 | δ′ (61) | 654.0 | δ′ (83) | |||
| 4 | 767.3 | δ (75) | 700.4 | δ (81) | |||
| 5 | 913.4 | rCH2 (43) | δ′ (19) | νCO(17) | 886.3 | rCH2 (80) | |
| 6 | 967.3 | νCC (73) | 901.4 | νCO (85) | |||
| 7 | 1004.9 | νCO (82) | 934.1 | rCH2 (74) | |||
| 8 | 1024.7 | νCO (87) | 984.7 | νCO (47) | |||
| 9 | 1147.7 | νCO (23) | rCH2 (17) | 1056.8 | rCH2 (74) | ||
| 10 | 1180.2 | rCH2 (55) | νCO (32) | 1077.5 | νCC (56) | ||
| 11 | 1198.9 | rCH2 (72) | 1179.4 | νCO (36) | |||
| 12 | 1239.8 | rCH2 (30) | tCH2 (21) | 1199.4 | νCO (51) | wCH2 (18) | |
| 13 | 1256.3 | tCH2 (68) | 1226.3 | tCH2 (96) | |||
| 14 | 1281.3 | tCH2 (74) | 1237.0 | tCH2 (77) | |||
| 15 | 1313.1 | tCH2 (80) | 1248.7 | tCH2 (86) | |||
| 16 | 1400.1 | wCH2 (96) | 1401.5 | wCH2 (45) | |||
| 17 | 1442.6 | wCH2 (93) | 1409.8 | wCH2 (64) | |||
| 18 | 1496.1 | wCH2 (81) | 1445.3 | wCH2 (61) | |||
| 19 | 1559.7 | βCH2 (83) | 1470.1 | βCH2 (64) | |||
| 20 | 1574.2 | βCH2 (82) | 1491.1 | βCH2 (64) | |||
| 21 | 1604.7 | βCH2 (93) | 1499.9 | βCH2 (87) | |||
| 22 | 2987.7 | νCH (97) | 2895.9 | νCH (98) | |||
| 23 | 2998.0 | νCH (96) | 2899.7 | νCH (97) | |||
| 24 | 3026.7 | νCH (98) | 2904.0 | νCH (100) | |||
| 25 | 3074.9 | νCH (95) | 2926.1 | νCH (98) | |||
| 26 | 3093.4 | νCH (91) | 2930.2 | νCH (96) | |||
| 27 | 3104.1 | νCH (86) | 2934.9 | νCH (95) |
Frequencies are in cm−1. Potential energy distributions were determined with the MOLVIB module in CHARMM (Brooks et al., 1983) and with Pulay's natural internal coordinates (Pulay et al., 1979; Fogarasi et al., 1992). τ and δ indicate ring deformations and ring torsions, respectively. ν, β, r, t, and w indicate stretching, bending, rocking, twisting, and wagging modes, respectively.
The internal geometry and the energy of the dioxolane ring and of the dioxolane dipeptide obtained from the HF/6-311G** and empirical force field calculations are described successively in Tables 3 and 4. In general, the calculated empirical bond lengths and bond angles are in satisfactory agreement with the ab initio geometries. In the final stages of the optimization, the fit of geometry and energy of the dipeptidic fragment received priority over that of the dioxolane molecule. Thus, while most of the torsional angles of dioxolane show significant deviations from the ab initio results (Table 3), the agreement improves in the dipeptide molecule (Table 4) with root-mean-square deviations of 0.021 Å for bond lengths, 2.4° for bond angles, and 7.1° for dihedral angles. Only three dihedral angles deviate from their QM values by more than 10°. These torsions involve the C1–C2 bond of the ring. Such discrepancies are a consequence of the fact that δ is a relatively soft degree of freedom with a broad energy minimum (see θ,δ contour map, Fig. 3, below), so that even though there is a significant deviation in the minimum-energy structures, the energy required to bring them into agreement is small.
TABLE 3.
Optimized internal coordinates of [1,3] dioxolane
| HF | MM | |
|---|---|---|
| Bond length (Å) | ||
| C1-C2 | 1.524 | 1.522 |
| C2-O3 | 1.407 | 1.406 |
| O3-C4 | 1.397 | 1.394 |
| C4-O5 | 1.386 | 1.395 |
| O5-C1 | 1.401 | 1.406 |
| Bond angle (degree) | ||
| C1-C2-O3 | 103.22 | 103.01 |
| C2-O3-C4 | 108.87 | 108.93 |
| O3-C4-O5 | 107.09 | 108.02 |
| C4-O5-C1 | 105.98 | 108.91 |
| O5-C1-C2 | 102.23 | 103.02 |
| Torsional angle (degree) | ||
| O5-C1-C2-O3 | −27.28 | −28.23 |
| C1-C2-O3-C4 | 9.24 | 23.43 |
| C2-O3-C4-O5 | 12.65 | −9.8 |
| O3-C4-O5-C1 | −31.32 | −9.7 |
| C4-O5-C1-C2 | 35.72 | 23.37 |
Atom numbering is defined in Fig. 1. Only the internal coordinates involving exclusively heavy atoms are shown.
TABLE 4.
Internal coordinates and relative energies of [1,3]dioxolane-4,5-dicarboxylic acid bis-methylamide
| Conformer 1
|
Conformer 2
|
|||
|---|---|---|---|---|
| QM | MM | QM | MM | |
| Relative energy* | 0.036 | 0.029 | 0.000 | 0.000 |
| Bond length (Å) | ||||
| C2-C1 | 1.519 | 1.528 | 1.555 | 1.535 |
| O3-C2 | 1.406 | 1.427 | 1.396 | 1.424 |
| C4-O3 | 1.397 | 1.396 | 1.398 | 1.392 |
| O5-C1 | 1.407 | 1.427 | 1.390 | 1.426 |
| O5-C4 | 1.397 | 1.395 | 1.385 | 1.390 |
| C10-C1 | 1.529 | 1.552 | 1.527 | 1.558 |
| O11-C10 | 1.197 | 1.229 | 1.193 | 1.229 |
| C12-C2 | 1.528 | 1.552 | 1.517 | 1.550 |
| O13-C12 | 1.197 | 1.229 | 1.204 | 1.231 |
| N14-C10 | 1.341 | 1.349 | 1.347 | 1.352 |
| N16-C12 | 1.342 | 1.349 | 1.332 | 1.347 |
| C18-N14 | 1.448 | 1.444 | 1.448 | 1.444 |
| C22-N16 | 1.448 | 1.444 | 1.448 | 1.444 |
| Bond angle (degree) | ||||
| O3-C2-C1 | 102.48 | 103.14 | 103.73 | 103.92 |
| C4-O3-C2 | 108.93 | 109.77 | 106.64 | 108.01 |
| C2-C1-O5 | 102.48 | 103.05 | 103.61 | 103.61 |
| C1-O5-C4 | 108.93 | 109.86 | 108.14 | 109.37 |
| O5-C4-O3 | 107.31 | 107.84 | 105.49 | 105.70 |
| C2-C1-C10 | 112.38 | 112.76 | 110.79 | 111.20 |
| O5-C1-C10 | 112.99 | 118.44 | 112.94 | 119.96 |
| O11-C10-C1 | 120.19 | 121.64 | 122.22 | 122.07 |
| C12-C2-C1 | 112.34 | 112.74 | 111.74 | 110.87 |
| O3-C2-C12 | 113.06 | 118.41 | 113.46 | 117.41 |
| O13-C12-C2 | 120.21 | 121.63 | 118.24 | 120.83 |
| N14-C10-C1 | 115.29 | 116.68 | 113.13 | 116.73 |
| O11-C10-N14 | 124.51 | 121.68 | 124.64 | 121.20 |
| N16-C12-C2 | 115.25 | 116.71 | 116.33 | 117.00 |
| O13-C12-N16 | 124.52 | 121.66 | 125.43 | 122.17 |
| C18-N14-C10 | 122.07 | 121.99 | 121.63 | 121.61 |
| C22-N16-C12 | 122.06 | 122.06 | 123.10 | 122.32 |
| Torsional angle (degree) | ||||
| O3-C2-C1-O5 | −31.03 | −24.79 | 3.27 | 5.80 |
| O3-C2-C1-C10 | 90.55 | 104.08 | 124.66 | 135.95 |
| C12-C2-C1-O5 | 90.61 | 104.09 | 125.86 | 132.82 |
| C12-C2-C1-C10 | −147.80 | −127.05 | −112.75 | −97.03 |
| C4-O3-C2-C1 | 25.82 | 20.88 | −23.27 | −22.83 |
| C4-O3-C2-C12 | −95.34 | −104.40 | −144.71 | −145.66 |
| O5-C4-O3-C2 | −10.75 | −8.67 | 35.67 | 31.76 |
| C4-O5-C1-C2 | 25.81 | 20.87 | 18.26 | 13.21 |
| C4-O5-C1-C10 | −95.35 | −104.39 | −101.67 | −111.46 |
| C1-O5-C4-O3 | −10.73 | −8.65 | −33.70 | −27.89 |
| O11-C10-C1-C2 | 47.59 | 47.30 | −100.84 | −109.57 |
| O11-C10-C1-O5 | 162.97 | 167.69 | 14.88 | 11.41 |
| N14-C10-C1-C2 | −133.78 | −133.02 | 77.94 | 69.86 |
| N14-C10-C1-O5 | −18.41 | −12.63 | −166.34 | −169.16 |
| O13-C12-C2-C1 | 47.65 | 47.31 | 57.06 | 53.17 |
| O13-C12-C2-O3 | 163.05 | 167.78 | 173.91 | 172.36 |
| N16-C12-C2-C1 | −133.85 | −133.06 | −123.64 | −126.61 |
| N16-C12-C2-O3 | −18.45 | −12.60 | −6.80 | −7.41 |
| C18-N14-C10-C1 | −174.01 | 179.94 | −173.73 | 178.58 |
| C18-N14-C10-O11 | 4.54 | −0.38 | 5.02 | −1.98 |
| C22-N16-C12-C2 | −174.06 | 179.95 | 179.21 | −179.82 |
| C22-N16-C12-O13 | 4.36 | −0.43 | −1.55 | 0.40 |
Energies are in kcal/mol. Atom numbering is shown in Fig. 1. Only the internal coordinates involving exclusively heavy atoms are shown.
QM and MM Energies of Conformer 1 are relative to those of Conformer 2, respectively.
FIGURE 3.

Potential energy surfaces for the conformational isomerization of the dioxolane dipeptide in (θ,δ) torsional space. Angular values are indicated in degrees. Contour spacing is 0.5 kcal/mol. Shading decreases with higher energy. (top) Ab initio quantum mechanics. (Bottom) Molecular mechanics calculations.
The potential energy of the RR-dioxolane dipeptide as a function of torsion θ is depicted in Fig. 2. An excellent fit was obtained in the region of the two energy minima. The barrier region (−180° < θ < −50°) is shifted by ∼10 degrees to higher values of θ in the MM curve relative to QM values. Because the barrier is due in large part to nonbonded interactions between peptide O atoms (see above), it is difficult to improve on the fit of energy in this region without compromising the fit of energies in the (δ,θ) space (see discussion of Fig. 3). For consistency with the force field used for the rest of the channel, we chose to retain the nonbonded parameters of the CHARMM force field for peptide bonds. This restriction limits the number of parameters that can be modified to improve the fit of potential energy maps.
FIGURE 2.

Potential energy as a function of torsion θ obtained from (solid) ab initio quantum mechanics and (dashed) molecular mechanics calculations.
Comparisons of QM and MM potential energy surfaces of the dipeptidic molecule in the space of θ and δ torsions are shown in Fig. 3. A good agreement is obtained between the force field and ab initio calculations. Despite the overestimate of the steepness of the energy in the MM potential energy profile at θ = −50° (Fig. 2), in the (δ,θ) contour plot the QM barrier is steeper and higher than its MM counterpart (Fig. 3). The discrepancies between QM and MM energy profiles in Figs. 2 and 3 are inherent to the MM approach, reflecting the compromise arising from the choice of a single set of MM parameters for the entire conformational space accessible to the linker. Finally, potential energy surfaces are depicted in Fig. 4 as a function of backbone torsions θ and θ′. The relative energy of the (trans,trans) and (trans,cis) dipeptide conformers differs by only 0.007 kcal/mol when computed by QM and MM (see Table 4). The agreement between QM and MM energy surfaces throughout (θ,θ′) torsional space is good. This comparison provides an important check on the quality of the parametrization, inasmuch as the MM energy map of Fig. 4 was not the result of a fit to its QM counterpart.
FIGURE 4.
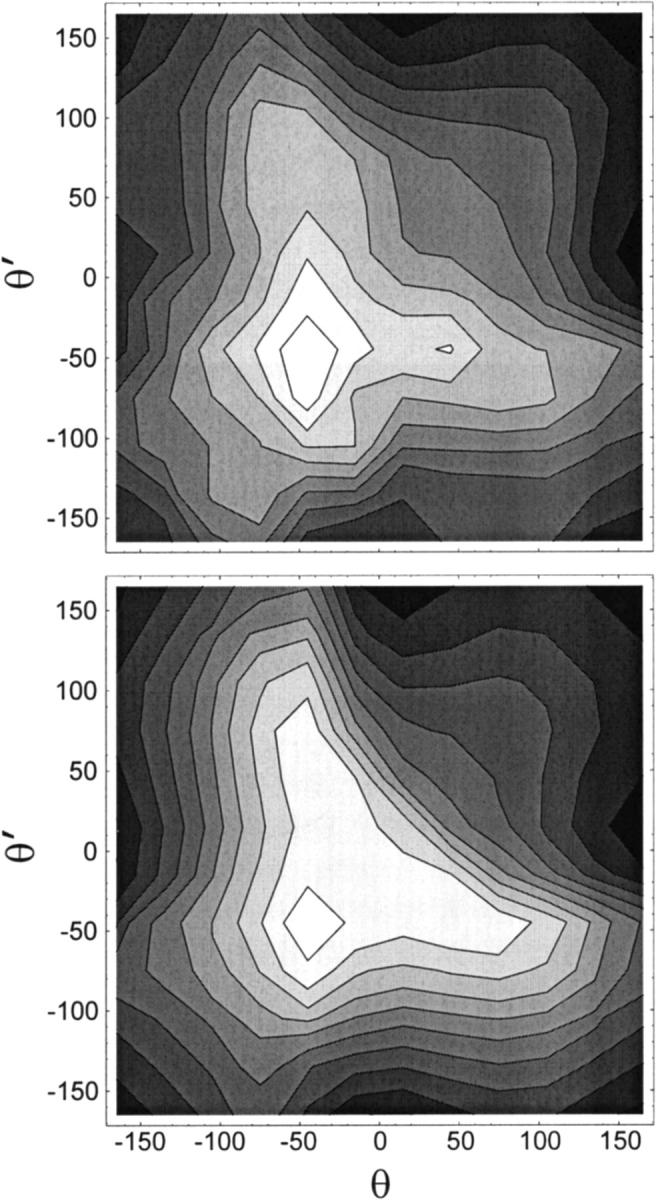
Potential energy surfaces for the conformational isomerization of the dioxolane dipeptide in (θ,θ′) torsional space. Angular values are indicated in degrees. Contour spacing is 0.5 kcal/mol. Shading decreases with higher energy. (Top) Ab initio quantum mechanics. (Bottom) Molecular mechanics calculations.
Dioxolane-linked gA channels
Simulated annealing
Forty uncorrelated structures of each of the two linked channels were obtained from an extensive search of conformational space using high-temperature simulated annealing. In all of these structures, the dioxolane ring remained outside the lumen of the channel. Because of the artificially high temperature used in the initial part of the search, eleven of the RR and four of the SS structures contain cis peptides. The isomerization of peptide bonds, which involves crossing an activation energy barrier of 21 kcal/mol in the CHARMM force field (MacKerell et al., 1998), is an artifact of high-energy simulations. Additionally, one RR and one SS structure contained a peptide bond flipped by 180° with respect to the channel axis compared to the native gA dimer. These 17 conformers, which compromise the secondary structure of the pore, were not considered in the subsequent analysis. The values of backbone torsions θ and θ′, which exhibit the widest spread among the remaining structures, are shown in Fig. 5. The symmetry of the linked dimers with respect to the linker is reflected in the map. The energy-minimized structures of the SS-linked dimer cluster together in a crescent extending around (θ, θ′) = (−100°, −100°). By contrast, the RR structures partition into several distinct conformational classes scattered in the lower left quadrant of the (θ, θ′) space.
FIGURE 5.

Backbone torsions of the conformations obtained from high-temperature simulated annealing of dioxolane-linked channels shown in (θ,θ′) torsional space. Angular values are indicated in degrees. (Circles) SS-. (Crosses) RR-linked channels. All the SS structures cluster together near (θ, θ′) = (−100°, −100°), whereas the RR structures partition into as many as seven classes centered near (−115°, −15°), (−15°, −115°), (−130°, −120°), (−120°, −130°), (−115°, −115°), (−120°, −165°), and (−120°, −180°), respectively.
Local backbone conformation
The equilibrium structure and fluctuations of SS and RR dioxolane-linked gA channels were obtained from molecular dynamics simulations at 298 K. Restraint relaxation led to small-amplitude adjustments in the four outer turns of the channel, allowing some relief of local strain in the middle turn of the helix. The linked dimers settled into well-defined conformations resembling that of the native gA dimer but retaining some distinctive features. The backbone of the SS linker remained in a single conformation, whereas the seven structures obtained from simulated annealing of the RR-linked dimer partitioned into four distinct conformational states.
The potential of mean force (PMF) surfaces governing the fluctuations of backbone torsions θ and θ′ in SS- and RR-linked channels are depicted in Fig. 6. The SS conformation occupies a broad basin in the PMF surface centered at (θ, θ′) = (−75°, −75°). The four conformations of the RR-linked backbone, henceforth labeled 1–4, are characterized by free energy wells centered at (θ, θ′) ≅ (−107°, −18°), (−37°, −37°), (−20°, −105°), and (−100°, −100°), respectively. Asymmetric conformations 1 and 3 correspond to free energy wells separated by the secondary minimum of conformer 4 and by the elongated basin of conformer 2. The relative free energies of the minima of the four basins are 0.0, 0.1, 0.0, and 0.7 kcal/mol. All four conformations are significantly occupied at 298 K, with populations of 30, 26, 33, and 11%, respectively. The activation energy barriers separating conformations 1 or 3 from conformations 2 and 4 are 0.6 and 1.3 kcal/mol, respectively. The fact that the asymmetry of the free energy contours with respect to the θ = θ′ diagonal is small attests to adequate statistical convergence of the simulations.
FIGURE 6.
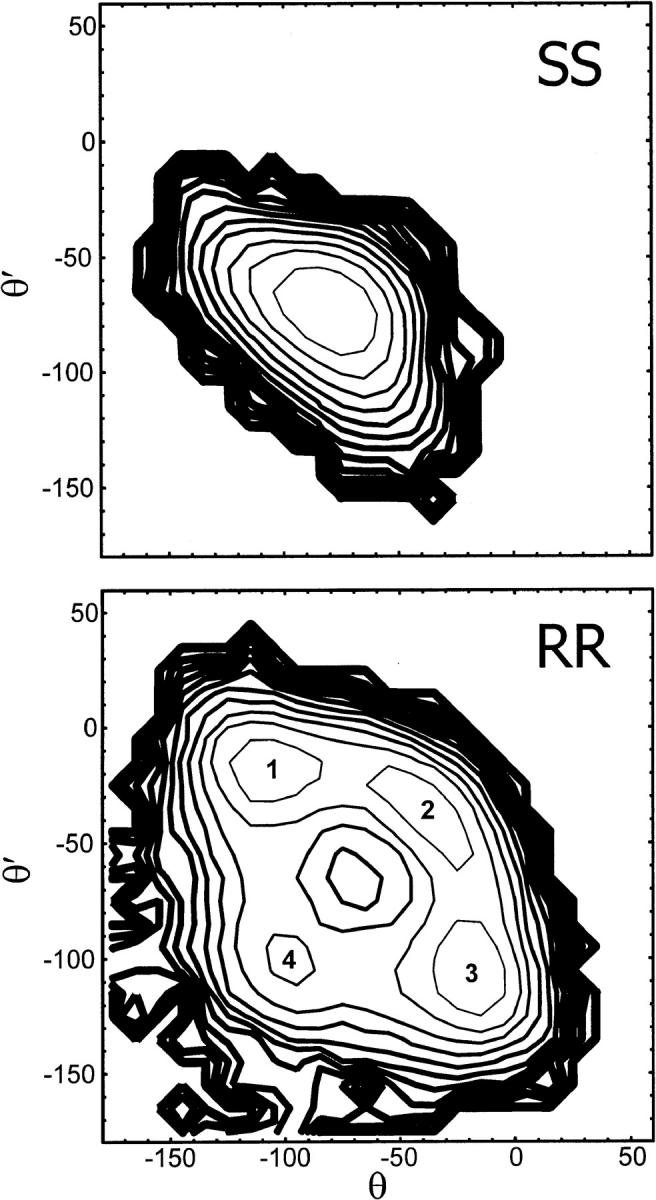
Potential of mean force for the structural fluctuations of the linked channels in (θ,θ′) torsional space. Angular values are indicated in degrees. Contour spacing is 0.5 kcal/mol. Line thickness increases with energy. (Top) SS channel. (Bottom) RR channel. Numbers 1–4 are the labels assigned to each of the conformers of the RR channel.
Overall pore structure
Table 5 lists the average length and rms fluctuations of the 26 peptide-to-peptide hydrogen bonds defining the β-helix. The overall secondary structure of the native gA pore is conserved in both SS and RR channels. In all three channels, most hydrogen bond lengths are between 2.86 and 3.05 Å on average, with rms fluctuations ranging from 0.13 to 0.19 Å. Notable exceptions include the C-termini of all three channels, where limited fraying results in the weakening of the outermost hydrogen bond. This is due in part to the greater looseness of the backbone at the end of the helix and to finite-size effects in our models, which neglect the presence of water molecules and of lipid headgroups near the mouths of the channel. In native gA, greater looseness in the secondary structure is also observed at the N-termini: the hydrogen bonds involving the two formyl carbonyl groups are elongated to nearly 3.1 Å and fluctuate more widely. In the SS-linked channel all but the outer two hydrogen bonds are stable. By contrast, the hydrogen bonds involving the two peptide bonds flanking the RR linker are perturbed significantly: N1–O5′ and N1′–O5 are dramatically weakened at 3.3 Å; inversely, O0–N7 and O0′–N7′ separations are notably shorter than in the other two channels. All other hydrogen bond lengths of the RR-linked channel are in close quantitative agreement with those of its native and SS counterparts. Thus, while the RR linker weakens the secondary structure of the pore, this effect remains confined to two intermonomeric H bonds in the middle half-turn of the helix.
TABLE 5.
Average O ⋯ N distances of hydrogen bonds in native, SS-linked, and RR-linked gA
| Peptide | Native | SS | RR |
|---|---|---|---|
| O15 N10 | 3.57 (0.62) | 3.35 (0.40) | 3.46 (0.52) |
| O13 N8 | 2.93 (0.16) | 2.95 (0.15) | 2.97 (0.17) |
| O11 N6 | 2.95 (0.15) | 2.96 (0.15) | 2.94 (0.15) |
| O9 N4 | 2.93 (0.15) | 2.92 (0.15) | 2.93 (0.15) |
| O8 N15 | 2.93 (0.19) | 2.92 (0.14) | 2.92 (0.15) |
| O7 N2 | 2.88 (0.14) | 2.90 (0.14) | 2.94 (0.16) |
| O6 N13 | 3.05 (0.18) | 3.09 (0.18) | 3.03 (0.18) |
| O5 N1′ | 2.86 (0.14) | 2.88 (0.14) | 3.30 (0.28) |
| O4 N11 | 3.01 (0.16) | 2.98 (0.16) | 3.02 (0.17) |
| O3 N3′ | 2.94 (0.15) | 2.96 (0.16) | 2.93 (0.15) |
| O2 N9 | 2.97 (0.19) | 2.98 (0.18) | 2.98 (0.19) |
| O1 N5′ | 2.92 (0.16) | 2.88 (0.13) | 2.91 (0.15) |
| O0 N7 | 3.09 (0.27) | 3.05 (0.17) | 2.89 (0.18) |
| O0′ N7′ | 3.07 (0.25) | 2.99 (0.17) | 2.90 (0.19) |
| O1′ N5 | 2.91 (0.15) | 2.87 (0.13) | 2.90 (0.15) |
| O2′ N9′ | 2.97 (0.19) | 3.01 (0.20) | 2.98 (0.19) |
| O3′ N3 | 2.94 (0.15) | 2.97 (0.17) | 2.94 (0.15) |
| O4′ N11′ | 3.01 (0.16) | 3.02 (0.17) | 3.02 (0.17) |
| O5′ N1 | 2.86 (0.14) | 2.88 (0.14) | 3.31 (0.29) |
| O6′ N13′ | 3.05 (0.18) | 3.01 (0.16) | 3.04 (0.18) |
| O7′ N2′ | 2.88 (0.14) | 2.87 (0.14) | 2.94 (0.16) |
| O8′ N15′ | 2.92 (0.17) | 2.91 (0.14) | 2.91 (0.14) |
| O9′ N4′ | 2.93 (0.15) | 2.92 (0.14) | 2.93 (0.15) |
| O11′ N6′ | 2.95 (0.15) | 2.94 (0.15) | 2.94 (0.15) |
| O13′ N8′ | 2.93 (0.15) | 2.93 (0.15) | 2.97 (0.16) |
| O15′ N10′ | 3.55 (0.59) | 3.38 (0.42) | 3.39 (0.40) |
Averages, in Å, obtained from 16-ns MD simulations. The value of standard deviations is indicated in parentheses. Significant differences are highlighted.
Origin of the distortions
The relationship between backbone distortions and strain in the secondary structure of the RR-linked channel is illustrated in Fig. 7. The differences in the conformations of SS and RR channels result from the orientation of the dioxolane ring with respect to the axis of the channel. In the SS linker, the dioxolane ring adopts an orientation in which its mean plane is approximately perpendicular to the axis of the channel. The three backbone C–C bonds resulting from covalent linkage go respectively up, down, and up together with the rise of the right-sided helix, satisfying its pitch naturally. By contrast, the chirality of the two backbone C atoms of RR dioxolane impose an orientation in which the mean plane of the ring is roughly perpendicular to the plane of the bilayer. In such an arrangement, all three backbone C–C bonds involving the chiral centers of the ring go up with the counterclockwise rise of the helix, which results in an overshoot of the helical pitch. Thus, the RR dioxolane ring tends to act as a wedge pushing the two gramicidin monomers away from each other and threatening the integrity of hydrogen bonds between the two monomers. To avert this effect, the two peptide bonds adjacent to the linker tilt out of alignment with the channel axis so as to restore the proper pitch of the helix. While these distortions fail to restore the proper donor–acceptor separation of two intermonomeric hydrogen bonds, hydrogen bonds involving nearby peptides in the middle turn (O of Val 1, O of Gly 2, and N of Ala 3 in each monomer) are all satisfied (see Table 5). Thus, the structure of the RR-linked channel reflects a compromise afforded by the intrinsic flexibility of the linked backbone: local structural distortions compensate for the perturbation of the helical pitch, sacrificing optimal alignment of peptides in the middle half-turn in order to retain the overall secondary structure of the pore.
FIGURE 7.
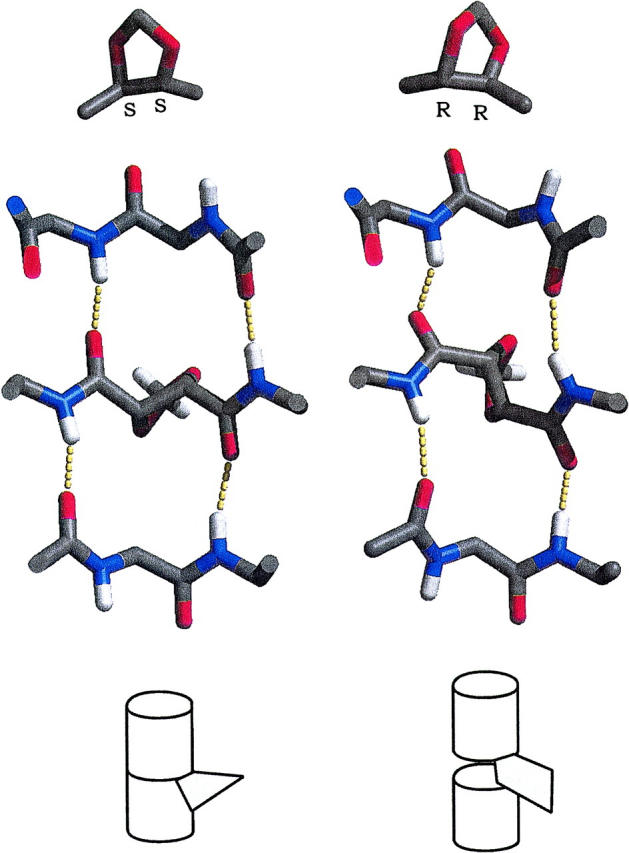
Dioxolane chirality and representative conformations of the main chain of dioxolane-linked gramicidin channels, looking from the center of the channel, perpendicularly to the channel axis. Amino acid side chains were omitted for clarity. The hydrogen bonds formed by the two peptide bonds flanking the linker are highlighted. (Left) SS linker. (Right) RR linker. Diagrams illustrating the mode of insertion of the ring are shown at the bottom.
Peptide tilts
Representative snapshots of the middle turn of the SS and RR channels are shown in Figs. 8 and 9, respectively. The peptide bonds flanking the SS linker are nearly aligned with the channel axis, and the backbone atoms forming the middle turn of the helix are disposed near the circumference of a cylinder. As mentioned above, the alignment of peptide bonds flanking the RR linker is compromised by significantly larger tilts than in both native and SS-linked channels. As shown in Fig. 9, there are two ways to tilt each of these two peptides, which results in four distinct conformations. Thus, conformers 1–4 differ by the topology of the peptide bonds with respect to the channel axis, projecting each of the two carbonyl O atoms either into the pore lumen or outwards, facing the lipid bilayer: respectively (in,out), (out,out), (out,in), and (in,in).
FIGURE 8.
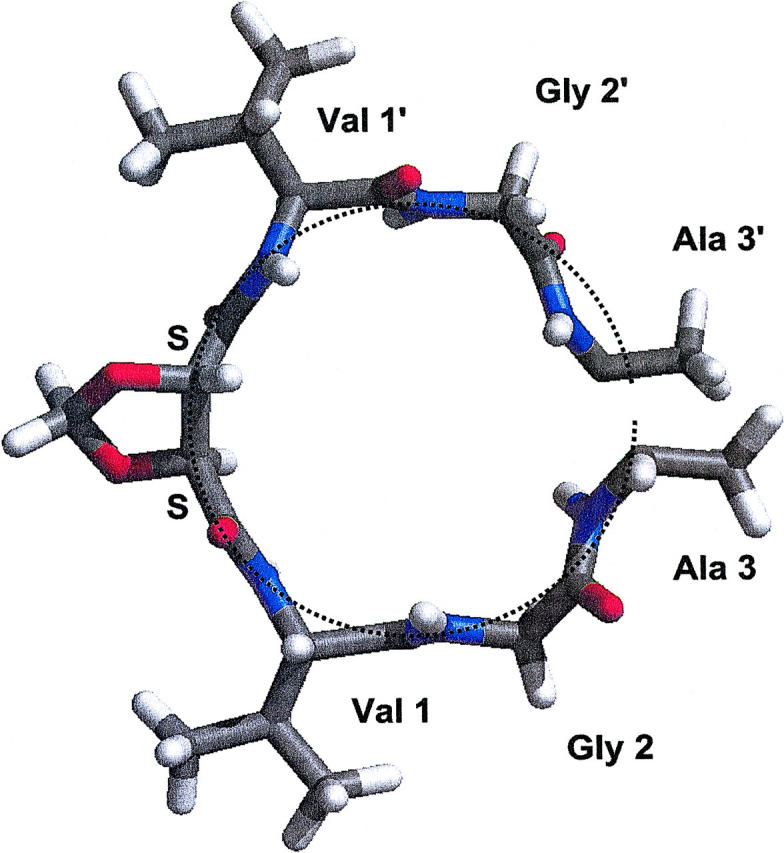
Middle turn of the SS-linked dimer, looking down the channel axis.
FIGURE 9.
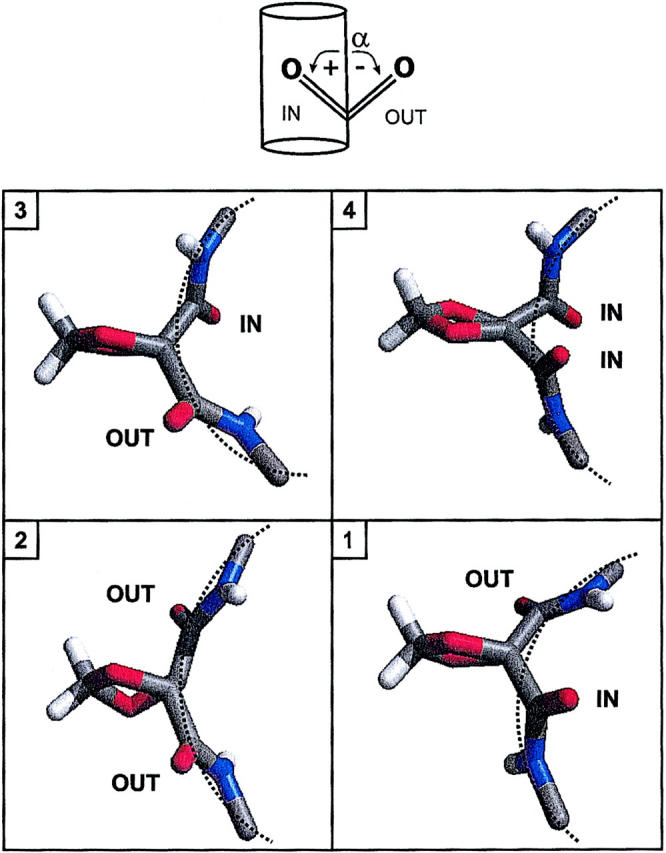
(Top) Definition of CO tilt angle α controlling the topology of carbonyl O atoms with respect to the channel lumen. (Bottom) Representative backbone conformations of the RR-linked dimer, looking down the channel axis. Conformations 1–4 differ by the topology of the two peptide bonds flanking the dioxolane ring.
The tilt angles of backbone carbonyl bonds with respect to the axis of the channel are listed in Table 6. The symmetry of each structure with respect to the center of the channel is reflected in the first three columns of Table 6. Within a given monomer, the average tilts vary both in sign and in magnitude from residue to residue. However, the tilts of most given residues are nearly identical in the native, SS, and RR channels. In general, the absolute magnitude of carbonyl tilts ranges from 0 to 20°, with rms fluctuations between 8 and 15°. Notable exceptions include peptides 15 and 15′ at the C-terminus, which exhibit greater deviations from alignment with the axis of the helix in all three channels (−27 to −31°). More importantly, significant differences between native, SS, and RR dimers are confined to the center of the channel. The two central CO groups face outward by 5° in native gA, inward by ∼3° in the SS channel. However, although they are also close to alignment on average, the two CO groups flanking the RR linker undergo uncharacteristically large rms fluctuations, twice as high as any other carbonyl group in the three channels. This discrepancy is a direct consequence of the conformational isomerization unique to the RR dimer. The central CO tilt angles are maximized in the individual RR conformers, with absolute magnitudes ranging from 20 to 27°. The only other CO groups to undergo a change in topology upon isomerization of the RR linker are those of residues 6 and 6′ one turn away in the helix. These changes, which are anticorrelated to those of the central peptide groups, preserve the intramonomeric H bond between O0 and N7. At ∼12°, the amplitude of these transitions is much smaller than corresponding flips of the central CO groups (∼50°). Finally, possibly as a local compensation to backbone distortions near the linker, the carbonyl groups of Val 1 and 1′, which point out by 19–22° in native and SS channels, only do so by 9–15° in the RR conformers.
TABLE 6.
CO tilt angles from simulations of native gA and SS- and RR-linked channels
| Peptide | Native | SS | RR | RR-1 | RR-2 | RR-3 | RR-4 |
|---|---|---|---|---|---|---|---|
| 15 | −31 (14) | −27 (13) | −29 (13) | −29 (13) | −29 (13) | −29 (13) | −31 (13) |
| 14 | 10 (9) | 11 (8) | 11 (8) | 10 (8) | 10 (8) | 11 (8) | 12 (8) |
| 13 | −3 (8) | −1 (8) | −3 (9) | −2 (9) | −4 (9) | −4 (9) | −3 (9) |
| 12 | 18 (8) | 18 (8) | 18 (8) | 19 (8) | 18 (8) | 17 (8) | 19 (8) |
| 11 | −4 (12) | −4 (12) | −3 (11) | −3 (12) | −4 (11) | −4 (11) | −1 (11) |
| 10 | 13 (10) | 13 (10) | 12 (10) | 12 (10) | 11 (10) | 12 (10) | 12 (10) |
| 9 | 15 (10) | 17 (9) | 18 (9) | 17 (9) | 17 (9) | 18 (9) | 18 (9) |
| 8 | −6 (11) | −12 (8) | −12 (9) | −11 (9) | −12 (9) | −13 (9) | −11 (10) |
| 7 | 9 (9) | 5 (7) | 6 (8) | 3 (8) | 7 (8) | 8 (8) | 5 (9) |
| 6 | −1 (8) | 0 (8) | 3 (10) | −4 (8) | 7 (8) | 7 (8) | −4 (8) |
| 5 | 12 (13) | 9 (9) | 10 (10) | 11 (8) | 13 (9) | 9 (11) | 3 (8) |
| 4 | −13 (8) | −12 (8) | −14 (8) | −14 (8) | −16 (8) | −13 (8) | −13 (8) |
| 3 | −12 (11) | −16 (8) | −15 (8) | −15 (8) | −15 (8) | −14 (9) | −15 (9) |
| 2 | 13 (15) | 19 (11) | 17 (12) | 14 (12) | 19 (11) | 19 (13) | 13 (13) |
| 1 | −22 (12) | −19 (13) | −13 (14) | −13 (12) | −9 (14) | −15 (14) | −13 (13) |
| 0 | −5 (14) | 6 (11) | −3 (27) | 26 (15) | −20 (9) | −25 (10) | 25 (15) |
| 0′ | −5 (14) | 1 (10) | 0 (28) | −26 (10) | −20 (9) | 27 (15) | 27 (14) |
| 1′ | −22 (12) | −21 (12) | −13 (13) | −15 (14) | −10 (14) | −14 (12) | −13 (13) |
| 2′ | 13 (15) | 18 (12) | 17 (12) | 19 (12) | 20 (11) | 15 (12) | 14 (13) |
| 3′ | −12 (11) | −15 (8) | −15 (8) | −15 (8) | −16 (7) | −15 (8) | −15 (8) |
| 4′ | −12 (8) | −13 (8) | −15 (8) | −14 (8) | −17 (7) | −15 (8) | −14 (7) |
| 5′ | 12 (13) | 9 (10) | 11 (10) | 10 (12) | 13 (9) | 12 (8) | 4 (8) |
| 6′ | 0 (8) | 2 (8) | 1 (11) | 7 (9) | 7 (8) | −6 (8) | −6 (8) |
| 7′ | 9 (9) | 9 (8) | 5 (8) | 8 (8) | 6 (8) | 3 (8) | 3 (8) |
| 8′ | −6 (10) | −10 (9) | −11 (9) | −12 (9) | −12 (8) | −11 (9) | −11 (9) |
| 9′ | 15 (10) | 16 (9) | 18 (9) | 18 (9) | 18 (9) | 18 (9) | 18 (9) |
| 10′ | 12 (10) | 12 (10) | 12 (10) | 12 (10) | 12 (10) | 13 (10) | 12 (10) |
| 11′ | −4 (12) | −3 (12) | −6 (12) | −6 (12) | −5 (11) | −6 (12) | −5 (12) |
| 12′ | 17 (8) | 17 (8) | 19 (8) | 18 (8) | 18 (8) | 20 (8) | 20 (8) |
| 13′ | −2 (8) | −3 (9) | −3 (9) | −4 (8) | −4 (8) | −2 (8) | −1 (8) |
| 14′ | 10 (9) | 10 (8) | 10 (8) | 10 (8) | 10 (8) | 10 (8) | 10 (8) |
| 15′ | −30 (14) | −27 (13) | −28 (12) | −28 (12) | −28 (12) | −28 (12) | −29 (13) |
Averages, in degrees, computed from the same set of simulations as in Table 5. Standard deviations are indicated in parentheses. CO tilt angles are calculated as the angle between the peptide plane of N–C(=O)−Cα and the axis of the channel (z-axis) and are defined as positive if the bond is pointing into the lumen. The CO tilt angles of RR dimer conformers 1–4 are listed in the last four columns. Significant differences are highlighted.
The PMF surfaces for tilt angles α and α′ of the central carbonyl groups in native and linked channels are depicted in Fig. 10. In the native and SS-linked dimers, the librations or oscillations of the two peptide planes occur around a single minimum near α = α′ = 0 and exhibit some anharmonic character. Tilt fluctuations are somewhat more pronounced in the native gA dimer due to the absence of chemical linkage between the two polypeptide chains. By contrast, the PMF of the RR linker is radically different from those of native and SS channels and strongly resembles that computed in (θ,θ′) space (compare to Fig. 6). The surface is characterized by four well-defined free energy wells, each of which lies well into its respective quadrant and corresponds to one of the four conformational states defined above as conformers 1–4. The activation free energy between conformers 1 or 3 and conformers 2 and 4 are respectively 0.7 and 1.4 kcal/mol. These thermally-activated transitions take place spontaneously in the nanosecond time range and occur stepwise, with only one of the two peptide bonds flipping in or out of the lumen at once. The concerted flip of both peptide bonds, which maximizes the local pitch of the helix, is unfavored. Accordingly, the fully-aligned structure (α = α′ = 0) lies not at the bottom of a well but near the top of a free energy peak culminating at 2.3 kcal/mol.
FIGURE 10.
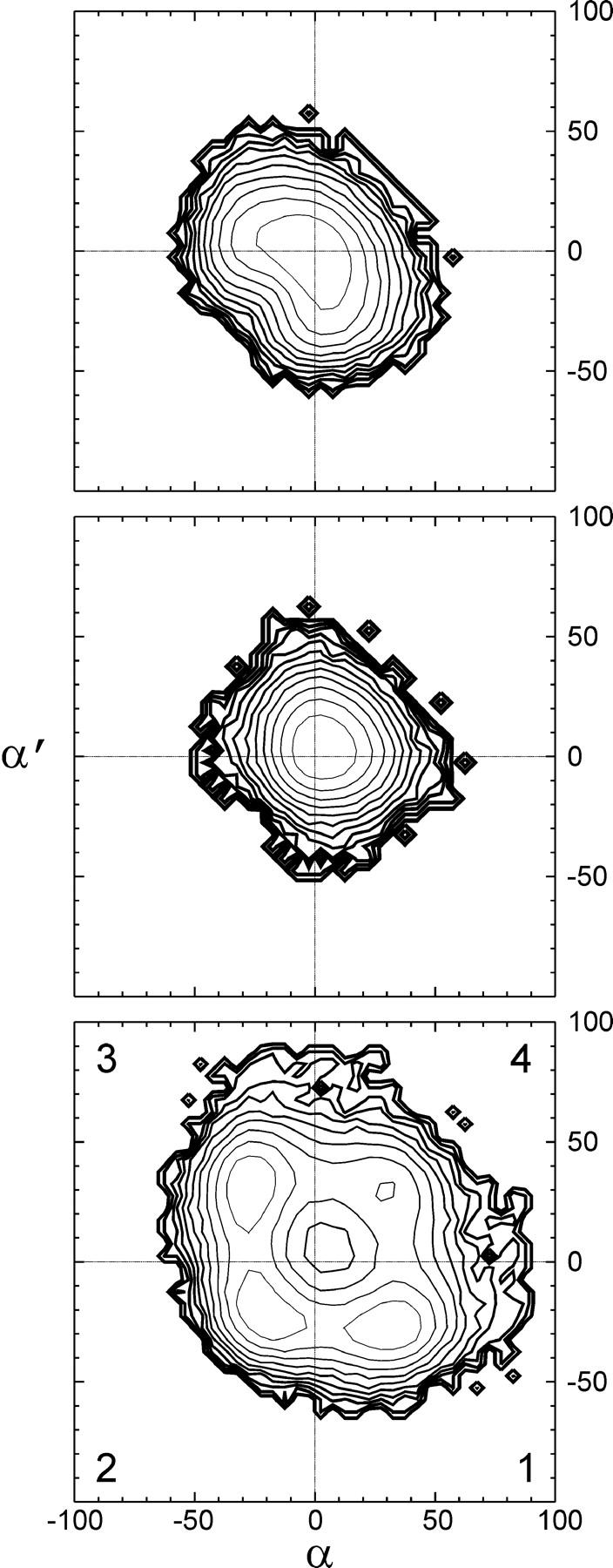
Potential of mean force for the structural fluctuations of native and linked channels in (α,α′) space. Tilt angles are indicated in degrees. Contour spacing is 0.5 kcal/mol. Line thickness increases with energy. (Top) Native gA. (Middle) SS dimer. (Bottom) RR dimer.
DISCUSSION
Quality of the force field
We presented detailed atomic models of dioxolane-linked gA channels using new MM parameters of the linker derived from QM calculations. In the design and refinement of these parameters, a special focus was placed on obtaining adequate fits of potential energy surfaces in the entire conformational space accessible to the torsions of the three C–C bonds added to the channel backbone by the insertion of the dioxolane linker. These properties affect not only the equilibrium conformations and the extent of backbone distortions but also the dynamic fluctuations governing conformational isomerizations of the linked channels. The dioxolane dipeptide was chosen as a molecular fragment of sufficient size to develop parameters for the linker region that are compatible with the force field used for the rest of the linked dimers. The transferability of parameters obtained for small fragments to models of polymers is one of the basic assumptions inherent to empirical biomolecular mechanics force fields. In the refinement of parameters for the dioxolane dipeptide fragment, we chose to retain all the CHARMM22 parameters pertaining to the amide bond so as to guarantee that the optimized parameters be consistent with CHARMM22. The accuracy of the torsional potential energy surfaces throughout the range of conformational space accessible to the dipeptide linker fragment is comparable to that of peptide and amino acid fragments in the CHARMM22 force field itself (MacKerell et al., 1998). The dependence of the potential energy on backbone torsions θ and θ′, which exhibit the largest changes among all variables describing the internal geometry of the linked channels, is well reproduced throughout the map. Because the (θ,θ′) map was not itself the result of a fit, these results attest to the consistency of the model. It is difficult to improve further upon the current fit of geometry and energies without modifying the parameters for the two peptide bonds flanking the dioxolane ring. In turn, such modifications would compromise the consistency of these parameters with the rest of the polypeptide in linked channels.
Comparison to earlier models of dioxolane-linked gA
Molecular models of dioxolane-linked gA channels were presented in three previous studies. In their seminal work, Stankovic et al. (1989, 1990) based the rational design of long-lived channels on structural models. The authors considered both left- and right-sided helix forms of the end-to-end dimer and discovered that the gap between the two desformylated N-termini could be bridged by monocyclic 1,2-trans-dicarboxylic acids. They went on to propose essential structural differences between SS and RR dioxolane-linked channels based on the respective ability of the two linkers to satisfy a basic stereochemical requirement of the channel backbone (i.e., that the carbonyl bonds of the two peptides flanking the ring point away from the bisecting plane of the homodimer). Using models of the linker assuming rigidity of the dioxolane ring, they observed that with the ring outside the lumen of the pore, the RR linker would induce strains in both left- and right-sided helical forms, whereas the backbone of both β-helix structures could be matched readily by the SS linker. This analysis is qualitatively confirmed by the present study in the case of a right-sided helix. However, a straightforward extension of our results to left-sided helices contradicts the structural models of Stankovic et al. (1989): because the backbones of left- and right-sided helices are mirror images of each other, and so are the SS and RR linkers, based on the present study the RR linker would fit in a left-sided gramicidin channel without significant distortions, with its plane perpendicular to the channel axis, exactly as the SS linker does in our right-sided helix. Inversely, the SS linker would act as a wedge and induce the very same distortions to a left-sided helix as the RR linker does to a right-sided helix. This refinement of the qualitative predictions made by Stankovic et al. (1989, 1990) underlines the need to account for the full conformational flexibility of the linked backbone.
Following the proposal of a closure mechanism by Stankovic et al. (1989), Crouzy et al. (1994) modeled the flip of the dioxolane ring into the channel lumen using molecular dynamics simulations. Parameters for the linker were based on similar chemical groups taken from the PARAM19 extended-atom CHARMM/XPLOR force field (Brooks et al., 1983). Crouzy et al. (1994) computed potential of mean-force profiles for the transition along several possible paths and found that the preferred pathway would be a strongly activated three-step process requiring the reversible trans-to-cis isomerization of one of the peptide bonds flanking the linker: from (ring out, trans peptide), to (out, cis), (in, cis), and finally (in, trans). Such conformational isomerizations were found to involve activation energy barriers of the order of 10 kcal/mol corresponding to rates of the order of a millisecond. No spontaneous intrusion of the dioxolane ring into the channel lumen was observed in the present simulations, and the conformations containing cis peptides obtained from high-temperature simulated annealing were not considered in our subsequent analysis of the linked channels. It was recently proposed that the brief closures of gramicidin channels are due to undulations of the lipid bilayer that obliterate the mouths of the channel's pore (Armstrong et al., 2001). More detailed discussions of closing flickers can be found elsewhere (Armstrong et al., 2001; Armstrong and Cukierman, 2002). The present study does not address possible mechanisms of channel gating or closure. Rather, we sought to characterize the structure and fluctuations of channels in their functional, open state, which was postulated to resemble that of native gA based on the overall functional similarities in native, SS- and RR-linked gA dimers (Stankovic et al., 1989; Quigley et al., 1999).
Finally, Quigley et al. (1999) constructed molecular models of SS- and RR-linked dimers using ad hoc linker parameters derived from CHARMM22 (MacKerell et al., 1998) and a rigid dioxolane ring. Based on energy-minimized structures of the “open” state of the channels, they noted an important qualitative difference in local pitch resulting from the orientation of RR- and SS-dioxolane rings with respect to the plane of the bilayer (respectively perpendicular and parallel), which is confirmed in the present study. However, the model of the RR channel constructed by Quigley et al. (1999) featured highly asymmetric backbone distortions. In the present study, the combination of a dedicated force field with full treatment of the internal flexibility of the linker and extensive search of the conformational space thermally accessible to the linked channels led to considerable refinement of these results.
Comparison to high-resolution structural and dynamic studies of native gA
Because the present results highlight differences in peptide carbonyl tilts and fluctuations, it is of interest to compare these properties to available experimental data. While no such data exist at present for linked channels, high-resolution structural and dynamic data pertaining to peptide orientation in native gA were the object of several nuclear magnetic resonance (NMR) studies by Cross and co-workers (Ketchem et al., 1993, 1997; Lazo et al., 1995; North and Cross, 1995).
Ketchem et al. (1997) constructed a molecular model of native gA incorporating peptide orientational constraints obtained by solid-state NMR of uniformly-oriented gA in a lamellar-phase lipid environment. By themselves, these constraints result in ambiguities in the sign of carbonyl group tilts with respect to the axis of the channel, a property which was defined as a chirality (Quine et al., 1997) and which turns out in the present study to be central to the structure of the RR dimer. A motionally averaged structure was obtained from simulated annealing based on a global penalty function combining all experimentally derived restraints with the CHARMM force field (Brooks et al., 1983) for the peptide in vacuo (Ketchem et al., 1997). In that structure, the chirality of 14 out of the 16 backbone carbonyl groups (in each monomer) is uniquely resolved, with 10 of them pointing into the lumen and four of them pointing out. Half of these (residues 1, 2, 6, 9, 10, 12, and 14) agree in sign with the present study, in which eight carbonyl groups point in and eight point out, with three of the latter close to perfect alignment (Table 6). These results suggest that the unique topology obtained by Ketchem et al. (1997) is primarily a result of NMR constraints that were not included in the present study. In addition, possible sources of discrepancy include systematic errors resulting from the neglect of water and lipid, as well as small inconsistencies in the starting structure and in the Trp side-chain restraints in the current study. Finally, a single, motionally averaged structure might not be representative of individual conformers differing in carbonyl topology in the ensemble probed by the NMR experiments. Although the topology of carbonyl groups in the native dimer is only in partial agreement between the two studies, the magnitude of the tilts obtained by Ketchem et al. (1997), which was found to range between −20 and +20°, is in good agreement with the present results.
The rms amplitudes of peptide librational tilt motions computed in the present study are between 8 and 15°, which falls precisely between the ranges obtained in two NMR studies of backbone dynamics, respectively by Lazo et al. (1995) and by North and Cross (1995). In the former study, the analysis of powder-pattern NMR spectra in hydrated dimyristoyl-phosphatidylcholine bilayer samples revealed that picosecond librations of peptide planes about the Cα–Cα axis extend from 14 to 20° at 263 K (Lazo et al., 1995). In the latter study, field-dependent 15N T1 relaxation measurements indicated librations of 6 to 8° in the nanosecond time range. The discrepancy between these results was interpreted as the effect of damping of rapid librations by slower correlated motions. North and Cross (1995) discussed possible mechanisms for the overdamping of peptide librations. Collective motions of the channel are likely to be affected by the lipid environment, which was neglected in the present study. In addition, previous computational studies suggest that picosecond librations of the peptide carbonyl groups are coupled to the fluctuation of water molecules and of ions in the channel lumen (Chiu et al., 1991; Roux and Karplus, 1991a; Roux and Karplus, 1994, and references therein). Detailed analyses of long simulations in the presence of water and lipid will be required to clarify the nature and the amplitude of collective nanosecond dynamics. Nevertheless, the overall consistency between the results reported here and the analysis of NMR spectroscopic data indicates that even a model in vacuo captures essential dynamic features of the channel in the picosecond-to-nanosecond time range.
Structural significance and functional relevance
The overall secondary structure of the gramicidin pore is conserved in both SS and RR channels. While global deformations of the pore were precluded by construction, the fluctuations and local distortions of the backbone do not appear to be overly affected by these artificial restraints, inasmuch as the structure and fluctuations of the backbone are consistent with high-resolution spectroscopic studies of the native dimer. Thus, the restrictions imposed in the present study on each of the outermost turns of the helix are at once strong enough to prevent unraveling of the C-termini and sufficiently gentle to allow peptide tilts and librational fluctuations comparable to experimental data (see above discussion).
The consistency of the three models over relatively long simulations (16 ns) suggests adequate statistical convergence. In turn, the overall similarity of the three models lends significance to differences in their structural and dynamic properties. Intramolecular hydrogen bonding defining the β-helix is best satisfied in the SS channel, because that chiral form of dioxolane fits well with the step and pitch of a right-sided helix. By contrast, significant distortions affect the RR linker. Both the origin and the nature of these distortions are local: the RR-dioxolane ring acts as a wedge resulting in an overshoot of the helical pitch which is compensated by tilting the planes of both adjacent peptide bonds out of alignment with the channel axis. There is no intrinsic preference for tilting either carbonyl inward or outward, as long as they tilt significantly. Thus, the four conformers of the RR linker, and the fact that the two peptide groups may only exchange in/out topology one at a time, reflect the need to avoid alignment. This “law of the excluded middle” is driven by the preservation of the secondary structure, as restoration of the helical pitch averts the rupture of intermonomeric hydrogen bonds.
This structural requirement gives rise to essential qualitative and quantitative differences in dynamic properties in the time scale of the simulations. All the peptide carbonyl groups of native gA and SS-linked channels undergo moderate librational motions around a single minimum, with rms fluctuations of up to ±15°. However, in the RR-linked channel, conformational isomerization of the backbone involves thermally activated transitions consisting of flipping or “switching” either one of the two central peptide carbonyl groups in and out of the lumen by 50°. Like these conformational switching motions themselves, the propagation of switching transitions reflects the inherent plasticity of the secondary structure. As discussed by North and Cross (1995), anticorrelated motion of the channel backbone may occur between peptide planes in the axial direction (“stripe” model), and between peptides adjacent to each other in the primary sequence. The dominant force underlying the stripe model is hydrogen bonding defining the secondary structure of the pore. Our results indicate that consistently with the stripe model, significant distortions of the backbone are partly compensated by moderate switching of peptide planes one turn away.
The results of the present study are compatible with our two initial hypotheses regarding underlying structure–function relationships in dioxolane-linked channels. The local character of the distortions in RR-linked channels, which are confined to the middle half-turn of the helix and compromise the secondary structure minimally, justify a posteriori our overall approach, which focused the conformational search on the middle turn of the helix. On the one hand, the integrity of the secondary structure of the SS channel is consistent with the similar magnitude of proton conductances measured in native and SS channels. On the other hand, local differences in the structure and dynamics of SS- and RR-linked channels point to a possible structural origin for the attenuation of proton currents in the RR channel relative to its native and SS counterparts.
Previous computational studies have revealed that the organization and dynamic fluctuations of the lumen contents of gramicidin are determined to a large extent by interactions with the channel backbone. The hydrogen-bonding coordination of water molecules and the local solvation of alkali metal ions by backbone carbonyl O atoms have been shown to play an important role in the stabilization of ions and of the water column in the single-file region in models of native gA (Jordan, 1990; Roux and Karplus, 1991a; 1994, and references therein; Duca and Jordan, 1997). Computational studies of gramicidin channel analogs pointed out that deformations involving significant tilts of peptide groups enhance solvation of cations by carbonyl oxygen atoms (Jordan, 1990; Roux and Karplus, 1991a). Roux and Karplus (1991a) observed that these distortions are controlled by local librations and concluded that functional dynamics of the channel are largely determined by the intrinsic conformational flexibility or “plasticity” of the backbone. Accordingly, the small-amplitude librations and net translational movement of water and alkali metal ions were shown to be coupled to backbone fluctuations (Chiu et al., 1991; Roux and Karplus, 1991b). Finally, water-channel interactions were also shown to play an essential role in the hop-and-turn or Grotthuss relay mechanism for the translocation of an excess proton in the single-file water chain of the gA dimer (Pomès and Roux, 1996). Hydrogen-bond donation from water to carbonyl O atoms was found not only to be well suited to both the hydration and the mobility of H+, but also to assist in the complementary turn step of the Grotthuss mechanism, the reorientation of water molecules (Pomès and Roux, 2002). In that perspective, the conformational equilibrium characterized in the present study, which involves significant spatial displacements of two carbonyl groups and occurs in a time scale similar to that of ion translocation, raises particularly interesting possibilities in terms of understanding structural and dynamic modulations of proton transport at the atomic level.
CONCLUSIONS
We have modeled the structure and fluctuations of dioxolane-linked gramicidin channel analogs. Parameters for the molecular mechanics force field governing the conformational properties of the linker were obtained from ab initio calculations. Particular care was taken to ensure that the novel MM force field reproduces the torsional dependence of the potential energy and that these parameters are compatible with the CHARMM potential energy functions used to describe the polypeptide moiety of the linked dimers. Consistently with the formation of ion channels with conductance properties similar to those of the native gA dimer, it was found that both SS and RR dioxolane-linked gramicidin dimers can satisfy the overall β-helix structure of native gA in its functional state. However, significant differences in the structure of the two linked channels, which arise from a different orientation of the two stereoisomeric forms of the dioxolane ring with respect to the axis of the pore, were identified. While the SS channel is found in a single conformation resembling that of the native dimer, the backbone of the RR-linked channel is significantly distorted in the vicinity of dioxolane linker. Consistently with previous modeling studies, this result supports the initial assumption that functional differences between SS and RR channels originate from local structural perturbations in the middle of the channel. A novel and unexpected result of the present study is that backbone distortions of the RR dimer are due to the perturbation of the helical pitch and result in four stable conformers of the open channel that differ in the tilt of the peptide bonds flanking the linker with respect to the channel axis.
While this study highlights important properties pertaining to the plasticity of the polypeptidic backbone of gA, it also suggests essential differences in the nature of structural fluctuations of the RR dimer. Backbone fluctuations were proposed to play a central role in the translocation of water and ions through gramicidin (Roux and Karplus, 1991a). In the SS dimer, as in native gA, our results indicate that dynamic fluctuations at 298 K are characterized by unimodal tilt librations of peptide planes with respect to the channel axis. The amplitude of these oscillations is comparable to that derived from high-resolution experimental studies of native gA over ps-to-ns time scales (Lazo et al., 1995; North and Cross, 1995). By contrast, backbone fluctuations of the RR dimer include activated transitions exchanging the topology of two carbonyl groups in and out of the lumen. The ns time scale for the conformational isomerization of the RR channel suggests that it could interfere with the passage of ions, raising the interesting possibility of direct modulation of ion transport by functional dynamics of the channel. The present study provides a theoretical framework for understanding the molecular basis for the modulation of proton conduction in gramicidin channels. The effect of structural and dynamic properties of linked gramicidin dimers on the molecular mechanism of proton relay will be considered in forthcoming studies.
Acknowledgments
We are grateful to Susan Newton for building Pulay's natural internal coordinates and to Lothar Schäfer for useful discussions.
This work was supported by National Institutes of Health grant RO1 GM59674 and by Research Training grants to S.C. and C.H.Y. from the Research Institute of the Hospital for Sick Children. R.P. is a CRCP Chairholder.
References
- Andersen, O. S., and R. O. Koeppe, II. 1992. Molecular determinants of channel function. Physiol. Rev. 72:S89–S158. [DOI] [PubMed] [Google Scholar]
- Armstrong, K. M., and S. Cukierman. 2002. On the origin of closing flickers in gramicidin channels: a new hypothesis. Biophys. J. 82:1329–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong, K. M., E. P. Quigley, D. S. Crumrine, and S. Cukierman. 2001. Covalently linked gramicidin channels: effects of linker hydrophobicity and alkaline metals on different stereoisomers. Biophys. J. 80:1810–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arseniev, A. S., I. L. Barsukov, V. F. Bystrov, A. L. Lomize, and Y. A. Ovchinnikov. 1985. 1H-NMR study of gramicidin-A transmembrane ion channel: head-to-head right handed single stranded helices. FEBS Lett. 186:168–174. [DOI] [PubMed] [Google Scholar]
- Breneman, C. M., and K. B. Wiberg. 1990. Determining atom-centered monopoles from molecular electrostatic potentials–The need for high sampling density in formamide conformational analysis. J. Comp. Chem. 11:361–373. [Google Scholar]
- Brooks, B. R., R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus. 1983. CHARMM: A program for macromolecular energy minimization and dynamics calculations. J. Comp. Chem. 4:187–217. [Google Scholar]
- Chiu, S. W., E. Jakobsson, S. Subramaniam, and J. A. McCammon. 1991. Time-correlation analysis of simulated water motion in flexible and rigid gramicidin channels. Biophys. J. 60:273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crouzy, S., T. B. Woolf, and B. Roux. 1994. A molecular dynamics study of gating in dioxolane-linked gramicidin A channels. Biophys. J. 67:1370–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cukierman, S. 2000. Proton mobilities in water and in different stereoisomers of covalently linked gramicidin-A channels. Biophys. J. 78:1825–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cukierman, S., E. P. Quigley, and D. S. Crumrine. 1997. Proton conduction in gramicidin A and its dioxolane-linked dimer in different lipid bilayers. Biophys. J. 73:2489–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Godoy, C. M. G., and S. Cukierman. 2001. Modulation of proton transfer in the water wire of dioxolane-linked gramicidin channels by lipid membranes. Biophys. J. 81:1430–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duca, K. A., and P. C. Jordan. 1997. Ion-water and water-water interactions in a gramicidinlike channel: effects due to group polarizability and backbone flexibility. Biophys. Chem. 65:123–141. [DOI] [PubMed] [Google Scholar]
- Florian, J., and B. G. Johnson. 1994. Comparison and scaling of Hartree-Fock and Density-functional harmonic force-fields. 1. Formamide monomer. J. Phys. Chem. 98:3681–3687. [Google Scholar]
- Fogarasi, G., X. Zhou, P. W. Taylor, and P. Pulay. 1992. The calculation of ab initio molecular geometries: efficient optimization by natural internal coordinates and empirical correction by offset forces. J. Am. Chem. Soc. 114:8191–8201. [Google Scholar]
- Frisch, M. J., G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, Jr., R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, J. L. Andres, M. Head-Gordon, E. S. Replogle, and J. A. Pople. 1998. Gaussian 98 User's Reference: Density Functional Methods. Gaussian, Inc., Pittsburgh, Pennsylvania.
- Hille, B. 2001. Ionic Channels of Excitable Membranes, 3rd ed. Sinauer, Sunderland, Massachusetts.
- Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD–Visual Molecular Dynamics. J. Molec. Graphics. 14:33–38. [DOI] [PubMed] [Google Scholar]
- Jordan, P. C. 1990. Ion-water and ion-polypeptide correlations in a gramicidin-like channel. A molecular dynamics study. Biophys. J. 58:1133–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketchem, R. R., W. Hu, and T. A. Cross. 1993. High-resolution conformation of gramicidin A in a lipid bilayer by solid-state NMR. Science. 261:1457–1460. [DOI] [PubMed] [Google Scholar]
- Ketchem, R. R., B. Roux, and T. A. Cross. 1997. High resolution polypeptide structure in a lamellar phase lipid environment from solid state NMR derived orientational constraints. Structure. 5:1655–1669. [DOI] [PubMed] [Google Scholar]
- Lazo, N. D., W. Hu, and T. A. Cross. 1995. Low-temperature solid-state 15N NMR characterization of polypeptide backbone librations. J. Magn. Reson. B. 107:43–50. [DOI] [PubMed] [Google Scholar]
- MacKerell, A. D., Jr., D. Bashford, M. Bellot, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reither III, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiórkiewicz-Kuczera, and M. Karplus. 1998. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- North, N. L., and T. A. Cross. 1995. Correlations between function and dynamics: time scale coincidence for ion translocation and molecular dynamics in the gramicidin channel backbone. Biochemistry. 34:5883–5895. [DOI] [PubMed] [Google Scholar]
- Pomès, R., and B. Roux. 1996. Structure and dynamics of a proton wire: a theoretical study of H+ translocation along the single-file water chain in the gramicidin A channel. Biophys. J. 71:19–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomès, R., and B. Roux. 2002. Molecular mechanism of H+ conduction in the single-file water chain of the gramicidin channel. Biophys. J. 82:2304–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulay, P., F. Fogarasi, F. Pang, and J. E. Boggs. 1979. Systematic ab initio gradient calculation of molecular geometries, force constants, and dipole moment derivatives. J. Am. Chem. Soc. 101:2550–2560. [Google Scholar]
- Quigley, E. P., P. Quigley, D. S. Crumrine, and S. Cukierman. 1999. The conduction of protons in different stereoisomers of dioxolane-linked gramicidin A channels. Biophys. J. 77:2479–2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley, E. P., D. S. Crumrine, and S. Cukierman. 2000. Gating and permeation in ion channels formed by gramicidin A and its dioxolane-linked dimer in Na+ and Cs+ solutions. J. Membr. Biol. 174:207–212. [DOI] [PubMed] [Google Scholar]
- Quine, J. R., M. Brenneman, and T. A. Cross. 1997. Protein structural analysis from solid-state NMR derived orientational constraints. Biophys. J. 72:2342–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux, B. 2002. Computational studies of the gramicidin channel. Acc. Chem. Res. 35:366–375. [DOI] [PubMed] [Google Scholar]
- Roux, B., and M. Karplus. 1991a. Ion transport in a model gramicidin channel: structure and thermodynamics. Biophys. J. 59:961–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux, B., and M. Karplus. 1991b. Ion transport in a gramicidin-like channel: dynamics and mobility. J. Phys. Chem. 95:4856–4868. [Google Scholar]
- Roux, B., and M. Karplus. 1994. Molecular dynamics simulations of the gramicidin channel. Annu. Rev. Biophys. Biomol. Struct. 23:731–761. [DOI] [PubMed] [Google Scholar]
- Ryckaert, J. P., G. Ciccotti, and H. J. C. Berendsen. 1977. Numerical integration of the Cartesian equation of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comp. Phys. 23:327–341. [Google Scholar]
- Stankovic, C. J., S. H. Heinemann, J. M. Delfino, F. J. Sigworth, and S. L. Schreiber. 1989. Transmembrane channels based on tartaric acid-gramicidin A hybrids. Science. 244:813–817. [DOI] [PubMed] [Google Scholar]
- Stankovic, C. J., S. H. Heinemann, and S. L. Schreiber. 1990. Immobilizing the gate of a tartaric acid-gramicidin A hybrid channel molecule by rational design. J. Am. Chem. Soc. 112:3702–3704. [Google Scholar]
- Tian, F., and T. A. Cross. 1999. Cation transport: an example of structural based selectivity. J. Mol. Biol. 285:1993–2003. [DOI] [PubMed] [Google Scholar]
- Woolf, T. B., and B. Roux. 1994. Molecular dynamics simulations of the gramicidin channel in a phospholipid bilayer. Proc. Natl. Acad. Sci. USA. 91:11631–11635. [DOI] [PMC free article] [PubMed] [Google Scholar]