

Abstract
Targeted molecular dynamics simulations were used to study the conformational transition of influenza hemagglutinin (HA) from the native conformation to putative fusogenic or postfusion conformations populated at low pH. Three pathways for this conformational change were considered. Complete dissociation of the globular domains of HA was observed in one pathway, whereas smaller rearrangements were observed in the other two. The fusion peptides became exposed and moved toward the target membrane, although occasional movement toward the viral membrane was also observed. The effective energy profiles along the paths show multiple barriers. The final low-pH structures, which are consistent with available experimental data, are comparable in effective energy to native HA. As a control, the uncleaved precursor HA0 was also forced along the same pathway. In this case both the final energy and the energy barrier were much higher than in the cleaved protein. This study suggests that 1) as proposed, the native conformation is the global minimum energy conformation for the uncleaved precursor but a metastable state for cleaved HA; 2) the spring-loaded conformational change is energetically plausible in full-length HA; and 3) complete globular domain dissociation is not necessary for extension of the coiled coil and fusion peptide exposure, but the model with complete dissociation has lower energy.
INTRODUCTION
Protein-mediated membrane fusion is a common process in living organisms (White, 1992). One of the best studied examples of a membrane fusion machine is the influenza virus Hemagglutinin (HA; Wiley and Skehel, 1987; Skehel and Wiley, 2000; Eckert and Kim, 2001). HA is a glycoprotein anchored in the viral lipid membrane by a single transmembrane domain near its C-terminus. It consists of the receptor-binding (HA1) and the membrane-anchoring (HA2) polypeptide chains, which are linked by a disulfide bond. The first 20 amino acid residues of the N-terminus of HA2 are highly conserved within the influenza virus family and appear to be crucial for virus-cell fusion as shown by site-directed mutagenesis (Gething et al., 1986; Durell et al., 1997; Qiao et al., 1999). They are referred to as the fusion peptide. The virus first binds to a sialylated cell surface receptor, and then enters the cell by endocytosis. The low pH of the endosome induces a large conformational change (Skehel et al., 1982), which is thought to trigger fusion between the viral and the endosomal membranes. The optimal pH range for membrane fusion by HA is between 5 and 5.5 (Skehel et al., 1982). However, activation of the HA-mediated membrane fusion has also been reported at neutral pH either at high temperature or in the presence of a chemical denaturant (Carr et al., 1997), which suggests that low pH merely serves to destabilize the native conformation.
The x-ray crystal structure of the ectodomain of HA (bromelain digested HA, BHA; Wilson et al., 1981) shows that the native HA is a rodlike homotrimer extending 135 Å in length. The N-terminus of the HA1 and the C-terminus of the HA2 domain are near the viral membrane. The HA2 chains are the major components of the α-helical stem region which forms the core of the HA homotrimer, whereas most of the HA1 subunits form globular domains located on top of the stem region. The fusion peptide is located 100 Å from the distal top and 35 Å from the viral membrane end of the molecule and cannot reach the viral or target membrane unless a major conformational change occurs within the protein.
Although the structure of BHA at low pH is not available, an enzymatically digested form of HA has been crystallized and solved (TBHA2) (Bullough et al., 1994). Each monomer of the TBHA2 consists of residues 38–175 of HA2 and 1–27 of the HA1, linked by disulfide bonds. Comparison between the neutral (BHA) and low pH (TBHA2) structures of hemagglutinin revealed a dramatic conformational change. The most remarkable observation is a growth of the triple-stranded, α-helical coiled coil due to incorporation of the random coil region (residues 56–75) and a second α-helix (residues 38–55) into the coiled coil. This growth of the coiled coil stem was predicted based on the amino acid sequence before the structure was solved (Carr and Kim, 1993). This conformational change in TBHA2 seems to allow the N-terminus of HA2 to interact directly with the target membranes. In addition, there is a helix-to-loop transition (HA2:105–112) which allows the polypeptide chain to reverse direction near its C-terminus. A somewhat longer portion of HA at low pH has also been solved (Chen et al., 1999). Similar trimeric coiled coils have been observed in the structures of other viral membrane fusion proteins (Hughson, 1997). It is not clear whether these correspond to the fusogenic conformation or the final conformation after fusion. In the case of HIV there is evidence that six-helix bundle formation occurs together with membrane fusion, i.e., the six-helix bundle is the postfusion conformation (Melikyan et al., 2000).
The structure of a mutant, uncleaved precursor (HA0) was also determined by x-ray crystallography (Chen et al., 1998). Only 19 residues bracketing the cleavage site are located differently in HA0 relative to BHA. The remaining residues in HA0 are essentially superimposable to BHA. Whereas the cleaved HA undergoes a low-pH induced conformational change (Bullough et al., 1994), the uncleaved precursor HA0 is stable at low pH (Chen et al., 1998).
Many studies have aimed to understand the role of the fusion peptide in viral fusion (Lear and DeGrado, 1987; Han and Tamm, 2000; Bechor and Ben-Tal, 2001). It is thought that the fusion peptide inserts into the cell membrane and destabilizes the bilayer (Skehel et al., 1982). Point mutations in this peptide can block membrane fusion (Durell et al., 1997). It has been proposed that the fusion peptide is obliquely inserted into the target membrane as a flexible monomer in an α-helical conformation (Brasseur et al., 1997). EPR studies suggest that the fusion peptide is an α-helix tilted ∼25° from the horizontal plane of the membrane (Macosko et al., 1997). More recent studies also found oblique orientations (Bradshaw et al., 2000; Han and Tamm, 2000). Recently, Tamm and co-workers determined the HA fusion domain structure in micelles (Cohen and Melikyan, 2001; Han et al., 2001) and their results suggested a helix-break-helix structural motif for the fusion domain of influenza HA at pH 5.
Several models have been proposed for the mechanism of membrane fusion. One model proposes a localized conformational change, resulting in the exposure of the fusion peptide, while maintaining the intact hairpin structure of the HA2 chain (Stegmann et al., 1990). In this model the fusion peptide can insert into the target membrane by tilting of the HA trimer. Another model (Bentz et al., 1990) proposed that the exposed fusion peptides from a cluster of HA trimers induce the formation of an inverted micelle in the space between them. The spring-loaded model (Carr and Kim, 1993; Bullough et al., 1994), which currently seems to be the most widely accepted, assumes a global conformation change in HA similar to the one observed in the x-ray crystal structure of TBHA2 (Bullough et al., 1994). Many variations of these models exist in the literature (Bentz, 1992; Chernomordik et al., 1998; Kozlov and Chernomordik, 1998; Shangguan et al., 1998; Bentz, 2000; Bonnafous and Stegmann, 2000).
The goal of this work is to test the spring-loaded model by calculating possible pathways for the conformational change in HA by computer simulations. We use the two crystal structures (1HGF and 1HTM) as initial and final states and try to find a path between them with targeted molecular dynamics (TMD; Schlitter et al., 1993; Apostolakis et al., 1999). This technique has been used to study the reaction paths for conformational changes in ras p21 (Diaz et al., 1997; Ma and Karplus, 1997), protein folding and unfolding (Ferrara et al., 2000a,b), conformational changes in DNA polymerase (Yang et al., 2002), and the rotation of the γ-stalk in F1-ATPase (Bockmann and Grubmuller, 2002; Ma et al., 2002). Because of the incomplete crystal structure of the low-pH HA, TMD is applied only on the residues available in the low-pH crystal structure while allowing the rest of the molecule to freely adjust to the conformational changes in the core.
METHODS
Molecular dynamics simulations were carried out using the program CHARMM (Brooks et al., 1983). In a large system like hemagglutinin, it is computationally prohibitive to treat water explicitly. In this work we use the effective energy function EEF1, based on the CHARMM 19 polar hydrogen force field (Neria et al., 1996), which provides a reasonable approximation for proteins in aqueous solution (Lazaridis and Karplus, 1999). In this method, the solvation free energy of a protein is assumed to be the sum of group contributions, which are determined from experimental data for small model compounds. In addition, all ionic side chains are neutralized and a distance-dependent dielectric constant is used to approximate the charge-charge interactions in solution. The van der Waals and electrostatic potentials are truncated at 9 Å with a switching function starting at 7 Å. Inasmuch as the HA conformational change occurs also at neutral pH (by increased temperature or denaturants; Carr et al., 1997), we do not consider changes in pH and the concomitant variations in the protonation state of ionizable residues. In this article, by energy we will refer to the effective energy, i.e., the sum of the intramolecular energy and the solvation free energy.
The pathway from native to putative low-pH model HA conformation was obtained by the method of targeted molecular dynamics (Schlitter et al., 1993; Apostolakis et al., 1999). In TMD, simulations are performed with a restraint aimed to force the system to sample conformations with a desired RMSD from a target conformation. This is accomplished by adding a harmonic potential energy term of the form
 |
where K is a force constant, RMSDf is the root-mean-square deviation from the target structure, and ρ is the desired RMSD value. In this work we use the implementation of TMD in CHARMM by Caflisch and co-workers (Apostolakis et al., 1999). The initial value of ρ is usually taken as the RMSD between the initial and final structures. During the simulation ρ is gradually decreased to zero, thus forcing the system from the initial to the final structure. To avoid rigid body translational and rotational motion, the Eckart conditions are applied to the system in the form of holonomic constraints (Apostolakis et al., 1999).
The starting point of the TMD simulations was the crystal structure of native Hemagglutinin (pdb entry 1HGF). Simulations were performed at a mean temperature of 300 K. Because the conformational change at hand is very large, it is not possible to force the whole trimer to its final conformation in one step. Instead, we carried out the TMD simulations gradually. In both the native HA and TBHA2 the residues 76–105 of HA2 have the same structure. Thus initially, only this region was included in the RMSD restraint for 100 ps. At the end of the simulation, the RMSD between the initial and final targeted peptide chain was less than 1.0 Å. In subsequent stages, the portion of TBHA2 included in the TMD restraint was gradually increased. This can be done in different ways, resulting to alternative paths.
In the present study, three pathways have been constructed. 1), For the first path, the low-pH structure was built simultaneously from both ends of the identical structure (HA2:76-105). Thus in each subsequent TMD step, four more amino acids from both ends of the previous segment were included in the RMSD restraint. It took 15 TMD steps to build the low pH model of HA from the neutral pH structure of BHA in this way. 2), An intermediate with the coiled coil extending toward the target membrane but without the reversal of direction at the C-terminal end of HA2 has been proposed (Hernandez et al., 1996; Hughson, 1997; Eckert and Kim, 2001). To incorporate such an intermediate in the reaction pathway, Path 2 was constructed in two stages. In the first stage, the coiled coil was extended in the N-terminal direction (10 steps) and in the second stage the reversal at the C-terminal end of HA2 was effected (15 steps). 3), Experimental studies suggested a dissociation of the globular domains of HA at low pH (Godley et al., 1992; Kemble et al., 1992). Therefore, in Path 3 the centers of mass of the globular domains were first moved to a distance of 50 Å, followed by 100-ps dynamics. The resulting structure was then subjected to TMD as in Path 1.
As a control, the same TMD simulations were performed on the uncleaved precursor HA0 (pdb entry 1HA0). Based on crystallographic symmetry, a trimer of HA0 was built using the program TINKER (Ponder, 2001). The simulation protocol used in Path 1 was used for the HA0 → 1HTM transformation. In all cases, each step of the TMD simulations consisted of 100 ps and the RMSDs between the final structures from the target was found to be less than 1.0 Å. The final structures obtained at the end of the complete TMD simulations are our model low-pH hemagglutinin structures. The models obtained from Path 1, Path 2, and Path 3 are referred to as HAlowpH1, HAlowpH2, and HAlowpH3, respectively, and the model obtained for HA0 is referred to as HA0lowpH.
The TMD restraints introduce substantial strain into the protein structures. As a result, the TMD structures may have artificially high energies. To obtain more realistic energetic estimates, all structures along the TMD paths were subjected to a 200-ps unconstrained dynamics simulation, checking that they remain close to their starting conformations. The resulting dynamics structures were once again minimized for 300 Adopted Basis Newton-Raphson (ABNR) steps. We refer to these structures as the relaxed structures and to the pathways composed by these structures as the relaxed pathways.
One major advantage of using implicit solvation is that the relative probability of different conformations is given by the Boltzmann factor of the difference in effective energy between the two conformations (Lazaridis and Karplus, 1999). In reality, protein conformational states consist not of one but of an ensemble of conformations. The probability of each state depends on the average effective energy, which is readily obtained using implicit solvation, and the conformational entropy of the state, i.e., the number of conformations that belong to that state (Lazaridis and Karplus, 2001). In this paper we evaluate effective energies and only qualitatively discuss possible differences in conformational entropy. We emphasize that only differences in energy between different conformations are meaningful, not the absolute values of the energy.
We also report the effective energy of the final structure after 300 energy minimization steps using the ABNR method. Although not rigorous, this is a fast and convenient way to assess the energetics of a conformational state. From our experience, minimized energies and average energies usually give the same trends. The reason for choosing 300 steps is that the largest decrease in energy in ABNR occurs in the first 200 steps. More extensive minimizations change the absolute values of the energy but not so much the relative energy of different conformations. For example, the difference between 1HGF and relHAlowpH1 in Table 3 using 300 steps is 144 kcal/mol. Using 500 steps this difference is 166 kcal/mol and using 1000 steps it is 187 kcal/mol. The difference between the average effective energies is 183 kcal/mol. Here the 1000-step result agrees best with the average energies but this is not always the case.
TABLE 3.
Energetics of the initial (1HGF) and final low-pH models of HA
| Energy (kcal/mol) | 1HGF | relHAlowpH1 | relHAlowpH2 | relHAlowpH3 |
|---|---|---|---|---|
| Total | −48634 (−39548) | −48490 (−39365) | −48697 (−39485) | −48571 (−39460) |
| van der Waals | −11707 (−10721) | −11252 (−10296) | −11360 (−10414) | −11608 (−10688) |
| Electrostatic | −27588 (−25654) | −27768 (−25687) | −27933 (−25791) | −27916 (−25851) |
| Solvation | −13143 (−13499) | −13285 (−13699) | −13188 (−13593) | −13006 (−13367) |
| Covalent | 3804 (10326) | 3815 (10316) | 3783 (10313) | 3959 (10445) |
The values in parentheses are the average energies over the last 5 ps of dynamics.
Average effective energies over an MD simulation are sensitive to the temperature. When the average temperatures in the simulations of two conformations are slightly different, a systematic bias is introduced in the results. The values reported in Tables 1, 3, and 4 have been corrected in the following way. Additional simulations were done at 295 K and 305 K and the results (〈W〉 versus 〈T〉) were fitted to a straight line. This line was then used to obtain the effective energy that corresponds to 〈T〉 = 300 K.
TABLE 1.
Comparison of the energies of 1HTM and 1HGF1HTM
| Energy (kcal/mol) | 1HTM | 1HGF1HTM | Difference |
|---|---|---|---|
| Total | |||
| EEF1 | −13469 (−11109) | −13291 (−10949) | −178 (−160) |
| GB | −28523 (−25207) | −28367 (−25070) | −156 (−137) |
| van der Waals | |||
| EEF1 | −3044 (−2757) | −2957 (−2697) | −87 (−60) |
| GB | −3450 (−3107) | −3258 (−2947) | −192 (−160) |
| Electrostatic | |||
| EEF1 | −7264 (−6737) | −7245 (−6712) | −19 (−25) |
| GB | −19807 (−18625) | −20111 (−19298) | 304 (673) |
| Solvation | |||
| EEF1 | −3942 (−4051) | −3872 (−3994) | −70 (−57) |
| GB | −5949 (−6816) | −5713 (−6186) | −236 (−630) |
| Covalent | |||
| EEF1 | 781 (2436) | 783 (2455) | −2 (−19) |
| GB | 683 (3341) | 715 (3360) | −32 (−19) |
The values in parentheses are average energies over the last 5 ps of dynamics.
TABLE 4.
Energetics of the initial (HA0) and final low-pH model of HA0
| Energy (kcal/mol) | HA0 | tmdHA0lowpH | relHA0lowpH |
|---|---|---|---|
| Total | −47605 (−38551) | −45270 (−32057) | −47332 (−38011) |
| van der Waals | −11416 (−10449) | −11016 (−10118) | −10960 (−10192) |
| Electrostatic | −27258 (−25235) | −26388 (−24721) | −27198 (−25194) |
| Solvation | −12742 (−13114) | −12625 (−12810) | −13016 (−13256) |
| Covalent | 3811 (10247) | 4759 (15592) | 3841 (10631) |
The values in parentheses are the average energies over the last 5 ps of dynamics.
RESULTS
Energetics of 1HTM relative to 1HGF
It has been proposed that the proteolyzed native form of HA is metastable. As a first step toward characterizing the energetics of the conformational change, MD simulations of 1HTM and the corresponding fragment from 1HGF were performed. 1HTM consists of residues 12–16:A, 40–153:B, 11–16:C, 40–162:D, 10–17:E, and 40–162:F. By deleting from 1HGF the residues that are not present in 1HTM we created a model named 1HGF1HTM. The two systems were initially energy-minimized for 300 ABNR steps, followed by 100 ps dynamics. The RMSDs of the heavy atoms in the final 1HTM and 1HGF1HTM structures from their initial minimized structures were 2.6 Å and 5.6 Å, respectively. The large RMSD of 1HGF1HTM is apparently due to the absence of the rest of the protein, which stabilizes its conformation. The 100-ps dynamics structures were again minimized for 300 ABNR steps and the energies of the final structures, as well as average energies over the last 5 ps of the 100 ps dynamics, were calculated. Simulations were also done using the Generalized Born (GB) model implemented in CHARMM (Dominy and Brooks, 1999). In this case, the heavy atoms RMSDs of 1HTM and 1HGF1HTM from their initial minimized structures were 2.6 Å and 6.9 Å, respectively. The results are shown in Table 1. Both the EEF1 and GB models show that the total energy of 1HTM (minimized or dynamic average) is lower than that of 1HGF1HTM, i.e., the low pH conformation of the coiled coil part of HA by itself is energetically more stable than the native conformation. This is probably due to the extended coiled coil structure in 1HTM. This result is consistent with the experimental observation that in the absence of HA1, HA2 folds into the low pH conformation (Carr and Kim, 1993; Chen et al., 1995). The energy components are also shown in Table 1. With EEF1, all energy terms are lower in 1HTM than in 1HGF1HTM. With GB all but the electrostatic energy term are lower for 1HTM.
Reaction pathways and final low pH models
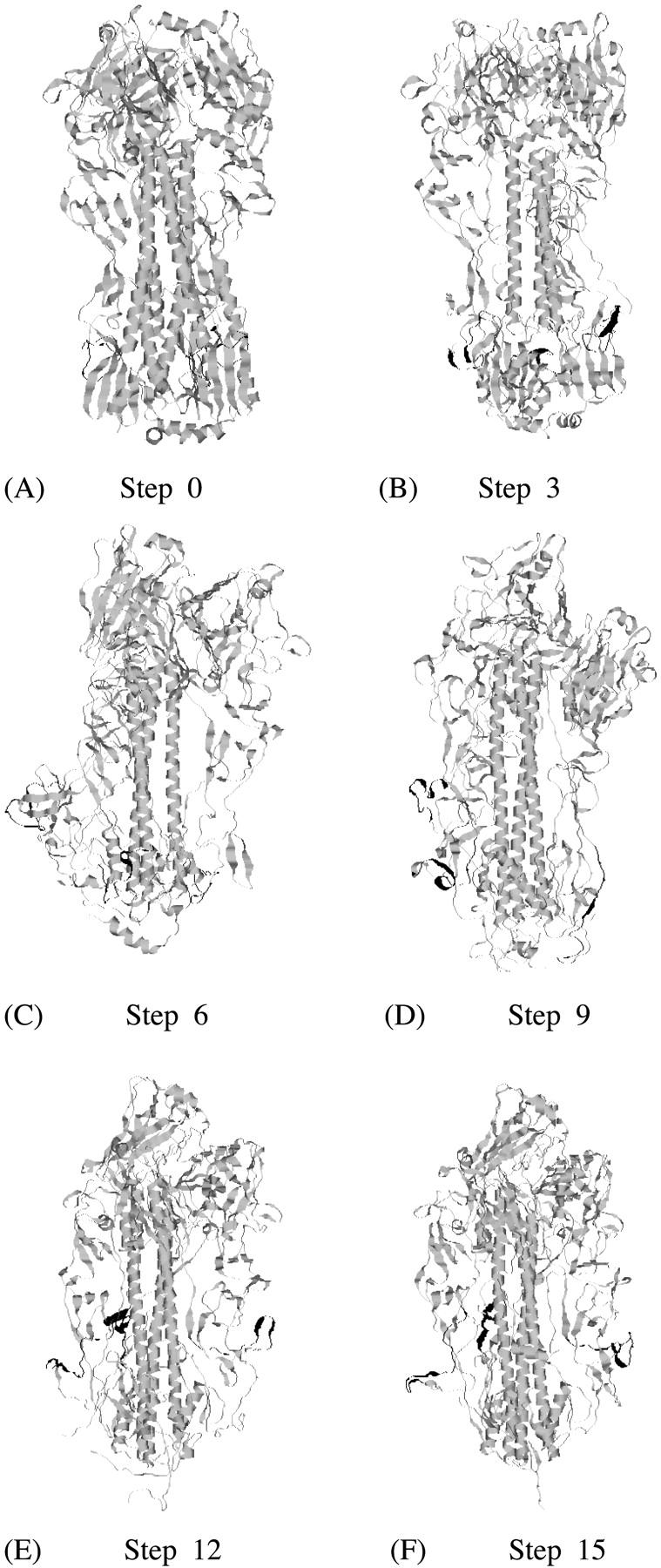
In the first pathway, the low-pH model was sequentially built simultaneously from both ends of the identical segments (HA2:76–105). After each TMD step, the RMSDs of the targeted peptide chains from the corresponding 1HTM fragment were less than 0.5 Å. The structure obtained at the end of all 15 steps of TMD is referred to as tmdHAlowpH1. The RMSD between 1HTM and the corresponding residues in tmdHAlowpH1 was 0.3 Å. Thus, the TMD method succeeded in converting 1HGF into a structure that contains the 1HTM core. Fig. 1 shows structures of HA along Path 1, highlighting the position of the fusion peptides (in black). In the native state of HA (Fig. 1 A), the fusion peptide is located ∼35 Å away from the viral membrane (bottom of the figure) buried in the trimeric coiled coil. At step 3 (Fig. 1 B) the location of the fusion peptide did not change much. The most significant change in structure occurs around residue HA2:106 where the coiled coil is forced to break and reverse direction. In the sixth step (Fig. 1 C), where HA2:56–125 residues were targeted, the loop between the inner and outer helices in the lower part of the molecule is formed. At this stage, the fusion peptides are displaced from their original position, with one fusion peptide moving toward the viral membrane. At the end of the ninth step (Fig. 1 D), this fusion peptide remained almost in the same region, another fusion peptide became exposed and the third fusion peptide moved upwards. In the 12th step (Fig. 1 E), the fusion peptide which was near the lower region of the HA moved upward and became exposed completely. The other two fusion peptides also moved upwards and into the interface between the globular domains and the coiled coil. This arrangement is preserved in the final structure (Fig. 1 F). Complete dissociation of the globular domains is not observed in this path.
FIGURE 1.

Ribbon representation of the structure of HA along TMD Path 1 for the 1HGF → 1HTM transformation: (A) at step 0, (B) at step 3, (C) at step 6, (D) at step 9, (E) at step 12, and (F) at step 15. The segments in black are fusion peptides.
The second pathway was generated in two stages. In the first stage, the coiled coil was extended toward the N-terminus of HA2 and in the second stage breaking of the long helix in the lower part of HA2 chain was effected. This pathway produced intermediate structures which are very different from those generated by Path 1. The structural changes of HA along this path are shown in Fig. 2. At the fourth step (Fig. 2 A) the globular domains moved only a few Å away from their original positions. However, by the end of the fifth step (Fig. 2 B), the globular domains became widely separated. At the end of the first stage, i.e., in the 10th TMD step (Fig. 2 C), the globular domains remained widely separated and the fusion peptides became stretched. Toward the end of the first stage tilting of the trimeric coiled coil with respect to the lower part of the coiled coil (HA2:105–125 in 1HGF) was observed. In the second stage (Fig. 2 D, 15th step), the fusion peptides started moving upward and one of the globular domains moved toward the viral membrane. All the fusion peptides were exposed at the 20th TMD step (Fig. 2 E). At the end of the TMD simulation (Fig. 2 F), one of the fusion peptides was at the tip of the coiled coil and the other two fusion peptides were almost completely exposed so that they could easily attach to a target membrane. The complete dissociation of the globular domains in the first stage of this path is probably due to the fact that the lower part remains intact and pulls the globular domains down and out as the coiled coil penetrates through their interface.
FIGURE 2.

Ribbon representation of the structure of HA along TMD Path 2 for the 1HGF → 1HTM transformation: (A) at step 4, (B) at step 5, (C) at step 10, (D) at step 15, (E) at step 20, and (F) at step 25. The segments in black are fusion peptides.
In Path 3, the centers of mass of the globular domains were first separated by 50 Å and continued to maintain this separation along the pathway. This conformation was ∼260 kcal/mol higher than native HA. After this initial displacement, the restraints proceeded as in Path 1. Conformational changes of HA during this pathway are presented in Fig. 3. Fig. 3 A is the initial structure of HA in Path 3, where the globular domains are separated by 50 Å. In the third step (Fig. 3 B), the fusion peptides have not moved much from their original position but the coiled coil has moved upward into the interface of the globular domains. In the sixth step (Fig. 3 C) the fusion peptides moved out of their original positions. In the ninth step (Fig. 3 D), the fusion peptides were still in the lower half of the HA. Fig. 3, E and F show one of the fusion peptides located at the upper region of HA. The other two fusion peptides became fully exposed; one of them moved toward the lower region of HA and the second remained in the lower half of HA but away from the viral membrane. Although the globular domains were initially separated, passage of the fusion peptide through their interface was not observed. However, unlike in the first pathway, full penetration of the coiled coil in the interface of globular domains was observed.
FIGURE 3.

Ribbon representation of the structure of HA along TMD Path 3 for the 1HGF → 1HTM transformation: (A) at step 0, (B) at step 3, (C) at step 6, (D) at step 9, (E) at step 12, and (F) at step 15. The segments in black are fusion peptides.
Because HA0 is stable at low pH (Chen et al., 1998) it can be used as a control experiment. Thus, the 1HTM core structure was built into the precursor HA0 in 15 steps of TMD as in the Path 1. The model obtained from HA0 is called tmdHA0lowpH. Structural changes of HA0 along this pathway are shown in Fig. 4. At step 3, even though the coiled coil breaks and reverses direction (Fig. 4 B), the fusion peptides remained close to their original positions. Some displacement of the fusion peptides was observed at the end of the sixth step (Fig. 4 C) and the ninth step (Fig. 4 D), with one fusion peptide moving toward the viral membrane. At step 12, (Fig. 4 E) the fusion peptide near the viral membrane returned near its original position. In the final step (Fig. 4 F), all the fusion peptides were near their original positions. In this pathway, a small separation of the globular domains was observed. A slight slanting of the globular domains was also seen, with one of the globular domains placed right on top of the coiled coil.
FIGURE 4.
Ribbon representation of the structure of HA0 along TMD path for the HA0 → 1HTM transformation: (A) at step 0, (B) at step 3, (C) at step 6, (D) at step 9, (E) at step 12, and (F) at step 15. The segments in black are fusion peptides.
Our final low pH models result from a 200-ps unconstrained simulation of the final TMD structures and they are called relHAlowpH1, relHAlowpH2, relHAlowpH3, and relHA0lowpH for Path 1, Path 2, Path 3, and the HA0 → 1HTM transformation, respectively. The RMSDs of the 1HTM core from the crystal structure in all the final TMD models were less than 1.0 Å. The RMSDs of the 1HTM core in the relaxed models were 2.03 Å, 3.07 Å, 4.0 Å, and 11.3 Å for relHAlowpH1, relHAlowpH2, relHAlowpH3, and relHA0lowpH structures, respectively. The RMSDs of the targeted residues of 1HTM core in the relaxed intermediate structures were also determined. They were less than 3 Å for all the intermediate structures of relaxed Path 1, Path 2, and Path 3, but between 8 Å and 10 Å for many of the intermediate structures of relaxed pathway for HA0 → 1HTM transformation. This is due to the large amount of strain in the HA0 intermediate structures which is relieved only by large deformations. During the conformational change the globular HA1 domains move more or less as rigid bodies (see below).
In the native state the fusion peptides are located ∼100 Å from the distal tip and ∼35 Å from the viral membrane. In the first low pH model, two of the fusion peptides moved toward the distal tip of the molecule (closer to the target membrane), and the third fusion peptide became exposed but remained close to its original position. The RMSD values of the globular domains (Table 2) indicate that two of them in relHAlowpH1 retained their overall structure but the third showed a larger deformation with an RMSD of 5.9 Å.
TABLE 2.
RMSDs of the HA1 globular domains in native HA from the final low-pH models of HA and HA0
| Globular domain | RMSD, Å relHAlowpH1 | RMSD, Å relHAlowpH2 | RMSD, Å relHAlowpH3 | RMSD, Å relHA0lowpH |
|---|---|---|---|---|
| HA1:60–260.A | 5.9 | 3.0 | 5.0 | 2.1 |
| HA1:60–260.C | 2.0 | 4.9 | 2.2 | 4.3 |
| HA1:60–260.E | 2.8 | 3.8 | 2.3 | 2.7 |
In the second low pH model the fusion peptides experienced large movements. In this model, one of the fusion peptides moved toward the distal tip of the fusogenic HA. The other two fusion peptides also moved in the upward direction and were exposed, becoming available for anchoring to the target membrane. A large separation of the HA1 globular domains was seen, with one of them reaching toward the viral membrane. Although the HA1 domains moved a long distance from their original positions, they largely retained their native structures (RMSDs were 3.0 Å, 3.8 Å, and 4.9 Å).
Because the globular domains were initially separated, the trimeric coiled coil in the third low pH model could pass through the interface of the globular domains. All the fusion peptides in this model were exposed. One of them moved toward the target membrane, going around one of the globular domains. Another fusion peptide moved toward the viral membrane and the third fusion peptide remained around the middle of the molecule but outside the long trimeric coiled coil. In this model two of the globular domains also retained their original structures and the third domain exhibited a larger distortion with an RMSD of 5.0 Å (Table 2).
The low pH model of HA0 was essentially denatured. The 1HTM structure built into the HA0 was destroyed during the relaxation process. The denaturation of the final low-pH model of HA0 is probably due to the presence of a large amount of strain developed during the TMD simulations. Despite this, the globular domains remained close to native (Table 2). The globular domains consist of several β-sheets and they are located in an isolated top region of HA. A drastic conformational change of this domain requires a lot of energy, which may be the reason why these domains retained their tertiary structures in our low pH models. The fusion peptides remained close to their original positions.
Fig. 5 shows the top view of the globular domains of the native, relHAlowpH1, and relHAlowpH3 states. In the native state (Fig. 5 A), the globular domains form a triangular interface with their centers of mass separated by ∼33 Å. In relHAlowpH1 most of the secondary structure is still present but one can see a slight opening up of the globular domains; one side of the triangle is ∼53 Å in length whereas the other two sides are ∼41 Å and 36 Å, respectively. Fig. 5 C shows that the secondary structures of two globular domains in relHAlowpH3 are well retained whereas some distortion (see Table 2) has occurred in the third globular domain.
FIGURE 5.

Top view of the ribbon diagram of the globular domains: (A) native HA, 1HGF; (B) low-pH HA of Path 1; and (C) low-pH HA of Path 3.
Energetics of the low-pH models and energy barriers
Fig. 6 shows the behavior of the minimized energy along the TMD pathways. Large increases in energy are observed as we move away from the native state with the final TMD models being higher in energy than the native HA by hundreds of kcal/mol. The barrier states of Path 1 and Path 3 are only slightly higher than the final states. These energies are artificially high, probably due to the strain introduced into the protein molecules by the TMD restraints.
FIGURE 6.

Energy profiles along the TMD and relaxed pathways of (A) Path 1, 1HGF → 1HTM transformation; (B) Path 2, 1HGF → 1HTM transformation; (C) Path 3, 1HGF → 1HTM transformation; and (D) HA0 → 1HTM transformation. Note the different energy scale in D.
Unrestrained MD simulations were used to relax the TMD structures. The energy profiles along these relaxed pathways have much lower energy barriers, of the order of 170–250 kcal/mol (dashed lines in Fig. 6). Since unrestrained MD simulations produce locally stable states, these barriers correspond to high energy basins and not transition states. Thus the energy barriers estimated this way are lower bounds for the true barriers. On the other hand, these paths are not optimal, minimum-energy paths and therefore may have barriers higher than those of the optimal paths. Also, these values may be too high because they do not include the usually favorable activation entropy contribution.
In Path 1, the highest barrier is found at the ninth TMD step and is 214 kcal/mol higher than the initial state. Along Path 2, two barriers are seen, one at the 10th step (242 kcal/mol) and another at the 20th step (252 kcal/mol). The low energy state at the 16th step, which is only 51 kcal/mol higher than the initial state, could be thought of as an intermediate along this path. The highest barrier of Path 3 is at the 12th step (∼170 kcal/mol). In all cases, analysis of the energy components shows that the van der Waals energy makes the largest contribution to the barriers.
Fig. 6 D shows the energy profile along the pathway of the HA0 → 1HTM transformation. In this case the TMD pathway has an energy barrier much higher than those of the HA pathways. Similarly, the final state is 2061 kcal/mol higher in energy than the initial state, almost three times higher than energy difference between the final and initial states of all the three pathways of the HA → 1HTM transformation. Unrestrained simulations of the structures along the TMD path led to large deviations from the target structure. The total energy of the relaxed low pH HA0 is ∼272 kcal/higher than the initial HA0.
The minimized energies as well as the average energies over the last 5 ps of dynamics of the initial and final low-pH models of HA are given in Table 3. Both the minimized and average energies of the low pH models are only slightly higher in energy than the native state (in fact, the minimized energy of model 2 is lower than that of the native state). Given the limited simulation time and extent of conformational search used in the simulations, the results indicate that the low pH models are very competitive in energy compared to the native state. Table 4 gives the energetics of the initial and final states of the HA0→1HTM transformation. In this case, both the minimized and dynamic average energies of the low-pH model of HA0 are significantly higher than those of the initial state. This supports the hypothesis that 1HGF is the global minimum energy conformation for the uncleaved precursor but a metastable state for cleaved HA.
Comparison with experimental data
In this section we examine the consistency of our low-pH structures with available experimental data, namely antigenicity changes, fusion activity of crosslinked mutants, electron microscopy, changes in proteolytic susceptibility, and other spectroscopic studies like circular dichroism and fluorescence.
Antigenicity changes at low pH
HA has four major antigenic sites labeled A, B, C, and D (Wiley et al., 1981), all located in the HA1 chain. Site A is a loop from amino acids 140 to 146, which protrudes 8 Å from the local molecular surface. Site B comprises the external residues 187–196 of an α-helix and adjacent residues along the upper edge of a pocket. Site C is a bulge in the tertiary structure at the disulfide bond between Cys 52 and Cys 277 which is 60 Å from the distal tip of the molecule. Site D is located in the interface of the globular domain ranging from amino acids 201 to 215. Experiments have shown that the antigenic sites A and C are not irreversibly affected antigenically (Daniels et al., 1983), whereas the antigenicity of sites B and D is lost (100- to 1000-fold) when hemagglutinin is subjected to low-pH treatment (Daniels et al., 1983; Webster et al., 1983). However, another study observed, with few exceptions, only a slight (5–10 fold) reduction in the reactivity of antigenic sites B and D with pH-5 treated virus (Yewdell et al., 1983).
For a given antigenic site to be active at low pH, the site must be available to the antibody in the same conformation as in the native state. Based on this assumption, the RMSDs of the antigenic sites in all three model low-pH HAs from their conformation in native HA were calculated (Table 5). Antigenic sites A and C from the three polypeptide chains are quite far from each other and probably bind to their antibodies individually. Sites B and D are located at the top and interface of the globular domains and all three sites of B or D can be accessed simultaneously by antibodies. Since sites A and C bind as monomers to their antibodies, their RMSDs are calculated from each chain of HA1 separately. For sites B and D, the RMSDs are calculated both individually from each chain and also from all the chains together.
TABLE 5.
RMSD of the antigenic sites A, B, C, and D in the final low-pH HA models from the native HA
| Antigenic site | RMSD, Å (Path 1) | RMSD, Å (Path 2) | RMSD, Å (Path 3) |
|---|---|---|---|
| A | HA1:A, 0.92 | HA1:A, 1.35 | HA1:A, 1.41 |
| HA1:C, 0.65 | HA1:C, 1.09 | HA1:C, 0.99 | |
| HA1:E, 0.93 | HA1:E, 0.59 | HA1:E, 0.51 | |
| B | HA1:A, 1.50 | HA1:A, 2.00 | HA1:A, 1.32 |
| HA1:C, 0.82 | HA1:C, 1.45 | HA1:C, 1.43 | |
| HA1:E, 2.50 | HA1:E, 1.57 | HA1:E, 1.07 | |
| HA1:A, C, and E, 9.6 | HA1:A, C, and E, 50.4 | HA1:A, C, and E, 17.9 | |
| C | HA1:A, 1.50 | HA1:A, 0.35 | HA1:A, 0.84 |
| HA1:C, 0.26 | HA1:C, 0.25 | HA1:C, 0.28 | |
| HA1:E, 0.28 | HA1:E, 0.269 | HA1:E, 1.33 | |
| D | HA1:A, 1.58 | HA1:A, 1.09 | HA1:A, 0.89 |
| HA1:C, 1.11 | HA1:C, 7.54 | HA1:C, 0.83 | |
| HA1:E, 1.39 | HA1:E, 3.35 | HA1:E, 1.43 | |
| HA1:A, C, and E, 7.3 | HA1:A, C, and E, 47.2 | HA1:A, C, and E, 13.7 |
In all three pathways, RMSDs of the antigenic sites A and C in each HA1 chain are less than 1.5 Å, which indicates that the conformations of these antigenic sites are well retained in their low pH models and can readily bind the antibodies. The individual RMSDs of antigenic sites B and D from each chain are less than 2.5 Å, except for the two antigenic D sites in the second low pH model which are >3.5 Å. However, when the RMSD was calculated with all the chains together, the antigenic sites B and D in all the low-pH models showed large deviations (>7.3 Å) from the native state. These large RMSDs indicate the degradation of these antigenic sites, which can abolish antigenicity. These experimental data are consistent with all three low-pH HA models. If sites B and D are involved in monomeric binding, then the RMSD values indicate a slight change in the antigenicity as observed by Yewdell et al. (1983). This result indicates that perhaps sites B and D are involved in trimeric or monomeric binding depending on the antibody.
White and Wilson (1987) compared the reactivity of antipeptide antibodies to neutral and low-pH treated BHA. At pH 5.0 and pH 4.6 they saw an increase in antibody reactivity for the antigenic sites A, B, C, and D, due to exposure of the individual peptide segments in conformations suitable for antibody binding. We have calculated the solvent-accessible surface area of these sites in the native and the low-pH HA using the MOLMOL program (Koradi et al., 1996). As the antigenic sites A and C are already in the peripheral region, the surface-accessible area did not change much for these sites. However, the surface-accessible area of the antigenic sites B and D increased in the low-pH model of HA. Average solvent-accessible surface area (SASA) of antigenic site B in the native state is ∼18%. In the low pH models it increased to 25%, 26%, and 23% for models 1, 2, and 3, respectively. Similarly, average SASA of antigenic site D in the native state is ∼23%. This value increased to 31%, 32%, and 29% for low pH models 1, 2, and 3, respectively. This increase in SASA is due to the partial or complete opening up of the globular domains in all the low-pH models. Increase in the surface accessible area of sites B and D is consistent with the experimentally observed increase in the reactivity of antipeptide antibody binding at low pH (White and Wilson, 1987).
Membrane fusion activity of crosslinked mutants
Introduction of intersubunit disulfide bonds, by mutating HA1:212 and HA1:216 to cysteines, in the membrane-distal region of HA inhibited the low-pH induced conformational changes and prevented the HA-mediated membrane fusion presumably by restricting the dissociation of the globular domains (Godley et al., 1992; Kemble et al., 1992). Conditions that reduced the novel disulfide bonds restored the membrane fusion activity. This experiment suggests that at least a partial dissociation of the distal globular domains is essential for membrane fusion activity.
In our model low-pH HAs, partial dissociation of the globular head domains is seen in tmdHAlowpH1 and a complete dissociation of the globular domains is seen in tmdHAlowpH2. In BHA the distance between the Cαs of the intersubunit residues HA1:212 and HA1:216 is ∼6.3 Å. In tmdHAlowpH1 one pair of intersubunit residues retained its original distance and the other two pairs were separated by 23.1 Å and 13.8 Å, indicating the partial opening up of the globular domains. In tmdHAlowpH2 these distances were very large for two pairs of intersubunit residues and the third pair was ∼8 Å away. In the third low-pH HA model, all the intersubunit residues HA1:212 and HA1:216 were separated by ∼33 Å. Thus, crosslinking between 212 and 216 is incompatible with all three putative low-pH models.
Electron microscopy
Electron microscopy on BHA at pH 7 showed a spike length of 13.7 nm, consistent with the x-ray structure (Ruigrok et al., 1986) Low pH leads to formation of rosettes presumably due to the extrusion of the fusion peptides (Skehel et al., 1982; Ruigrok et al., 1986) and an increment of 3.7 nm in spike length (Ruigrok et al., 1986). It has been suggested that the fusion peptides are either extruded sideways or toward the membrane proximal end of the molecule (Ruigrok et al., 1986). Thermolytic digestion of the amino terminal region resulted in shortening of BHA2 and suggested that the amino terminus of HA2 moves to the membrane proximal region of the molecule (Ruigrok et al., 1988). In our low-pH HA models we observe some of the fusion peptides moving to the distal tip of the trimeric stem and some extruded sideways. In these models an increment of HA length is also seen compared with the length of native HA, mainly due to the extension of the trimeric coiled coil. Electron cryomicroscopy on HA at fusogenic pH has shown flattening of the distal top HA1 domains and formation of a continuous central cavity through the whole trimer (Böttcher et al., 1999), which does not seem to be consistent with 1HTM and our models, which are based on 1HTM.
Analysis of the rosettes at low pH suggested that a single molecule would have a radius of gyration of 51 Å (Ruigrok et al., 1986). Fig. 7 shows the radius of gyration of the molecule along each pathway. Along the first pathway, the radius of gyration of the molecule fluctuated a little but in the final low-pH model was similar to that of the native HA. In Path 2, it increased along the pathway and the final low-pH HA had a radius of gyration of 50.4 Å, which is very close to the value predicted for a single molecule at low pH. In contrast, the radius of gyration of the molecule decreased along the third pathway. This observation shows that among the three models, model 2 is in best agreement with these data.
FIGURE 7.

Radius of gyration of HA along the reaction pathway of HA → 1HTM transformation.
Changes in proteolytic susceptibility
Incubation of BHA at pH 5.0 showed changes in its susceptibility to trypsin and produced components of soluble (BHA1:28-328) and aggregate fractions (BHA2 and probably BHA1:1-27; Skehel et al., 1982; Doms et al., 1985). The tryptic cleavage of BHA in the low-pH conformation occurs first at HA1:27 and then at HA1:224. Examination of these sites of tryptic cleavage in the native BHA structure shows that the peptide bond at residue 27 is buried behind residues 32 and 33 near the trimer interface. The peptide bond at residue 224 may be inaccessible to trypsin due to its proximity to the trimer interface and the carbohydrate side chain at residue 165 of the adjacent subunit. A conformational change affecting subunit interaction could, therefore, render both residues susceptible to tryptic attack. We have examined the accessibility of the peptide bond at residue HA1:27 by measuring the solvent-accessible surface area in all the three low-pH models and native HA. In all models these peptide bonds were well exposed compared to BHA. Average SASA of residues HA1:26–28 in the native state is ∼30%. In the low-pH models the exposure of these residues increased to 51%, 36%, and 43% in models 1, 2, and 3, respectively. In model 2 the peptide bond at residue 27 of one of the chains was less accessible than the other two chains because of its proximity to its globular domain in the final conformation (see Fig. 2). Similarly, the accessibility of the residue HA1:224 in all low-pH models increased substantially compared to 1HGF. In the native state the SASA of HA1:224 is ∼42% and it increased to 57%, 57%, and 50% in the low-pH models 1, 2, and 3, respectively. This result is in good agreement with the experimental observations.
Other spectroscopic studies: CD and fluorescence
Circular dichroism (CD) spectra as a function of pH in both the near and far UV region have shown distinct conformational features of HA. The far-UV CD spectrum of HA showed that HA in rosettes maintained its overall secondary structure throughout the pH range of 7.4–4.5 (Wharton et al., 1988; Remeta et al., 2002). This indicates that even though the crystal structures of native and low-pH HA show dramatic conformational changes, especially in HA2 chains, the overall secondary structure was retained at low pH. We have examined the secondary structures of HA in our low-pH models. As a measure of how much the secondary structures are retained in our models we use the ratio between the number of residues in secondary structures in the low-pH model and the native state. The number of residues in the secondary structures are obtained from the secondary structure list created during the ribbon diagram representation of the molecule by the MOLMOL program. In the native state the number of residues in α-helices is ∼318 and in β-strands ∼429. The ratio between the total number of residues in the α-helices in the low-pH and the native HA are 0.99, 0.99, and 0.91 for models 1, 2, and 3, respectively. Similarly, the ratio for β-strands are 0.90, 0.90, and 0.82 for models 1, 2, and 3, respectively This shows that the overall secondary structure in our low-pH models is comparable to the secondary structure of native HA, in agreement with the far-UV CD studies.
Intrinsic tryptophan (Trp) fluorescence is generally used for monitoring structural properties and conformational changes of proteins. In the native state at pH 7.4, the fluorescence spectrum of HA reflected the characteristic apolar environment of tryptophan. Acidification of HA in rosette structures resulted in exposure of the aromatic residues (Krumbiegel et al., 1994; Remeta et al., 2002). We have calculated the solvent-accessible surface area of Trp in HA1 and HA2 chains in all our low-pH models. We found that most of the Trp residues that were buried in the native HA at pH 7.5 got exposed in our low-pH models. The SASA of all the Trp residues in the native state and low-pH models are as follows: HA1:Trp84 native = 3.5%, model 1 = 8%, model 2 = 9%, model 3 = 18%; HA1:Trp127 native = 5%, model 1 = 22%, model 2 = 17%, model 3 = 21%; HA1:Trp153 native = 8%, model 1 = 19%, model 2 = 18%, model 3 = 21%; HA1:Trp 180 native = 4%, model 1 = 14%, model 2 = 15%, model 3 = 9%; HA1:Trp222 native = 57%, model 1 = 50%, model 2 = 43%, model 3 = 55%; HA1:Trp234 native = 18%, model 1 = 30%, model 2 = 24%, model 3 = 30%; HA2:Trp14 native = 33%, model 1 = 66%, model 2 = 68%, model 3 = 75%; HA2:Trp21 native = 39%, model 1 = 61%, model 2 = 45%, model 3 = 65%. Fluorescence spectroscopy revealed that HA2:Trp92 is more buried in the low-pH conformation than the native HA conformation (Wharton et al., 1988). Solvent-accessible surface area calculation on this residue also showed that HA2:Trp92 in all three low-pH models is more buried than in the native conformation. In the native state, the SASA of Trp92 was ∼51%. In the low-pH models it decreased to 24%, 25%, and 28% for models 1, 2, and 3, respectively. Thus, our low-pH models are in agreement with the fluorescence and CD spectroscopic studies.
DISCUSSION
In the present study the conformational transition of HA from the native to putative fusogenic or postfusion conformations was examined using TMD simulations. Based on some of the hypotheses proposed in the literature, three pathways were considered. The main findings of the present work, which are consistent with most experimental observations, are as follows.
The effective energy (both EEF1 and GB) of the 1HTM conformation is lower than that of the 1HGF conformation for the residues present in 1HTM. This is in agreement with the experimental observation that in the absence of HA1, HA2 folds into the 1HTM conformation (Carr and Kim, 1993; Chen et al., 1995).
The low-pH model produced by Path 2 is energetically more probable than the models produced by Paths 1 and 3. The radius of gyration of this model is comparable with the experimentally suggested value. The main features of low-pH model 2 are i) the HA1 globular domains are widely dissociated; ii) the overall structure of the head domains is maintained; and iii) the fusion peptides are exposed and some of them placed toward the tip of trimeric coiled coil (where the target membrane is presumably located). The other two models show only partial separation of the globular domains.
Our model low-pH HA0 was higher in energy than the precursor HA0, supporting the idea that the native conformation is the global effective energy minimum for the uncleaved precursor.
The most probable model (Path 2) has two barriers, one at the 10th and another at the 20th TMD step. The low energy state between these two barriers (16th step) can be considered an intermediate along this path. In this intermediate state the coiled coil is fully extended toward the target membrane with a partial reversal of the C-terminal end of HA2. This intermediate is similar to the one proposed based on the spring-loaded mechanism (Hernandez et al., 1996; Hughson, 1997; Eckert and Kim, 2001). Model 2 has all the features of the spring-loaded mechanism, i.e., 1) an intermediate as discussed above; 2) dissociation of the globular domains, maintaining the overall structure; and 3) presence of the fusion peptide at the tip of the molecule. This model is also in agreement with some of the experimental findings, such as antigenicity changes at low pH, membrane-fusion activity of crosslinked mutants, electron microscopy, etc.
In models 1 and 3, which are energetically less favorable, extension of the coiled coil and exposure of fusion peptides occurred with only a partial separation of the globular domains. This indicates that complete dissociation of the globular domains is not essential for extension of the coiled coil and exposure of the fusion peptides. In the simulations the fusion peptides were displaced only when the helix-to-loop transition (HA2:105-112), which reverses the direction of the C-terminal portion of HA2, occurs in the reaction pathway. In the native state the fusion peptides are hydrogen-bonded to the above-mentioned helix region. Thus, local destabilization in this region may trigger the extrusion of the fusion peptide. The absence of complete dissociation of the globular domains in Path 1 may be a result of the short time scale of the simulation. Longer simulations might produce dissociation of the partially opened globular domains and allow the fusion peptides to move closer to the target membrane.
Our simulations have a number of limitations. First, sampling of the system was limited to a total of 4.5 ns for each TMD path (7.5 ns for the second path). This may not provide enough time for the unrestrained part of the molecule to find an optimal configuration with respect to the restrained part. Secondly, in this study we considered only three paths for obtaining the low-pH models. Although reasonable, these may not correspond to the optimal path. Thirdly, no membranes were present in our study, therefore any protein-membrane interactions that could be important in the conformational change have been neglected. Finally, this study has not addressed the question why low pH triggers the conformational transition. This could be addressed in the future by calculations of the pKa of titratable residues in HA.
Acknowledgments
We are grateful to Dr. A. Caflisch for the TMD code. This work is dedicated to the memory of Professor Don C. Wiley.
We thank the National Science Foundation (DBI-9974621) for financial support. Partial support was also provided by a grant from the City University of New York PSC-CUNY Research Award Program.
References
- Apostolakis, J., P. Ferrara, and A. Caflisch. 1999. Calculation of conformational transitions and barriers in solvated systems: application to the alanine dipeptide in water. J. Chem. Phys. 110:2099–2108. [Google Scholar]
- Bechor, D., and N. Ben-Tal. 2001. Implicit solvent model studies of the interactions of the influenza hemagglutinin fusion peptide with lipid bilayers. Biophys. J. 80:643–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentz, J. 1992. Intermediates and kinetics of membrane fusion. Biophys. J. 63:448–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentz, J. 2000. Membrane fusion mediated by coiled coils: a hypothesis. Biophys. J. 78:886–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentz, J., H. Ellens, and D. Alford. 1990. An architecture for the fusion site of influenza hemagglutinin. FEBS Lett. 276:1–5. [DOI] [PubMed] [Google Scholar]
- Bockmann, R. A., and H. Grubmuller. 2002. Nanoseconds MD simulation of primary mechanical energy transfer steps in F1-ATP synthase. Nature Str. Biol. 9:198–202. [DOI] [PubMed] [Google Scholar]
- Bonnafous, P., and T. Stegmann. 2000. Membrane perturbation and fusion pore formation in influenza hemagglutinin-mediated membrane fusion. J. Biol. Chem. 275:6160–6166. [DOI] [PubMed] [Google Scholar]
- Böttcher, C., K. Ludwig, A. Herrmann, M. V. Heel, and H. Stark. 1999. Structure of influenza hemagglutinin at neutral and at fusogenic pH by electron cryo-microscopy. FEBS Lett. 463:255–259. [DOI] [PubMed] [Google Scholar]
- Bradshaw, J. P., M. J. M. Darkes, T. A. Harroun, J. Katsaras, and R. M. Epand. 2000. Oblique membrane insertion of viral fusion peptide probed by neutron diffraction. Biochemistry. 39:6581–6585. [DOI] [PubMed] [Google Scholar]
- Brasseur, R., T. Pillot, L. Lins, J. Vandekerckhove, and M. Rosseneu. 1997. Peptides in membranes: tipping the balance of membrane stability. TIBS. 22:167–171. [DOI] [PubMed] [Google Scholar]
- Brooks, B. R., R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus. 1983. CHARMM: a program for macromolecular energy minimization and dynamics calculations. J. Comput. Chem. 4:187–217. [Google Scholar]
- Bullough, P. A., F. M. Hughson, J. J. Skehel, and D. C. Wiley. 1994. Structure of influenza hemagglutinin at the pH of membrane fusion. Nature. 371:37–43. [DOI] [PubMed] [Google Scholar]
- Carr, C. M., C. Chaudhry, and P. S. Kim. 1997. Influenza hemagglutinin is spring-loaded by a metastable native configuration. Proc. Natl. Acad. Sci. USA. 94:14306–14313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr, C. M., and P. S. Kim. 1993. A spring-loaded mechanism for the conformational change of influenza hemagglutinin. Cell. 73:823–832. [DOI] [PubMed] [Google Scholar]
- Chen, J., K. H. Lee, D. A. Steinhauer, D. J. Stevens, J. J. Skehel, and D. C. Wiley. 1998. Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell. 95:409–417. [DOI] [PubMed] [Google Scholar]
- Chen, J., J. J. Skehel, and D. C. Wiley. 1999. N- and C-terminal residues combine in the fusion-pH influenza hemagglutinin HA2 subunit to form an N-cap that terminates the triple-stranded coiled coil. Proc. Natl. Acad. Sci. USA. 96:8967–8972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J., S. A. Wharton, W. Weissenhorn, L. J. Calder, F. M. Hughson, J. J. Skehel, and D. C. Wiley. 1995. A soluble domain of the membrane-anchoring chain of influenza virus hemagglutinin (HA2) folds in Escherichia coli into the low-pH-induced conformation. Proc. Natl. Acad. Sci. USA. 92:12205–12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernomordik, L. V., E. Leikina, V. Frolov, P. Bronk, and J. Zimmerberg. 1998. The pathway of membrane fusion catalyzed by influenza hemagglutinin: restriction of lipids, hemifusion, and lipidic fusion pore formation. J. Cell Biol. 140:1369–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen, F. S., and G. B. Melikyan. 2001. Implications of a fusion peptide structure. Nat. Struct. Biol. 8:653–655. [DOI] [PubMed] [Google Scholar]
- Daniels, R. S., A. R. Douglas, J. J. Skehel, and D. C. Wiley. 1983. Analyses of the antigenicity of influenza hemagglutinin at the pH optimum for virus-mediated membrane fusion. J. Gen. Virol. 64:1657–1662. [DOI] [PubMed] [Google Scholar]
- Diaz, J., J. Wroblowski, J. Schlitter, and Y. Engelborghs. 1997. Calculation of pathways for the conformational transition between the GTP- and GDP-bound states of the HA-ras-p21 protein: calculations with explicit solvent simulations and comparison with calculations in vacuum. Proteins. 28:434–451. [PubMed] [Google Scholar]
- Dominy, B. N., and C. L. Brooks, III. 1999. Development of a generalized Born model parameterization for proteins and nucleic acids. J. Phys. Chem. B. 103:3765–3773. [Google Scholar]
- Doms, R. W., A. Helenius, and J. White. 1985. Membrane fusion activity of influenza virus hemagglutinin. J. Biol. Chem. 260:2973–2981. [PubMed] [Google Scholar]
- Durell, S. R., I. Martin, J. M. Ruysschaert, Y. Shai, and R. Blumenthal. 1997. What studies of fusion peptides tell us about viral envelope glycoprotein-mediated membrane fusion. Mol. Membr. Biol. 14:97–112. [DOI] [PubMed] [Google Scholar]
- Eckert, D. M., and P. S. Kim. 2001. Mechanisms of viral membrane fusion and its inhibition. Annu. Rev. Biochem. 70:777–810. [DOI] [PubMed] [Google Scholar]
- Ferrara, P., J. Apostolakis, and A. Caflisch. 2000a. Computer simulations of protein folding by targeted molecular dynamics. Proteins. 39:252–260. [DOI] [PubMed] [Google Scholar]
- Ferrara, P., J. Apostolakis, and A. Caflisch. 2000b. Targeted molecular dynamics simulations of protein unfolding. J. Phys. Chem. B. 104:4511–4518. [Google Scholar]
- Gething, M. J., R. W. Doms, D. York, and J. White. 1986. Studies on the mechanism of membrane fusion: site-specific mutagenesis of the hemagglutinin of influenza virus. J. Cell Biol. 102:11–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godley, L., J. Pfeifer, D. Steinhauer, B. Ely, G. Shaw, R. Kaufmann, E. Suchanek, C. Pabo, J. J. Skehel, D. C. Wiley, and S. Wharton. 1992. Introduction of intersubunit disulfide bonds in the membrane-distal region of the influenza hemagglutinin abolishes membrane fusion activity. Cell. 68:635–645. [DOI] [PubMed] [Google Scholar]
- Han, X., J. H. Bushweller, D. S. Cafiso, and L. K. Tamm. 2001. Membrane structure and fusion-triggering conformational change of the fusion domain from influenza hemagglutinin. Nat. Struct. Biol. 8:715–720. [DOI] [PubMed] [Google Scholar]
- Han, X., and L. K. Tamm. 2000. A host-guest system to study structure-function relationships of membrane fusion peptides. Proc. Natl. Acad. Sci. USA. 97:13097–13102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez, L. D., L. R. Hoffman, T. G. Wolfsberg, and J. M. White. 1996. Virus-cell and cell-cell fusion. Annu. Rev. Cell Dev. Biol. 12:627–661. [DOI] [PubMed] [Google Scholar]
- Hughson, F. M. 1997. Enveloped viruses: a common mode of membrane fusion? Curr. Biol. 7:R565–R569. [DOI] [PubMed] [Google Scholar]
- Kemble, G. W., D. L. Bodian, J. Rose, I. A. Wilson, and J. M. White. 1992. Intermonomer disulfide bonds impair the fusion activity of influenza virus hemagglutinin. J. Virol. 66:4940–4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koradi, R., M. Billeter, and K. Wüthrich. 1996. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 14:51–55. [DOI] [PubMed] [Google Scholar]
- Kozlov, M. M., and L. V. Chernomordik. 1998. A mechanism of protein-mediated fusion: coupling between refolding of the influenza hemagglutinin and lipid rearrangements. Biophys. J. 75:1384–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumbiegel, M., A. Herrmann, and R. Blumenthal. 1994. Kinetics of the low pH-induced conformational changes and fusogenic activity of influenza hemagglutinin. Biophys. J. 67:2355–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaridis, T., and M. Karplus. 1999. Effective energy function for proteins in solution. Proteins. 35:133–152. [DOI] [PubMed] [Google Scholar]
- Lazaridis, T., and M. Karplus. 2001. Microscopic basis of macromolecular thermodynamics. In Thermodynamics in Biology. E. DiCera, editor. Oxford University Press, Oxford. pp3–48.
- Lear, J. D., and W. F. DeGrado. 1987. Membrane binding and conformational properties of peptides representing the NH2 terminus of influenza HA2. J. Biol. Chem. 262:6500–6505. [PubMed] [Google Scholar]
- Ma, J., T. C. Flynn, Q. Cui, A. G. W. Leslie, J. E. Walker, and M. Karplus. 2002. A dynamic analysis of the rotation mechanism for conformational change in F1-ATPase. Structure. 10:921–931. [DOI] [PubMed] [Google Scholar]
- Ma, J., and M. Karplus. 1997. Molecular switch in signal transduction: reaction paths of the conformational changes in ras p21. Proc. Natl. Acad. Sci. USA. 94:11905–11910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko, J. C., C.-H. Kim, and Y.-K. Shin. 1997. The membrane topology of the fusion peptide region of influenza hemagglutinin determined by spin-labeling EPR. J. Mol. Biol. 267:1139–1148. [DOI] [PubMed] [Google Scholar]
- Melikyan, G. B., R. M. Markosyan, H. Hemmati, M. K. Delmedico, D. M. Lambert, and F. S. Cohen. 2000. Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J. Cell Biol. 151:413–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neria, E., S. Fischer, and M. Karplus. 1996. Simulation of activation free energies in molecular systems. J. Chem. Phys. 105:1902–1921. [Google Scholar]
- Ponder, J. W. 2001. TINKER, Version 3.9. Software Tools for Molecular Design. [DOI] [PMC free article] [PubMed]
- Qiao, H., R. T. Armstrong, G. B. Melikyan, F. S. Cohen, and J. M. White. 1999. A specific point mutant at position 1 of the influenza hemagglutinin fusion peptide displays a hemifusion phenotype. Mol. Biol. Cell. 10:2759–2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remeta, D. P., M. Krumbiegel, C. A. S. A. Minetti, A. Puri, A. Ginsbur, and R. Blumenthal. 2002. Acid-induced changes in thermal stability and fusion activity of influenza hemagglutinin. Biochemistry. 41:2044–2054. [DOI] [PubMed] [Google Scholar]
- Ruigrok, R. W. H., A. Aitken, L. J. Calder, S. R. Martin, J. J. Skehel, S. A. Wharton, W. Weis, and D. C. Wiley. 1988. Studies on the structure of the influenza virus hemagglutinin at the pH of membrane fusion. J. Gen. Virol. 69:2785–2795. [DOI] [PubMed] [Google Scholar]
- Ruigrok, R. W. H., N. G. Wrigley, L. J. Calder, S. Cusack, S. A. Wharton, E. B. Brown, and J. J. Skehel. 1986. Electron microscopy of the low pH structure of influenza virus hemagglutinin. EMBO J. 5:41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlitter, J., M. Engels, P. Krüger, E. Jacoby, and A. Wollmer. 1993. Targeted molecular dynamics simulation of conformational change application to the T → R transition in insulin. Mol. Simul. 10:291–308. [Google Scholar]
- Shangguan, T., D. P. Siegel, J. D. Lear, P. H. Axelsen, D. Alford, and J. Bentz. 1998. Morphological changes and fusogenic activity of influenza virus hemagglutinin. Biophys. J. 74:54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skehel, J. J., P. M. Bayley, E. B. Brown, S. R. Martin, M. D. Waterfield, J. M. White, I. A. Wilson, and D. C. Wiley. 1982. Changes in the conformation of influenza virus hemagglutinin at the pH optimum of virus-mediated membrane fusion. Proc. Natl. Acad. Sci. USA. 79:968–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skehel, J. J., and D. C. Wiley. 2000. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 69:531–569. [DOI] [PubMed] [Google Scholar]
- Stegmann, T., J. M. White, and A. Helenius. 1990. Intermediates in influenza induced membrane fusion. EMBO J. 9:4231–4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster, R. G., L. E. Brown, and D. C. Jackson. 1983. Changes in the antigenicity of the hemagglutinin molecule of H3 influenza virus at acidic pH. Virology. 126:587–599. [DOI] [PubMed] [Google Scholar]
- Wharton, S. A., R. W. H. Ruigrok, S. R. Martin, J. J. Skehel, P. M. Bayley, W. Weis, and D. C. Wiley. 1988. Conformational aspects of the acid-induced fusion mechanism of influenza virus hemagglutinin. J. Biol. Chem. 263:4474–4480. [PubMed] [Google Scholar]
- White, J. M. 1992. Membrane fusion. Science. 258:917–924. [DOI] [PubMed] [Google Scholar]
- White, J. M., and I. A. Wilson. 1987. Anti-peptide antibodies detect steps in a protein conformational change: low-pH activation of the influenza virus hemagglutinin. J. Cell Biol. 105:2887–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley, D. C., and J. J. Skehel. 1987. The structure and function of the hemagglutinin membrane glycoprotein of influenza virus. Annu. Rev. Biochem. 56:365–394. [DOI] [PubMed] [Google Scholar]
- Wiley, D. C., I. A. Wilson, and J. J. Skehel. 1981. Structural identification of the antibody-binding sites of Hong Kong influenza hemagglutinin and their involvement in antigenic variation. Nature. 289:373–378. [DOI] [PubMed] [Google Scholar]
- Wilson, I. A., J. J. Skehel, and D. C. Wiley. 1981. Structure of the hemagglutinin membrane glycoprotein of influenza virus at 3 Å resolution. Nature. 289:366–373. [DOI] [PubMed] [Google Scholar]
- Yang, L., W. A. Beard, S. H. Wilson, S. Broyde, and T. Schlick. 2002. Polymerase β simulations suggest that Arg258 rotation is a slow step rather than large subdomain motions Per Se. J. Mol. Biol. 317:651–671. [DOI] [PubMed] [Google Scholar]
- Yewdell, J. W., W. Gerhard, and T. Bachi. 1983. Monoclonal anti-hemagglutinin antibodies detect irreversible antigenic alterations that coincide with the acid activation of influenza virus A/PR/834-mediated hemolysis. J. Virol. 48:239–248. [DOI] [PMC free article] [PubMed] [Google Scholar]