

Abstract
It is shown that the averaged chemical shift (ACS) of a particular nucleus in the protein backbone empirically correlates well to its secondary structure content (SSC). Chemical shift values of more than 200 proteins obtained from the Biological Magnetic Resonance Bank are used to calculate ACS values, and the SSC is estimated from the corresponding three-dimensional coordinates obtained from the Protein Data Bank. ACS values of 1Hα show the highest correlation to helical and sheet structure content (correlation coefficient of 0.80 and 0.75, respectively); 1HN exhibits less reliability (0.65 for both sheet and helix), whereas such correlations are poor for the heteronuclei. SSC estimated using this correlation shows a good agreement with the conventional chemical shift index-based approach for a set of proteins that only have chemical shift information but no NMR or x-ray determined three-dimensional structure. These results suggest that even chemical shifts averaged over the entire protein retain significant information about the secondary structure. Thus, the correlation between ACS and SSC can be used to estimate secondary structure content and to monitor large-scale secondary structural changes in protein, as in folding studies.
INTRODUCTION
Since the first observation of the chemical shift in NMR spectra in 1957 by Gutowsky et al., it has been used as a powerful indicator of the type of secondary structure that a biopolymer can adopt. Thus, most development of modern experimental methods is driven by the goal to increase the resolution and sensitivity by which the chemical shift of a nucleus can be measured. In addition to structural information (Dalgarno et al., 1983; Pastore and Saudek, 1990; Williamson, 1990; Wishart et al., 1991a; Laws et al., 1993; Oldfield, 1995; Cornilescu et al., 1999), chemical shifts provide detailed information about the nature of hydrogen exchange dynamics, ionization and oxidation states, ring current influence of aromatic residues, and hydrogen bonding interactions (Szilagyi, 1995). Several recent and excellent review articles describe a variety of experimental and computational methods to correlate chemical shifts to protein three-dimensional structural information (Szilagyi, 1995; Case et al., 1994; Wishart and Nip, 1998; Ando et al., 2001; Wishart and Case, 2001).
Here, we would like to explore whether any meaningful structural information could still be obtained from chemical shifts before completion of resonance assignments. Thus, the extensive information found in the Biological Magnetic Resonance Bank and Protein Data Bank was used to determine whether there is a correlation between protein secondary structure content (SSC) and the average chemical shift (ACS) value for a particular type of nucleus. We have determined that the highest correlation with secondary structure content is found with the 1Hα ACS value, followed by 1HN ACS. No reliable correlations could be determined for backbone heteronuclei 13Cα and 15N. The correlation between 1Hα and 1HN ACS values and SSC was used to estimate the percentage of helical and sheet content for a set of proteins that have complete chemical shift information available in the BMRB, but the NMR or x-ray determined three-dimensional structures of which are not yet available. The estimates are then compared with estimates calculated using a more conventional method, the chemical shift index (CSI), which uses the individual chemical shifts of each resonance instead of an average. The results show a good agreement for the helical content between the two estimates, whereas the agreement is only moderate for the sheet content. Though determination of CSI values is superior in obtaining SSC estimates in cases where the resonances can be readily assigned, estimates of SSC could be obtained from a simple ACS value as well, especially in cases where assignments would be difficult if not impossible to obtain (e.g., under denaturing conditions). Though circular dichroism also be used to estimate SSC, and requires less sample and experimental time, circular dichroism cannot be used in cases where the signal is masked by the solvent signal, such as when high concentrations of urea are used.
METHODS
Chemical shift values corresponding to the protein backbone atoms (1HN, 15N, 1Hα, and 13Cα) were obtained from the Biological Magnetic Resonance Bank (BMRB, http://www.bmrb.wisc.edu/) star files (Seavey et al., 1991). If the information on the structure of the protein was also present in the star file, it was extracted, as was the information on the amino acid sequence. Structure files obtained from the Rutgers Center for Structural Biology (PDB format, http://www.rcsb.org/pdb/) (Berman et al., 2000) were cross-checked against the corresponding BMRB star file manually to ensure a correct match. NMR determined structures were used whenever possible, but if they were not available, the corresponding x-ray structure was used instead. Only proteins with more than 50 amino acid residues were considered, inasmuch as these were expected to contain a significant amount of secondary structure.
The averaged chemical shift (ACS) of a nucleus “i” is defined by:
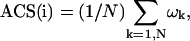 |
(1) |
where N is the total number of observed crosspeaks (typically in a single bond correlated spectrum, such as a heteronuclear single quantum correlation (HSQC)) and ωk is the corresponding chemical shift of the kth resonance (referenced using recommended procedures (Wishart et al., 1995)).
To evaluate the secondary structure content for the set of proteins, the program probability-based protein secondary structure identification (PSSI) was used (Wang and Jardetzky, 2002). In this method, CSI of the set of backbone atoms are used to define the probability with which the secondary structure (sheet or helix) is assigned. Secondary structure content in percentage is then calculated with respect to the total number of residues in the sequence.
The structure-based percentage of sheet and helix (sum of α and 310) was determined using the program PROMOTIF (http://www.biochem.ucl.ac.uk/gail/promotif/promotif.html) (Hutchinson and Thornton, 1996), which uses the atomic coordinate files obtained from the RCSB. All the analyses were performed using codes written using C++ and other scripts (awk or perl) on a Silicon Graphics UNIX work station (copies of the code are available from the authors). A complete list of the 213 proteins (comprised of 1HN, 15N, 1Hα, and 13Cα chemical shifts) and 25 additional proteins (with only 1HN and 15N chemical shifts), their individual ACS values and the structure based secondary structure content estimates are available from the authors.
RESULTS
Correlations between averaged chemical shifts and secondary structure content
Fig. 1 shows the ACS values of 1HN and 15N nuclei (top and bottom rows, respectively) plotted against the respective sheet and helical content (left and right columns, respectively). Fig. 2 shows a similar plot for 1Hα and 13Cα nuclei. A total of 238 and 213 proteins were used in Figs. 1 and 2, respectively (Supporting Information Table S1). The continuous lines in Figs. 1 and 2 correspond to linear regression analyses of the data, and Table 1 lists the results of the analyses. ACS values of both 1HN and 1Hα are much more indicative of the overall secondary structure content than those of the heavy atoms. Correlation coefficients for the plots of 1HN, 15N, 1Hα, and 13Cα versus percent sheet content are 0.64, 0.44, 0.75, and 0.44, respectively, whereas the coefficients obtained in the plots versus percent helix content are 0.65, 0.40, 0.80, and 0.58, respectively (Table 1). Although 13Cα ACS values show a wider dispersion with respect to helical content (Fig. 2 d) than the corresponding 15N data (Fig. 1 d), the correlation coefficients for the plots of heteronuclei are equally poor. Overall, the best correlations were obtained with the 1HN and 1Hα data.
FIGURE 1.

Plots of ACS values versus the secondary structure contents estimated from the three-dimensional structures using the program PROMOTIF. (a) and (b) show the correlation of 1HN and 15N ACS values, respectively, to the percentage of β-sheet structures, whereas (c) and (d) correlate the same ACS values with respect to the percentage of helical structure (sum of α and 310 helices). Continuous lines represent the linear regression analysis results.
FIGURE 2.

Plots of ACS values versus the secondary structure contents estimated from the three-dimensional structures using the program PROMOTIF. (a) and (b) show the correlation 1Hα and 13Cα ACS values, respectively, to the percentage of β-sheet structures, whereas (c) and (d) correlate the same ACS values with respect to the percentage of helical structure (sum of α and 310 helices). Results of the linear regression analysis are shown by continuous lines.
TABLE 1.
Linear correlation of ACS to secondary structure content
|
*Sheet (%)
|
*Helix (%)
|
|||||
|---|---|---|---|---|---|---|
| ACS (ppm) | †CC | ‡Slope | ‡Intercept | †CC | ‡Slope | ‡Intercept |
| 1HN | 0.64 | 60.9 ± 4.4 | −488.0 ± 36.3 | 0.65 | −95.0 ± 6.7 | 818.9 ± 55.4 |
| 15N | 0.44 | 5.1 ± 0.63 | −598.4 ± 75.5 | 0.40 | −7.2 ± 1.0 | 899.1 ± 118.6 |
| 1Hα | 0.75 | 66.2 ± 3.7 | −273.1 ± 16.0 | 0.80 | −102.8 ± 5.2 | 482.1 ± 22.6 |
| 13Cα | 0.44 | −6.27 ± 0.9 | 377.6 ± 49.8 | 0.58 | 12.4 ± 1.2 | −680.0 ± 67.5 |
Secondary structures defined based on PROMOTIF.
CC: Correlation coefficient for linear regression analysis.
Slope and intercept are defined based on a linear equation ACS (ppm) = Slope× Secondary Structure Content + Intercept.
A notable feature of these results is that the slopes of the lines for the ACS values versus helix and sheet content are opposite to each other (most clearly seen in panels a and c of Figs. 1 and 2). The change in the sign of the slope indicates that changes in ACS values can allow differentiation of increasing or decreasing helical or sheet secondary structural elements upon changes in environment. The ACS values increase with an increase in the total sheet content and decrease with an increase in the total helical content.
Estimation of SSC for proteins having no determined three-dimensional structure
A set of 36 proteins obtained from the BMRB for which complete assignments of the backbone atoms are known, but for which structures have not yet been determined, were used to estimate SSC by using the empirical correlation between SSC and 1Hα or 1HN ACS values. SSC was also calculated using the consensus chemical shift indices using the program PSSI (Methods) using all the backbone atoms. The list of all the proteins and their estimated SSC, using the correlation and CSI-based methods is given in Table 2. There is an overall agreement between the SSCs estimated using these two methods (Fig. 3). Larger deviations were observed in the 1HN ACS values compared to the 1Hα ACS values. The correlation between 1HN ACS and secondary structure (Table 1) is worse than that for the 1Hα ACS, as shown in Fig. 3, b and d.
TABLE 2.
Estimated SSC using ACS- and CSI-based methods for proteins with no three-dimensional structural information
| Helix (%) from
|
Sheet (%) from
|
||||||
|---|---|---|---|---|---|---|---|
| BMRB | Protein name | *ACS (1Hα) | *ACS (1HN) | †CSI | *ACS (1Hα) | *ACS (1HN) | †CSI |
| 4840 | Adenylate kinase | 15.31 | 12.65 | 12.70 | 38.11 | 34.58 | 20.40 |
| 4834 | Staphylococcus aureus peptide deformylase | 25.56 | 18.69 | 21.70 | 25.54 | 20.70 | 36.50 |
| 4825 | Recombinant RC-RNase 2 | 27.28 | 18.45 | 15.10 | 25.70 | 19.60 | 39.60 |
| 4821 | DcuS | 17.62 | 15.87 | 20.40 | 33.70 | 30.37 | 19.10 |
| 4795 | Human D187N gelsolin domain 2 | 20.83 | 18.67 | 37.10 | 28.73 | 26.01 | 24.10 |
| 4794 | Human wild-type gelsolin domain 2 | 25.69 | 19.45 | 20.70 | 25.06 | 20.62 | 35.30 |
| 4787 | Apical membrane antigen 1 | 26.10 | 12.95 | 13.10 | 29.25 | 20.36 | 21.30 |
| 4784 | Tyrosine repressor | n.a. | 6.38 | 6.60 | n.a. | 47.90 | 57.40 |
| 4776 | Sud dimer | 14.03 | 11.31 | 19.70 | 40.21 | 36.56 | 42.30 |
| 4771 | Tola3 | 8.18 | 4.57 | 21.70 | 50.72 | 45.64 | 36.80 |
| 4752 | gpnu1-E68 | 11.92 | 4.32 | 16.20 | 51.62 | 39.25 | 39.70 |
| 4735 | Olfactory marker protein | 23.33 | 12.27 | 21.50 | 29.68 | 22.13 | 41.10 |
| 4722 | Shikimate kinase | 9.34 | 6.80 | 19.00 | 47.78 | 43.28 | 32.10 |
| 4716 | Auxilin | 3.53 | 2.78 | 7.70 | 54.01 | 52.33 | 39.00 |
| 4712 | Newt acidic FGF | 16.76 | 10.94 | 5.30 | 30.54 | 26.34 | 45.50 |
| 4711 | RNA-binding protein | n.a. | 13.99 | 31.70 | n.a. | 36.02 | 20.80 |
| 4698 | Transforming growth factor beta type II receptor | n.a. | 13.86 | 4.90 | n.a. | 28.66 | 41.00 |
| 4688 | L18 | 22.46 | 14.66 | 32.40 | 34.98 | 23.48 | 31.50 |
| 4670 | PIN1At | 30.71 | 17.80 | 29.20 | 25.68 | 17.80 | 33.30 |
| 4664 | Lipocalin Q83 | 25.54 | 11.77 | 17.20 | 30.01 | 20.71 | 51.00 |
| 4579 | FYVE domain of EEA1 | n.a. | 20.16 | 18.60 | n.a. | 27.05 | 11.60 |
| 4567 | Catalytic domain of yUBC1 | 19.30 | 13.23 | 24.50 | 37.21 | 28.38 | 38.40 |
| 4558 | YopH-NT monomer | 17.48 | 12.64 | 25.70 | 38.13 | 31.21 | 37.50 |
| 4463 | Ras-binding domain of Byr2 | 21.74 | 18.02 | 31.90 | 30.37 | 23.95 | 33.60 |
| 4447 | p23fyp | 20.93 | 19.80 | 33.30 | 26.97 | 25.85 | 29.80 |
| 4353 | p13 C-terminal domain | 23.81 | 16.69 | 15.90 | 26.84 | 21.82 | 38.90 |
| 4335 | Calerythrin | 5.88 | 2.21 | 11.40 | 54.90 | 48.68 | 60.20 |
| 4313 | E2 | 24.09 | 17.73 | 41.70 | 30.19 | 20.94 | 21.10 |
| 4294 | Human MBF1(57-148) core domain | 5.96 | 4.99 | 15.20 | 50.59 | 48.55 | 42.40 |
| 4271 | Calcium binding protein from Entamoeba histolytica | 11.00 | 8.70 | 13.40 | 44.27 | 41.27 | 48.50 |
| 4239 | f29-SSB bacteriophage | 23.69 | 14.46 | 15.30 | 28.27 | 21.90 | 43.50 |
| 4147 | Cold shock domain | 14.95 | 6.93 | 8.90 | 32.65 | 27.95 | 44.30 |
| 4136 | Escherichia coli multidrug Resistance protein E | n.a. | 3.46 | 1.80 | n.a. | 52.96 | 68.20 |
| 4132 | Human ubiquitin-conjugating enzyme | 18.15 | 12.68 | 19.90 | 38.07 | 30.17 | 33.10 |
| 4027 | S. aureus DHFR(F98Y)-NADPH-TMP ternary complex | 17.36 | 17.00 | 22.20 | 26.64 | 25.96 | 44.90 |
| 1583 | Micrococcal nuclease | n.a. | 20.59 | 7.30 | n.a. | 23.89 | 48.90 |
Secondary structure content estimated using the correlation listed in Table 1.
Secondary structure content estimated using probability-based protein secondary structure identification (Wang and Jardetzky, 2002).
n.a.: not determined due to absence of chemical shift information.
FIGURE 3.

Comparison of helical and sheet content percent calculated using 1Hα or 1HN ACS values to that obtained using a consensus chemical shift index-based method for a set of proteins for which no three-dimensional structures are available. (a) and (b) correspond to the helical content using the 1Hα and 1HN ACS values, respectively, whereas (c) and (d) are the corresponding sheet content using the same ACS values. The dashed lines correspond to a perfect correlation between these two methods.
DISCUSSION
Previously, Wishart and co-workers (Wishart et al., 1991b; Wishart and Sykes, 1994) have suggested two methods to estimate secondary structure content from two-dimensional NMR data. In one of their methods, the total number of crosspeaks over a preselected region of a homonuclear two-dimensional correlation spectrum is counted to estimate the SSC. Here we show that even the averaged chemical shift values of a backbone atom of a protein can retain significant information about the proteins secondary structure. In particular, the 15N-1HN chemical shift values, which are often underutilized as indicators of secondary structure because they are liable to change with small variations in temperature and pH (Glushka et al., 1989; Le and Oldfield, 1994), can be used to estimate SSC from 1HN ACS values as a function of varying buffer conditions. In addition to the backbone atoms 1HN, 15N, 1Hα, and 13Cα, ACS versus SSC correlations for the other backbone and side-chain nuclei 13CO, 13Cβ, and 1Hβ were also evaluated (data not shown). The correlation coefficients for the plots of ACS values for these nuclei versus SSC were good, in particular for the carbon atoms. However, estimates of SSC using the 13CO, 13Cβ, and 1Hβ ACS were not considered further because residue-specific secondary structure determination can readily be accomplished with chemical shift assignments by using other empirical methods, such as TALOS, developed by Cornilescu et al. (1999).
The statistical analysis of the correlation between ACS and SSC is relatively good for the 1Hα ACS values (75–80%), whereas a moderate correlation (65%) is obtained with the 1HN ACS values. As the number of proteins that can be added into the correlations of ACS with secondary structure increases, the correlation coefficients should improve significantly. However, certain factors may result in lowering the correlation coefficient. ACS values were based on the total number of crosspeaks that were observed, and not on the total number of residues in the protein. For example, a 15N-HSQC spectrum will not contain resonances from a proline residue, which will consequently not be included in the ACS value, though it is present in the sequence. Significant contributions in lowering the correlation are expected from the residues that are present in the turns that will contribute to the ACS value as a sheet or helix. For example, residues that are part of a β-turn will be considered as β-sheet when the average values are calculated. The distribution of chemical shifts for each of the amino acids found in the BMRB database suggests that no particular amino acid dominates the ACS values, and hence the chemical shifts, for a particular type of amino acid. Therefore no particular amino acid expected to bias the correlation. Moreover, Sharman et al. (2001) have used rigorous statistical analyses of 1Hα chemical shifts to show that there is no correlation between amino acid type and propensity to fall within helical or sheet regions. However, it is possible that certain proteins will contain a large number of one type of residue (or a preponderance of a few types of residues) that may skew the ACS value. The relatively low correlation coefficients (0.64–0.8) for the ACS versus SSC correlations may result from these and other factors. However, estimating SSC from ACS values may still be a way to detect secondary structural changes, especially increases or decreases in helical content.
From a practical point of view, this correlation would be most useful if a sufficient number of individual crosspeaks are observed in an HSQC spectrum. Although the correlations were not evaluated by systematically eliminating a certain percentage of peaks from the data, it is recommended that a minimum of 70% of the total number of peaks expected should be observed in the spectra to determine a reliable ACS value. Experimental methods based on transverse relaxation optimized spectroscopy (Pervushin et al., 1997, 1998) can provide an additional advantage for estimating SSC from ACS values.
In summary, the observed correlation between ACS and SSC can be used to monitor structural changes in real time, such as in protein folding experiments, to detect large-scale structural changes in complex formation and to identify initial protein folds in high throughput proteomics applications.
SUPPORTING MATERIAL
Table S1: List of all the proteins, BMRB and pdb codes, ACS values and PROMOTIF estimates of secondary structure content is available from the authors. (15 pages, data used in Figs. 1 and 2).
Acknowledgments
Thanks to Dr. R. Balhorn and S.P. Mielke for critical reading of the manuscript.
A.B.S. was supported by a summer student fellowship from Department of Energy, Defense Programs, and Office of University Partnerships. This work was performed under the auspices of the U.S. Department of Energy by the University of California, Lawrence Livermore National Laboratory, under Contract No. W-7405-Eng-48 and Laboratory Wide Director's Initiative Grant LW-068 (V.V.K.).
Dedicated with admiration and affection to Professor Anil Kumar, Department of Physics, Indian Institute of Science, Bangalore, India who is superannuating in 2003.
References
- Ando, I., S. Kuroki, H. Kurosu, and T. Yamanobe. 2001. NMR chemical shift calculations and structural characterizations of polymers. Progress in Nuclear Magnetic Resonance Spectroscopy. 39:79–133. [Google Scholar]
- Berman, H. M., J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov, and P. E. Bourne. 2000. The Protein Data Bank. Nucleic Acids Res. 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case, D. A., H. J. Dyson, and P. E. Wright. 1994. Use of chemical shifts and coupling constants in nuclear magnetic resonance structural studies of peptides and proteins. Methods Enzymol. 239:392–416. [DOI] [PubMed] [Google Scholar]
- Cornilescu, G., F. Delaglio, and A. Bax. 1999. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR. 13:289–302. [DOI] [PubMed] [Google Scholar]
- Dalgarno, D. C., B. A. Levine, and R. J. Williams. 1983. Structural information from NMR secondary chemical shifts of peptide alpha C-H protons in proteins. Biosci. Rep. 3:443–452. [DOI] [PubMed] [Google Scholar]
- Glushka, J., M. Lee, S. Coffin, and D. Cowburn. 1989. 15N chemical shifts of backbone amides in bovine pancreatic trypsin inhibitor and apamin. J. Am. Chem. Soc. 111:7716–7722. [Google Scholar]
- Gutowsky, H. S., A. Saika, M. Takeda, and D. E. Woessner. 1957. Proton magnetic resonance studies on natural rubber. II. Line shape and T1 measurements. J. Chem. Phys. 27:534–542. [Google Scholar]
- Hutchinson, E. G., and J. M. Thornton. 1996. PROMOTIF—a program to identify and analyze structural motifs in proteins. Protein Sci. 5:212–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laws, D. D., A. C. Dedios, and E. Oldfield. 1993. NMR chemical shifts and structure refinement in proteins. J. BIOMOL. NMR. 3:607–612. [DOI] [PubMed] [Google Scholar]
- Le, H. B., and E. Oldfield. 1994. Correlation between 15N NMR chemical shifts in proteins and secondary structure. J. Biomol. NMR. 4:341–348. [DOI] [PubMed] [Google Scholar]
- Oldfield, E. 1995. Chemical shifts and three-dimensional protein structures. J. Biomol. NMR. 5:217–225. [DOI] [PubMed] [Google Scholar]
- Pastore, A., and V. Saudek. 1990. The relationship between chemical shift and secondary structure in proteins. J. Magn. Reson. 90:165–176. [Google Scholar]
- Pervushin, K., R. Riek, G. Wider, and K. Wuthrich. 1997. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc. Natl. Acad. Sci. USA. 94:12366–12371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pervushin, K., R. Riek, G. Wider, and K. Wuthrich. 1998. Transverse relaxation-optimized spectroscopy (TROSY) for NMR studies of aromatic spin systems in C13-labeled proteins. J. Am. Chem. Soc. 120:6394–6400. [Google Scholar]
- Seavey, B. R., E. A. Farr, W. M. Westler, and J. L. Markley. 1991. A relational database for sequence-specific protein NMR data. J. Biomol. NMR. 1:217–236. [DOI] [PubMed] [Google Scholar]
- Sharman, G. J., S. R. Griffiths-Jones, M. Jourdan, and M. S. Searle. 2001. Effects of amino acid phi,psi propensities and secondary structure interactions in modulating H alpha chemical shifts in peptide and protein beta-sheet. J. Am. Chem. Soc. 123:12318–12324. [DOI] [PubMed] [Google Scholar]
- Szilagyi, L. 1995. Chemical shifts in proteins come of age. Progress in Nuclear Magnetic Resonance Spectroscopy. 27:325–443. [Google Scholar]
- Wang, Y. J., and O. Jardetzky. 2002. Probability-based protein secondary structure identification using combined NMR chemical-shift data. Protein Sci. 11:852–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson, M. P. 1990. Secondary-structure dependent chemical shifts in proteins. Biopolymers. 29:1428–1431. [DOI] [PubMed] [Google Scholar]
- Wishart, D. S., C. G. Bigam, J. Yao, F. Abildgaard, H. J. Dyson, E. Oldfield, J. L. Markley, and B. D. Sykes. 1995. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J. Biomol. NMR. 6:135–140. [DOI] [PubMed] [Google Scholar]
- Wishart, D. S., and D. A. Case. 2001. Use of chemical shifts in macromolecular structure determination. Methods Enzymol. 338:3–34. [DOI] [PubMed] [Google Scholar]
- Wishart, D. S., and A. M. Nip. 1998. Protein chemical shift analysis: a practical guide. Biochem Cell Biol. 76:153–163. [DOI] [PubMed] [Google Scholar]
- Wishart, D. S., and B. D. Sykes. 1994. Chemical shifts a tool for structure determination. Methods Enzymol. 239:363–392. [DOI] [PubMed] [Google Scholar]
- Wishart, D. S., B. D. Sykes, and F. M. Richards. 1991a. Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J. Mol. Biol. 222:311–333. [DOI] [PubMed] [Google Scholar]
- Wishart, D. S., B. D. Sykes, and F. M. Richards. 1991b. Simple techniques for the quantification of protein secondary structure by 1H NMR spectroscopy. FEBS Lett. 293:72–80. [DOI] [PubMed] [Google Scholar]