

Abstract
Earlier studies using atomic force microscopy (AFM) demonstrated the presence of fusion pores at the cell plasma membrane in a number of live secretory cells, revealing their morphology and dynamics at nm resolution and in real time. Fusion pores were stable structures at the cell plasma membrane where secretory vesicles dock and fuse to release vesicular contents. In the present study, transmission electron microscopy confirms the presence of fusion pores and reveals their detailed structure and association with membrane-bound secretory vesicles in pancreatic acinar cells. Immunochemical studies demonstrated that t-SNAREs, NSF, actin, vimentin, α-fodrin and the calcium channels α1c and β3 are associated with the fusion complex. The localization and possible arrangement of SNAREs at the fusion pore are further demonstrated from combined AFM, immunoAFM, and electrophysiological measurements. These studies reveal the fusion pore or porosome to be a cup-shaped lipoprotein structure, the base of which has t-SNAREs and allows for docking and release of secretory products from membrane-bound vesicles.
INTRODUCTION
The fusion of membrane-bound secretory vesicles at the cell plasma membrane (PM) and consequent expulsion of vesicular contents is a fundamental cellular process regulating basic physiological functions such as neurotransmission, enzyme secretion, or hormone release. Secretory vesicles dock and fuse at specific PM locations after secretory stimuli. Earlier electrophysiological studies on mast cells suggested the existence of fusion pores at the cell PM, which became continuous with the secretory vesicle membrane after stimulation of secretion (Monck et al., 1995). Atomic force microscopy (AFM) has confirmed the existence of the fusion pore and its structure and dynamics in both exocrine (Schneider et al., 1997) and neuroendocrine cells (Cho et al., 2002a,b) at near nm resolution and in real time. Fusion pores in NG108-15 nerve cells have also been reported (Tojima et al., 2000).
Isolated live pancreatic acinar cells in physiological buffer, when imaged with the AFM (Cho et al., 2002c; Schneider et al., 1997), reveal at the apical PM a group of circular pits measuring 0.4–1.2 μm in diameter which contain smaller “depressions” (see Fig. 1 a). Each depression averages between 100 and 150 nm in diameter, and typically 3–4 depressions are located within a pit. The basolateral membrane of acinar cells is, however, devoid of either pits or depressions. High resolution AFM images of depressions in live cells further reveal a cone-shaped morphology (see Fig. 1 d). The depth of each depression cone measures ∼15–30 nm. Similarly, both growth hormone (GH) secreting cells of the pituitary gland and chromaffin cells possess pits and depression structures in their PM (Cho et al., 2002a,b), suggesting their universal presence in secretory cells.
FIGURE 1.

AFM and immunoAFM micrographs of the fusion pore, demonstrating pore morphology and the release of secretory products at the site. (a) A pit with four fusion pores within, found at the apical surface in a live pancreatic acinar cell; (b) After stimulation of secretion, amylase-specific immunogold localizes at the pit and fusion pores within, demonstrating them to be secretory release sites; (c) Some fusion pores demonstrate greater immunogold localization, suggesting more release of amylase from them; (d) AFM micrograph of a single fusion pore in a live acinar cell.
Exposure of pancreatic acinar cells to a secretagogue (mastoparan) results in a time-dependent increase (20–35%) in depression diameter, followed by a return to resting size after completion of secretion (Cho et al., 2002a,c; Schneider et al., 1997). No demonstrable change in pit size, however, is detected during this time (Schneider et al., 1997). Enlargement of depression diameter and an increase in its relative depth after exposure to secretagogues correlated with increased secretion. Conversely, exposure of pancreatic acinar cells to cytochalasin B, a fungal toxin that inhibits actin polymerization, resulted in a 15–20% decrease in depression size and a consequent 50–60% loss in secretagogue-induced secretion. Results from these studies suggested that depressions are the fusion pores in pancreatic acinar cells. Furthermore, these studies demonstrated the involvement of actin in regulation of the structure and function of depressions. Analogous to pancreatic acinar cells, examination of resting GH secreting cells of the pituitary (Cho et al., 2002a) and chromaffin cells of the adrenal medulla (Cho et al., 2002b) also revealed the presence of pits and depressions on the cell PM. Depressions in resting GH cells measured 154 ± 4.5 nm (mean ± SE) in diameter. Exposure of the GH cell to a secretagogue resulted in a 40% increase (215 ± 4.6 nm; p < 0.01) in depression diameter but no appreciable change in pit size.
The enlargement of depression diameter during secretion and the known effect that actin depolymerizing agents decrease depression size and inhibit secretion (Schneider et al., 1997) suggested the depressions to be the fusion pores. However, a more direct determination of the function of depressions was required. Localization of a gold conjugated antibody to a specific vesicular secretory protein with AFM provided the ability to determine whether secretion occurred at depressions (Cho et al., 2002a,c). The membrane-bound secretory vesicles in exocrine pancreas contain the starch digesting enzyme amylase. The AFM was used to localize amylase-specific antibodies tagged with colloidal gold at “depressions” after stimulation of secretion (Cho et al., 2002c). These studies confirm that “depressions” are the fusion pores in pancreatic acinar cells where membrane-bound secretory vesicles dock and fuse to release vesicular contents. Similarly, in somatotrophs of the pituitary, gold-tagged GH-specific antibody was selectively localized at depressions after stimulation of secretion (Cho et al., 2002a), again identifying the depressions in GH cells as the fusion pores.
Although the molecular composition of the fusion pore is unknown, studies on the role of actin in the regulation of depression structure and dynamics (Schneider et al., 1997) clearly suggest that actin is a component of the fusion pore complex. Target membrane proteins SNAP-25 and syntaxin (t-SNARE) and secretory vesicle associated membrane protein (v-SNARE) are part of the conserved protein complex involved in fusion of opposing bilayers (Rothman, 1994; Weber et al., 1998). Because membrane-bound secretory vesicles dock and fuse at depressions to release vesicular contents, it is reasonable to suggest that PM-associated t-SNAREs are part of the fusion pore complex. In the last decade, a number of studies demonstrated the involvement of cytoskeletal proteins in exocytosis, some directly interacting with SNAREs (Bennett, 1990; Cho et al., 2002c; Faigle et al., 2000; Goodson et al., 1997; Nakano et al., 2001; Ohyama et al., 2001). The actin and microtubule-based cytoskeleton has been implicated in intracellular vesicle traffic (Goodson et al., 1997). Fodrin, which was previously implicated in exocytosis (Bennett, 1990), has recently been shown to interact directly with SNAREs (Nakano et al., 2001). Recent studies demonstrated that α-fodrin regulates exocytosis through its interaction with the syntaxin family of proteins (Nakano et al., 2001). The C-terminal coiled-coil region of syntaxin interacts with α-fodrin, a major component of the sub membranous cytoskeleton. Similarly, vimentin filaments interact with SNAP-23/25 and control the availability of free SNAP-23/25 for assembly of the SNARE complex (Faigle et al., 2000). Results from these studies suggest that vimentin, α-fodrin, actin, and SNAREs may all be part of the fusion pore complex. Additional proteins such as v-SNARE (VAMP or synaptobrevin), synaptophysin, and myosin may associate when the fusion pore establishes continuity with the secretory vesicle membrane. The globular tail domain of myosin V contains a binding site for VAMP which is bound in a calcium independent manner (Ohyama et al., 2001). Further interaction of myosin V with syntaxin requires both calcium and calmodulin. It has been suggested that VAMP acts as a myosin V receptor on secretory vesicles and regulates formation of the SNARE complex (Ohyama et al., 2001). Interaction of VAMP with synaptophysin and myosin V was also observed by Prekeris and Terrian (1997). To understand the fusion pore in greater detail, its structure and biochemistry were further examined in the present study by using a combination of AFM, immunoAFM, electron microscopy (EM), and immunochemical analysis.
MATERIALS AND METHODS
Isolation of pancreatic acinar cells
Acinar cells for secretion experiments, light microscopy, AFM, and EM were isolated using minor modification of a published procedure (Jena et al., 1991). For each experiment, a male Sprague Dawley rat weighing 80–100 g was euthanized by CO2 inhalation. The pancreas was dissected and diced into 0.5 mm3 with a razor blade, mildly agitated for 10 min at 37°C in a siliconized glass tube with 5 ml of oxygenated buffer A (98 mM NaCl, 4.8 mM KCl, 2 mM CaCl2, 1.2 mM MgCl2, 0.1% bovine serum albumin, 0.01% soybean trypsin inhibitor, 25 mM Hepes, pH 7.4) containing 1000 units of collagenase. The suspension of acini was filtered through a 224 μm Spectra-Mesh (Spectrum Laboratory Products, Rancho Dominguez, CA) polyethylene filter to remove large clumps of acini and undissociated tissue. The acini were washed six times, 50 ml per wash, with ice-cold buffer A. Isolated rat pancreatic acini and acinar cells were plated on Cell-Tak-coated (Collaborative Biomedical Products, Bedford, MA) glass cover slips. Two to three hours after plating, cells were imaged with the AFM before and during stimulation of secretion. Isolated acinar cells and hemiacinar preparations were used in the study because fusion of secretory vesicles at the PM in these cells occurs at the apical region facing the acinar lumen.
Pancreatic plasma membrane preparation
Rat pancreatic PM fractions were isolated using a modification of a published method (Rosenzweig et al., 1983). Male Sprague Dawley rats weighing 70–100 g were euthanized by CO2 inhalation. Pancreata were removed and placed in ice-cold phosphate-buffered saline (PBS), pH 7.5. Adipose tissue was removed, and the pancreata were diced into 0.5 mm3 pieces using a razor blade in a few drops of homogenization buffer A (1.25 M sucrose, 0.01% trypsin inhibitor, 25 mM Hepes, pH 6.5). The diced tissue was homogenized in 15% (w/v) ice-cold homogenization buffer A using four strokes at maximal speed of a motor-driven pestle (using a Wheaton Overhead Stirrer). A total of 1.5 ml of the homogenate was layered over a 125 μl cushion of 2M sucrose, and 500 μl of 0.3 M sucrose was layered onto the homogenate in Beckman centrifuge tubes. After centrifugation at 145,000 × g for 90 min in a Sorvall AH-650 rotor, the material banding between the 1.2 and 0.3 M sucrose interface was collected and the protein concentration (Bradford, 1976) was determined. For each experiment, fresh PM was prepared and used the same day in all AFM experiments.
Atomic force microscopy
Pits and fusion pores at the PM in live pancreatic acinar secreting cells in PBS, pH 7.5, were imaged with the AFM (BioScope III, Digital Instruments, Santa Barbara, CA) using both contact and “tapping” modes. All images presented in this manuscript were obtained in the tapping mode in fluid, using silicon nitride tips with a spring constant of 0.06 N/m, and an imaging force of <200 pN. Images were obtained at line frequencies of 1 Hz, with 512 lines per image, and constant image gains. Topographical dimensions of pits and fusion pores at the cell PM were analyzed using the software NanoScope IIIa version 4.43r8 supplied by Digital Instruments.
ImmunoAFM on live cells
Immunogold localization in live pancreatic acinar cells was assessed after 5 min stimulation of secretion with 10 μM of the secretagogue mastoparan. After stimulation of secretion, the live pancreatic acinar cells in buffer were exposed to a 1:200 dilution of α-amylase-specific antibody (Biomeda, Foster City, CA) and 30 nm gold conjugated secondary antibody for 1 min and were washed in PBS before AFM imaging in PBS at room temperature. Pits and fusion pores within, at the apical end of live pancreatic acinar cells in PBS pH 7.5, were imaged by the AFM (BioScope III, Digital Instruments) using both contact and tapping mode. All images presented were obtained in the tapping mode in fluid, using silicon nitride tips as described previously.
ImmunoAFM on fixed cells
After stimulation of secretion with 10 μM mastoparan, the live pancreatic acinar cells were fixed for 30 min using ice-cold 2.5% paraformaldehyde (PFA) in PBS. Cells were then washed in PBS, followed by labeling with 1:200 dilution of α-amylase-specific antibody (Biomeda) and 10 nm gold conjugated secondary antibody for 15 min, fixed, washed in PBS, and imaged in PBS with AFM at room temperature.
Isolation of zymogen granules
Zymogen granules (ZGs) were isolated by using a modification of the method of Jena et al. (1991). Male Sprague Dawley rats weighing 80–100 g were euthanized by CO2 inhalation for each ZG preparation. The pancreata were dissected and diced into 0.5-mm3 pieces. The diced pancreata were suspended in 15% (w/v) ice-cold homogenization buffer (0.3 M sucrose, 25 mM Hepes, pH 6.5, 1 mM benzamidine, 0.01% soybean trypsin inhibitor) and homogenized with a Teflon glass homogenizer. The resultant homogenate was centrifuged for 5 min at 300 × g at 4°C to obtain a supernatant fraction. One volume of the supernatant fraction was mixed with 2 volumes of a Percoll–sucrose–Hepes buffer (0.3 M sucrose, 25 mM Hepes, pH 6.5, 86% Percoll, 0.01% soybean trypsin inhibitor) and centrifuged for 30 min at 16,400 × g at 4°C. Pure ZGs were obtained as a loose white pellet at the bottom of the centrifuge tube.
Transmission electron microscopy
Isolated rat pancreatic acini and ZGs were fixed in 2.5% buffered PFA for 30 min, and the pellets were embedded in Unicryl resin and were sectioned at 40–70 nm. Thin sections were transferred to coated specimen transmission electron microscopy (TEM) grids, dried in the presence of uranyl acetate and methyl cellulose, and examined in a transmission electron microscope.
Immunoprecipitation and Western blot analysis
Immunoblot analysis was performed on pancreatic PM and total homogenate fractions. Protein in the fractions was estimated by the Bradford method (Bradford, 1976). Pancreatic fractions were boiled in Laemmli reducing sample preparation buffer (Laemmli, 1970) for 5 min, cooled, and used for SDS-PAGE. PM proteins were resolved in a 12.5% SDS-PAGE and electrotransferred to 0.2 μm nitrocellulose sheets for immunoblot analysis with a SNAP-23 specific antibody. The nitrocellulose was incubated for 1 h at room temperature in blocking buffer (5% non-fat milk in PBS containing 0.1% Triton X-100 and 0.02% NaN3), and immunoblotted for 2 h at room temperature with the SNAP-23 antibody (ABR, Golden, CO). The primary antibodies were used at a dilution of 1:10,000 in blocking buffer. The immunoblotted nitrocellulose sheets were washed in PBS containing 0.1% Triton X-100 and 0.02% NaN3 and were incubated for 1 h at room temperature in horseradish peroxidase-conjugated secondary antibody at a dilution of 1:2,000 in blocking buffer. The immunoblots were then washed in the PBS buffer, processed for enhanced chemiluminescence, and exposed to X-OMAT-AR film. To isolate the fusion complex for immunoblot analysis, SNAP-23 specific antibody conjugated to protein A-sepharose was used. One gram of total pancreatic homogenate solubilized in Triton/Lubrol solubilization buffer (0.5% Lubrol; 1 mM benzamidine; 5 mM ATP; 5 mM EDTA; 0.5% Triton X-100, in PBS) supplemented with protease inhibitor mix (Sigma, St. Louis, MO) was used. SNAP-23 antibody conjugated to the protein A-sepharose was incubated with the solubilized homogenate for 1 h at room temperature followed by washing with wash buffer (500 mM NaCl, 10 mM Tris, 2 mM EDTA, pH = 7.5). The immunoprecipitated sample attached to the immunosepharose beads was incubated in Laemmli sample preparation buffer, before 12.5% SDS-PAGE, electrotransfer to nitrocellulose, and immunoblot analysis using specific antibodies to actin (Sigma), fodrin (Santa Cruz Biotechnology, Santa Cruz, CA), vimentin (Sigma), syntaxin 2, Ca2+-β3, and Ca2+-α1c (Alomone Labs, Jerusalem, Israel).
RESULTS AND DISCUSSION
Fusion pore revealed from AFM studies
As reported earlier (Cho et al., 2002c; Schneider et al., 1997), pancreatic acinar cells in physiological buffer reveal the presence of pits measuring 0.4–1 μm in diameter which contain “depressions” or fusion pores. Each fusion pore measured ∼125–185 nm in diameter and 19–25 nm in relative depth (Fig. 1 a). Localization of gold-labeled anti-amylase with the AFM confirmed that amylase is located at the fusion pore after stimulation of secretion (Fig. 1, b–d and Cho et al., 2002c). To determine the morphology of the fusion pore at the cytosolic side, a pancreatic PM preparation was used. Isolated PM in buffer, when placed on freshly cleaved mica, tightly adheres to the mica surface, allowing imaging by the AFM. The PM preparations contain scattered, circular disks measuring 0.5–1 μm in diameter, with inverted cup-shaped structures within (Fig. 2). The inverted cups range in height from 10–15 nm. On several occasions, ZGs of 0.4–0.1 μm in diameter are found associated with one or more of the inverted cups (Fig. 2, a–c). Because ZGs are the next largest structures after the nucleus in pancreatic acinar cells, this suggests the circular disks are pits, and the inverted cup-like structures are the fusion pores. To determine if the cup-shaped structures in isolated PM preparations are the fusion pores, immunoAFM studies were performed. Because our studies (Fig. 1, b–d; and Cho et al., 2002c; Schneider et al., 1997) demonstrated that ZGs dock and fuse at the fusion pore to release vesicular contents, it is reasonable to suggest that PM-associated t-SNAREs may be located at the base of the fusion pore, i.e., the tip of the inverted cup-shaped structure. The t-SNARE protein, SNAP-23, has been identified and implicated in secretion from pancreatic acinar cells (Gaisano et. al., 1997). A polyclonal monospecific SNAP-23 antibody, recognizing a single 23 kDa band in the Western blot analysis of a pancreatic homogenate (Fig. 2 d), was used in all immunoAFM studies. When the SNAP-23 specific antibody was added to the PM preparation during imaging with the AFM, the antibody specifically localized to the base of the cup-shaped structure which is the tip of the inverted cup (Fig. 2, e and f). No antibody labeling of the structure was detected when preimmune serum was applied (data not shown). These results demonstrate that the inverted cup-shaped structures in isolated PM preparations are the fusion pores observed from its cytosolic side. To further confirm our AFM studies, and to have a better understanding of the fusion pore, TEM was done.
FIGURE 2.

Morphology of the cytosolic side of the fusion pore revealed in AFM studies on isolated pancreatic plasma membrane preparations. (a) This AFM micrograph of isolated plasma membrane preparation reveals the cytosolic end of a pit with inverted cup-shaped structures, the fusion pores. Note the 600 nm in diameter ZG at the left hand corner of the pit; (b) Higher magnification of the same pit showing clearly the 4–5 fusion pores within; (c) The cytosolic end of a single fusion pore is depicted in this AFM micrograph; (d) Immunoblot analysis of 10 μg and 20 μg of pancreatic plasma membrane preparations, using SNAP-23 antibody, demonstrates a single 23 kDa immunoreactive band; (e and f) The cytosolic side of the plasma membrane demonstrates the presence of a pit with a number of fusion pores within, shown (e) before and (f) after addition of the SNAP-23 antibody. Note the increase in height of the fusion pore cone base revealed by section analysis (bottom panel), demonstrating localization of SNAP-23 antibody to the base of the fusion pore.
Transmission electron microscopy
Because fusion pores are relatively small structures (125–185 nm in diameter at its wide end and 10–40 nm at the base) and are present only at the apical PM of pancreatic acinar cells, it would be extremely rare to be able to take a cross section through the fusion pore along with any associated secretory vesicles. Even if rare, such structures were observed in electron micrographs of isolated cells or tissue preparations (Fig. 3). TEM studies confirm the fusion pore to have a cup-shaped structure, with similar dimensions as determined in all our AFM studies (Cho et al., 2002a,b,c,d; Schneider et al., 1997). Additionally, TEM results reveal that the fusion pore has a basket-like morphology, with three lateral and a number of vertically arranged ridges. A ring-like structure is also observed at the base of the fusion pore cup (Fig. 3).
FIGURE 3.
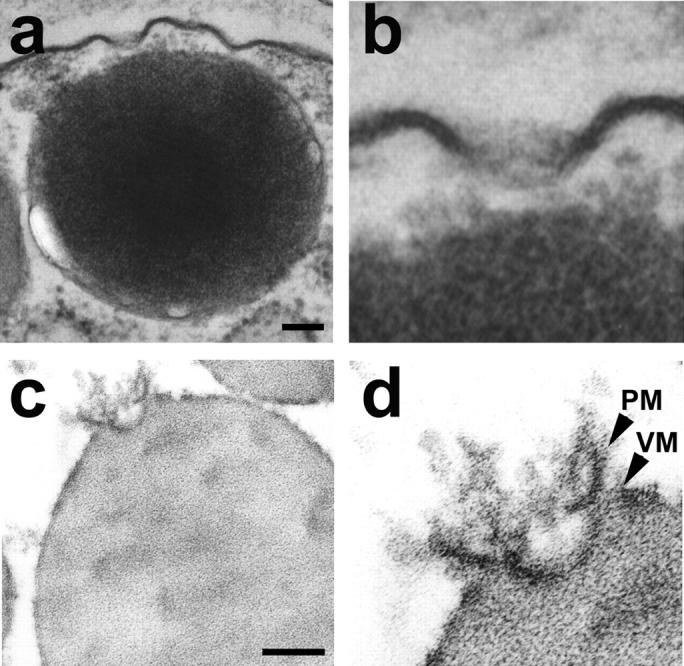
Transmission electron micrograph of the fusion pore showing vertical and lateral structures, giving it a basket-like appearance. (a) Two fusion pores at the apical plasma membrane of a pancreatic acinar cell, with a docked zymogen granule; (b) High resolution image of one of the fusion pores clearly shows three lateral and a number of vertical ridges, giving it a basket-like appearance; (c) An isolated zymogen granule associated with a fusion pore, reveals (d) clearly the lateral and vertical structures of the fusion pore complex. Scale = 100 nm.
Because these PM structures are stable, we hypothesized that, if membrane-bound secretory vesicles (ZGs) were to fuse at the base of the fusion pore, it should be possible to prepare isolated ZG-associated fusion pore complexes. To test this hypothesis, ZGs were isolated and the preparation was processed for TEM. Our TEM studies confirmed this hypothesis and reveal the isolation of the fusion pore associated with docked vesicles (Fig. 3). The PM and the ZG membrane are distinct and clearly visible in the isolated fusion pore-ZG complex. As observed in whole cells (Fig. 3, a and b), vertical structures originate from within the fusion pore complex (Fig. 3, c and d). These vertical structures appear attached to the fusion pore membrane (Figs. 3 and 4). The presence of such vertical ridges lining the fusion pore in NG108-15 nerve cells has been previously reported (Tojima, et al., 2000). In our EM micrograph the ridges or vertical rods appear supported by a central core rod. This arrangement may be similar to the nuclear pore having a plug at the center. In our AFM micrograph, however, the central rod is undetectable (Fig. 1 d), possibly due to its molecular size and softness. Thus, the combination of AFM and EM studies provides a better understanding of the fusion pore morphology. It needs to be mentioned that earlier studies report the association of t-SNAREs with secretory vesicles (Otto et al., 1997). This could be due to the co-isolation of the fusion pore (containing t-SNAREs) during secretory vesicle preparation.
FIGURE 4.
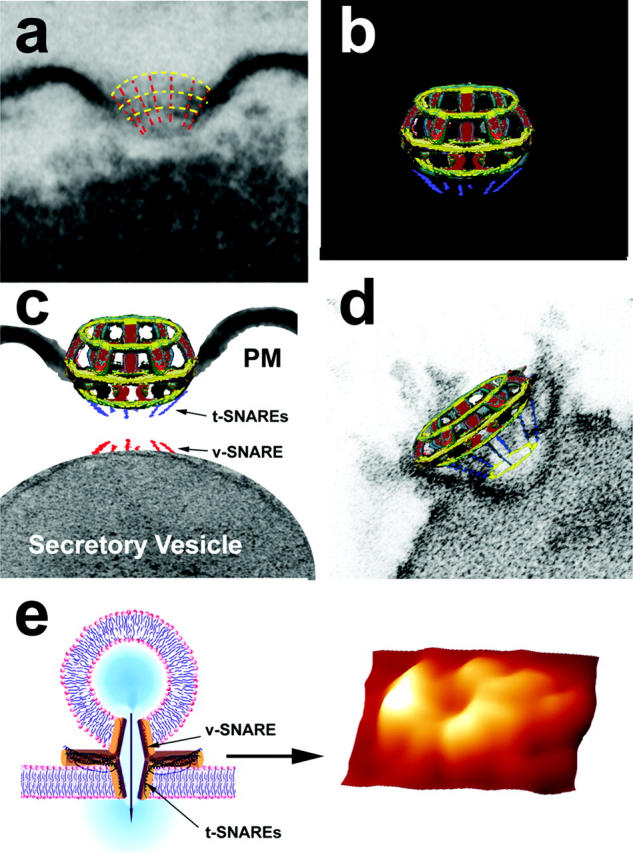
The fusion pore complex. (a) Electron micrograph of a fusion pore, with positions of the vertical and lateral structures depicted by red and yellow dashed lines for clarity; (b) A schematic model of the fusion pore showing (c) the presence of t-SNAREs at the base of the basket, where the v-SNARE associated ZG can dock; (d) Thus, the t- and v-SNAREs associated in opposing bilayers can interact in a circular array (yellow ring) to establish continuity between ZG contents and the fusion pore; (e) When v-SNARE-reconstituted artificial lipid vesicles are allowed to interact with a t-SNARE-reconstituted lipid support, the t-/v-SNAREs interact in a circular array as shown in the AFM micrograph on the right. The pore formed by t-/v-SNARE interaction is conducting, as demonstrated by electrophysiological measurements (Cho et al., 2002e).
Composition of the fusion pore complex
In a recent study, Cho et al. (2002e) used purified SNARE proteins and artificial lipid membranes to demonstrate that t- and v-SNAREs, located in opposing bilayers, interact in a circular array to form conducting pores (Fig. 4 e; Cho et al., 2002e). The 45–50 nm circular structure that is observed at the base of the fusion pore (Fig. 3 b) and the SNAP-23 immunoreactivity that is also localized at this site (Fig. 2 e) demonstrate the presence of t-SNAREs at the base of the fusion pore cup. To determine the association of other proteins with the fusion pore complex, immunoprecipitation studies on total pancreatic homogenates using SNAP-23 specific antibody were performed. The precipitated sample was resolved using 12.5% SDS-PAGE, transferred to a nitrocellulose membrane, and probed using various antibodies. In agreement with earlier findings in other tissues (Bennett, 1990; Cho et al., 2002c; Faigle et al., 2000; Goodson et al., 1997; Nakano et al., 2001; Ohyama et al., 2001; Rothman, 1994; Weber et al., 1998), our study demonstrates the association of SNAP-23 with syntaxin 2; with cytoskeletal proteins actin, α-fodrin, and vimentin; and with calcium channels β3 and α1c, together with the SNARE regulatory protein, NSF (Fig. 5). These studies demonstrate that the fusion pore is a cup-shaped lipoprotein basket at the cell PM where secretory vesicles dock and fuse to release vesicular contents. The base of the fusion pore complex is where t- and v-SNAREs interact in a circular array to form a pore, and hence we name the structure the “porosome”. Purification and further characterization of the porosome is required to determine its complete biochemical composition. Furthermore, immunoTEM combined with immunoAFM will help determine the specific arrangement and localization of the various porosome-associated proteins.
FIGURE 5.

SNAP-23 associated proteins in pancreatic acinar cells. Total pancreatic homogenate was immunoprecipitated using the SNAP-23 specific antibody. The precipitated material was resolved using 12.5% SDS-PAGE, electrotransferred to nitrocellulose membrane, and then probed using antibodies to a number of proteins. Association of SNAP-23 with syntaxin 2; with cytoskeletal proteins actin, α-fodrin, and vimentin; and with calcium channels β3 and α1c, together with the SNARE regulatory protein NSF, is demonstrated (arrowheads). Lanes showing more than one arrowhead suggest presence of isomers or possible proteolytic degradation of the specific protein.
Acknowledgments
This work was supported in part by research grants DK56212 and NS39918 to B.P.J. from the National Institutes of Health. S.-J.C. is a recipient of a National Institutes of Health postdoctoral fellowship award (DK60368).
References
- Bennett, V. 1990. Spectrin-based membrane skeleton: A multipotential adaptor between plasma membrane and cytoplasm. Physiol. Rev. 70:1029–1065. [DOI] [PubMed] [Google Scholar]
- Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254. [DOI] [PubMed] [Google Scholar]
- Cho, S.-J., K. Jeftinija, A. Glavaski, S. Jeftinija, B. P. Jena, and L. L. Anderson. 2002a. Structure and dynamics of the fusion pores in live GH-secreting cells revealed using atomic force microscopy. Endocrinology. 143:1144–1148. [DOI] [PubMed] [Google Scholar]
- Cho, S.-J., A. Wakade, G. D. Pappas, and B. P. Jena. 2002b. New structure involved in transient membrane fusion and exocytosis. New York Acad. Sci. 971:254–256. [DOI] [PubMed] [Google Scholar]
- Cho, S.-J., A. S. Quinn, M. H. Stromer, S. Dash, J. Cho, D. J. Taatjes, and B. P. Jena. 2002c. Structure and dynamics of the fusion pore in live cells. Cell Biol. Int. 26:35–42. [DOI] [PubMed] [Google Scholar]
- Cho, S.-J., J. Cho, and B. P. Jena. 2002d. The number of secretory vesicles remains unchanged following exocytosis. Cell Biol. Int. 26:29–33. [DOI] [PubMed] [Google Scholar]
- Cho, S.-J., M. Kelly, K. T. Rognlien, J. Cho, J. K. H. Hoerber, and B. J. Jena. 2002e. SNAREs in opposing bilayers interact in a circular array to form conducting pores. Biophys. J. 83:2522–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faigle, W., E. Colucci-Guyon, D. Louvard, S. Amigorena, and T. Galli. 2000. Vimentin filaments in fibroblasts are a reservoir for SNAP-23, a component of the membrane fusion machinery. Mol. Biol. Cell. 11:3485–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaisano, H. Y., L. Sheu, P. P. Wong, A. Klip, and W. S. Trimble. 1997. SNAP-23 is located in the basolateral plasma membrane of rat pancreatic acinar cells. FEBS Lett. 414:298–302. [DOI] [PubMed] [Google Scholar]
- Goodson, H. V., C. Valetti, and T. E. Kreis. 1997. Motors and membrane traffic. Curr. Opin. Cell Biol. 9:18–28. [DOI] [PubMed] [Google Scholar]
- Jena, B. P., P. J. Padfield, T. S. Ingebritsen, and J. D. Jamieson. 1991. Protein tyrosine phosphatase stimulates Ca(2+)-dependent amylase secretion from pancreatic acini. J. Biol. Chem. 266:17744–17746. [PubMed] [Google Scholar]
- Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 227:680–685. [DOI] [PubMed] [Google Scholar]
- Monck, J. R., A. F. Oberhauser, and J. M. Fernandez. 1995. The exocytotic fusion pore interface: a model of the site of neurotransmitter release. Mol. Membr. Biol. 12:151–156. [DOI] [PubMed] [Google Scholar]
- Nakano, M., S. Nogami, S. Sato, A. Terano, and H. Shirataki. 2001. Interaction of syntaxin with α-fodrin, a major component of the submembranous cytoskeleton. Biochem. Biophys. Res. Commun. 288:468–475. [DOI] [PubMed] [Google Scholar]
- Ohyama, A., Y. Komiya, and M. Igarashi. 2001. Globular tail of myosin-V is bound to vamp/synaptobrevin. Biochem. Biophys. Res. Commun. 280:988–991. [DOI] [PubMed] [Google Scholar]
- Otto, H., P. I. Hanson, and R. Jahn. 1997. Assembly and disassembly of a ternary complex of synaptobrevin, syntaxin, and SNAP-25 in the membrane of synaptic vesicles. Proc. Natl. Acad. Sci. USA. 94:6197–6201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prekeris, R., and D. M. Terrian. 1997. Brain myosin V is a synaptic vesicle-associated motor protein: Evidence for a Ca2+-dependent interaction with the synaptobrevin-synaptophysin complex. J. Cell Biol. 137:1589–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig, S. A., L. J. Miller, and J. D. Jamieson. 1983. Identification and localization of cholecystokinin-binding sites on rat pancreatic plasma membranes and acinar cells: a biochemical and autoradiographic study. J. Cell Biol. 96:1288–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman, J. E. 1994. Mechanism of intracellular protein transport. Nature. 372:55–63. [DOI] [PubMed] [Google Scholar]
- Schneider, S. W., K. C. Sritharan, J. P. Geibel, H. Oberleithner, and B. P. Jena. 1997. Surface dynamics in living acinar cells imaged by atomic force microscopy: identification of plasma membrane structures involved in exocytosis. Proc. Natl. Acad. Sci. USA. 94:316–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tojima, T., Y. Yamane, H. Takagi, T. Takeshita, T. Sugiyama, H. Haga, K. Kawabata, T. Ushiki, K. Abe, T. Yoshioka, and E. Ito. 2000. Three-dimensional characterization of interior structures of exocytotic apertures of nerve cells using atomic force microscopy. Neuroscience. 101:471–481. [DOI] [PubMed] [Google Scholar]
- Weber, T., B. V. Zemelman, J. A. McNew, B. Westermann, M. Gmachl, F. Parlati, T. H. Söllner, and J. E. Rothman. 1998. SNAREpins: minimal machinery for membrane fusion. Cell. 92:759–772. [DOI] [PubMed] [Google Scholar]