

Abstract
The main function of the transmembrane light-harvesting complexes in photosynthetic organisms is the absorption of a light quantum and its subsequent rapid transfer to a reaction center where a charge separation occurs. A combination of freeze-thaw and dialysis methods were used to reconstitute the detergent-solubilized Light Harvesting 2 complex (LH2) of the purple bacterium Rhodopseudomonas acidophila strain 10050 into preformed egg phosphatidylcholine liposomes, without the need for extra chemical agents. The LH2-containing liposomes opened up to a flat bilayer, which were imaged with tapping and contact mode atomic force microscopy under ambient and physiological conditions, respectively. The LH2 complexes were packed in quasicrystalline domains. The endoplasmic and periplasmic sides of the LH2 complexes could be distinguished by the difference in height of the protrusions from the lipid bilayer. The results indicate that the complexes entered in intact liposomes. In addition, it was observed that the most hydrophilic side, the periplasmic, enters first in the membrane. In contact mode the molecular structure of the periplasmic side of the transmembrane pigment-protein complex was observed. Using Föster's theory for describing the distance dependent energy transfer, we estimate the dipole strength for energy transfer between two neighboring LH2s, based on the architecture of the imaged unit cell.
INTRODUCTION
Photosynthesis is the process where solar energy is transferred into useful chemical energy. In purple photosynthetic bacteria the photosynthetic unit is localized in a system of intracytoplasmic membranes. It consists of transmembrane pigment-protein complexes: light-harvesting (LH) complexes and reaction centers (RC). In short, photons are absorbed by LH2 complexes and the excitation energy is transferred to a LH1 complex, which is closely associated with the RC (LH1-RC). The excitation energy is then funneled to the RC where a charge separation takes places followed by electron transport across the membrane. The process ultimately results in the production of adenosine triphosphate.
All known LH2 complexes of purple bacteria display a similar architecture (Zuber, 1985). The basic unit is a heterodimer consisting of two polypeptides (α and β), each ∼50 amino acids long, binding a total of three bacteriochlorophyll (BChl) molecules. An aggregation of the subunits in a ring shape gives the overall structure of the LH2 complex. Structural studies with x-ray diffraction, transmission electron microscopy (TEM), and recently performed atomic force microscopy (AFM) have shown that LH2 complexes are formed from eight or nine αβ-subunits (McDermott et al., 1995; Koepke et al., 1996; Savage et al., 1996; Scheuring et al., 2001).
X-ray diffraction, TEM, and AFM are the most frequently used techniques for obtaining the structure of transmembrane proteins. Each of these techniques has its advantages and disadvantages. X-ray diffraction has been quite successful in resolving protein structures with atomic resolution, but it requires highly ordered, heavily derivated three-dimensional (3D) crystals. Transmembrane proteins are often not stable enough under such nonphysiological conditions, and high-quality crystals are difficult to produce. TEM has been used in some cases where two-dimensional (2D) crystallization was easier to achieve than 3D crystallization. The disadvantages of TEM are, however, that specifically prepared, frozen samples are needed and that, due to the inherent low contrast, a good signal-to-noise ratio is difficult to obtain in a single TEM image. AFM is a relatively new surface technique, suitable to image lipid bilayers and membrane organization, which has the advantage of working in aqueous solutions, an environment in which biomolecules are fully functional. Furthermore, the orientation and organization of transmem-brane proteins in the membrane can be investigated. The disadvantages of AFM are, however, the dependence of resolution on the specific tip-sample interacting forces and on the mechanical stability of the sample. Although TEM and AFM have been proven to be powerful techniques, the resolution obtained with x rays has not yet been achieved. A significant improvement can be achieved by applying AFM on two-dimensional, more or less crystalline, arrays of proteins.
The most frequently used strategy for reconstitution and crystallization of transmembrane proteins in lipid bilayers is comicellization of the proteins and lipids, both solubilized with detergent, which is removed after mixing the separate solutions (e.g., Jap et al., 1992; Kühlbrandt, 1992; Rigaud et al., 1995; Mosser, 2001). Extra chemical agents are usually added to the solution: detergents such as octyl thioglucoside (Scheuring et al., 2001), octyl glucoside (Montoya et al., 1995), octyl glucopyranoside (Montoya et al., 1995), organic solvents such as pentane, hexane (Szoka and Papahadjopoulos, 1978), or other chemicals such as glucose (Walz et al., 1998) or glycerol (Ikeda-Yamasaki et al., 1998). The concentration of the detergent molecules in these solutions is then gradually reduced, either by dialysis or by the addition of Bio-Beads (Rigaud et al., 1997). As the concentration of the detergent decreases from these lipid-detergent and lipid-protein-detergent micellar solutions, lipid bilayers are progressively formed in which the transmembrane proteins are incorporated. Usually, the morphology of the resulting 2D crystals depends on several poorly defined factors, and depending on the circumstances, various structures can be obtained such as planar sheets, proteo-liposomes, multilayered stacked sheets, thin three-dimensional crystals, and tubes (Lacapere et al., 1998; Mosser, 2001). High-resolution imaging with AFM has only been performed so far on purely planar sheets and opened-up proteo-liposomes.
We have used a combination of methods for reconstituting the LH2 complex from Rhodopseudomonas (Rps.) acidophila, strain 10050, in natural preformed lipid bilayers, without the need of chemical agents other than the detergent with which the LH2 was solubilized. Using this method, 2D quasicrystalline LH2 liposomes were obtained. Imaging the LH2 bilayers with AFM allowed us to investigate the orientation of the transmembrane complex in the membrane. Two modes of AFM operation were used: tapping mode AFM (TMAFM) in air and contact mode (CMAFM) in an aqueous solution. In TMAFM the tip is being excited in resonance oscillation while being scanned across the surface. In this way the lateral forces are significantly reduced and biomolecules adsorbed on a surface can be easily imaged in air. In CMAFM the tip is simply scanned across the surface. Using this mode of operation high-resolution images of biomolecules in liquid have been obtained, and the ring structure of the LH2 complex was resolved. Finally, the energy transfer between neighboring LH2s for this system is described using a simple model.
MATERIALS AND METHODS
Materials
Egg phosphatidylcholine (egg PC) was purchased from Avanti Polar Lipids (Alabaster, AL), Lauryldimethylamine N-oxide (LDAO) from Fluka (Buchs, Switzerland). 500-μl Slide-A-Lyzer dialysis cassettes with a cutoff of 10 kDa were purchased from Pierce (Rockford, IL). All other chemicals were purchased from Sigma Chemicals (Western, Australia).
Reconstitution of LH2 in liposome
Egg PC (20 mg) was dissolved in 1 ml of chloroform. The solvent was evaporated under a flow of dry nitrogen gas for several hours. After adding buffer A (10 mM Tris-HCl (pH 8.0), 400 mM NaCl), resulting in a lipid concentration of 2.5 mg/ml, the hydrated films were sonicated in a bath sonicator for at least 15 min. The solution was freeze-thawed and sonicated with a microtip sonicator (Sonics & Materials Inc., Danbury, CT) three or more times until it was almost transparent.
LH2 from Rps. acidophila, strain 10050, complexes were diluted to ∼5 mg/ml in buffer B (10 mM Tris-HCl (pH 8.0), 400 mM NaCl, 0.6% LDAO). The protein solution was added to the lipid solution at a lipid-to-protein ratio of 0.4 (w/w). The solution was further diluted twofold with detergent-free buffer, buffer A. The mixed solution was freeze-thawed once again and transferred to dialysis cassettes. It was dialyzed against 1.5 liters with detergent-free buffer for 4 days at 4°C in the dark. The dialysis buffer was renewed two times. The protein concentration was determined from the absorbance at 850 nm, using an extinction coefficient per Bchl of 382 mM−1 × cm−1 and a value of 129 kDa for the molecular weight of the LH2 complex.
Atomic force microscopy
Freshly cleaved mica was used as a support. The reconstituted solutions were diluted in buffer C (10 mM Tris-HCl (pH 7.2), 150 mM KCl, 25 mM MgCl2). A small droplet of the diluted solution was applied on mica and let to adsorb at room temperature. After 30–45 min the samples were gently washed with the imaging buffer, buffer D, (10 mM Tris-HCl (pH 7.2), 150 mM KCl) to remove weakly attached membranes. For measurements done in ambient conditions the samples were gently blown dry under a flow of nitrogen gas.
A commercial AFM (Nanoscope III, Digital Instruments, Santa Barbara, CA) equipped with a 14-μm scanner (E-scanner) was employed. For tapping mode AFM, Si probes were used with a resonance frequency of 75 kHz and a nominal spring constant of 2.8 N/m (Nanosensors GmbH, Wetzlar-Blankenfeld, Germany). For contact mode AFM, oxide sharpened Si3N4 probes with a nominal spring constant of 0.06 N/m (Digital Instruments) were used. Before use, the liquid-cell-tip holder was cleaned with 1% sodium dodecyl sulfate, washed extensively with ultrapure water and blown dry under a flow of nitrogen gas. The AFM was placed on an active antivibration table (Halcyonics GmbH, Goettingen, Germany), and in an isolating box (Burleigh, Vanier, Canada). Scans in liquid were recorded in buffer D with typical scan line rates of 2–3 Hz. An open liquid cell was used. The minimal force was applied, of the order of 100 pN, which was manually adjusted to compensate for thermal drifts. The particle and lattice dimensions were measured with WS×M (Nanotec, Spain) or with the Nanoscope program.
Optical measurements
The absorbance of egg PC liposomes was monitored at 524 nm with a UV-Vis spectrophotometer (Shimadzu, Columbia, MD) as LDAO was added. The absorbance was recorded after addition of the detergent until a constant value was obtained (usually within a few minutes).
RESULTS AND DISCUSSION
Three-dimensional structure of LH2 Rps. acidophila strain 10050
Fig. 1 shows schematic representations of the structure of LH2 of Rps. acidophila strain 10050 obtained by Cogdell and co-workers with x-ray diffraction on 3D crystals, with a resolution of 2.5 Å (McDermott et al., 1995). The complex is a nonameric circular aggregate of two polypeptides, α and β, (Fig. 1 B) with associated pigments, three Bchls, and one (possibly two) carotenoid molecules. The overall structure (Fig. 1 A) is rather like a hollow cylinder, believed to be filled with lipids. The inner walls of the cylinder are formed by the α-apoproteins and the outer walls are formed by the β-apoproteins. Within these protein walls the pigment molecules are located.
FIGURE 1.

(A) Topology model (top view, periplasmic side) of the LH2 of Rps. acidophila strain 10050. It consists of a concentric arrangement of two hollow cylinders built from nine α- and nine β-apoproteins. The outer surface of the cylinder is formed by the β-subunits, and its inner surface by the α-subunits. Each αβ-apoprotein pair binds four pigment molecules, three bacteriochlorophylls, and two carotenoid molecules. The spheres in various shades of gray represent the 18 apoproteins and the black spheres represent the pigment molecules. (B) Topology model of an αβ-apoprotein pair without the associated pigment molecules. Model A has been scaled down five times with respect to Model B.
Reconstitution of LH2 complexes in lipid liposomes
Formation of liposomes
For AFM imaging, the stability of the biomolecules is crucial; the biomolecule that is being imaged has to withstand the strong lateral forces exerted by the tip. Immobilization of highly aggregated biomolecules via adsorption on a flat substrate has proven to meet the requirements for high-resolution AFM imaging. When the biomolecules are closely packed, the neighboring molecules stabilize the biomolecule under the AFM tip (Shao, 1999). For transmembrane proteins, this can be achieved by using aggregates of reconstituted proteins in lipid bilayers. To be able to image individual LH2 complexes, LH2 bilayers were prepared.
Here, we chose to incorporate the transmembrane protein, LH2, in preformed natural egg PC liposomes. A combination of strategies found in the literature for preparing liposomes was utilized. In detail, first multilamellar lipid liposomes were formed by adding aqueous buffer to a dry lipid film. To produce small, unilamellar liposomes, high energies of subsequent sonic radiation are required. For the formation of large unilamellar liposomes, the solution of small unilamellar liposomes is exposed to multiple freeze-thaw cycles. In practice, the solution is rapidly frozen in liquid nitrogen and upon slowly thawing large unilamellar liposomes are formed. It has been suggested (Pick, 1981) that upon the rapid freezing, water molecules crystallize on the lipid interface, forming two frozen planes separated by the hydrophobic core of the membrane. This causes the lipid bilayer to fracture. During the slow thawing fractured bilayers fuse together.
Lipid-detergent interactions
To reconstitute the LH2 complex into the egg PC liposomes, a solution of detergent-solubilized (LDAO) LH2 and the egg PC liposomes was dialysized against a detergent-free buffer. Upon dialysis the detergent was slowly removed from the solution, passing through the dialysis membrane, and the LH2 inserted in the lipid bilayer. Important in this mixture are the liposome-detergent interactions. In general, liposomes can readily interact with detergents. Depending on the nature of the detergent and the detergent-to-lipid ratio, the structure of the liposome can be altered, varying from the formation of holes to the complete disruption of the bilayer structure at high ratios.
An indirect indicator of the microstructural changes of liposomes (Lasch, 1995; Knol et al., 1998) are the variations in the optical density (turbidity). The absorbance at 524 nm (OD524) was measured as a function of the detergent-to-lipid ratio (w/w) (Fig. 2), to check the stability of the liposomes. The curve can be divided in three main stages. In stage I the OD524 has a high value. During stage I, detergent molecules partition between the aqueous buffer and the bilayer forming detergent-doped liposomes. In stage II the OD524 decreases with increasing detergent concentration. In this stage liposomes are saturated with detergent and this process continues upon further increase of the detergent concentration. In stage III the OD524 remains constant. In our procedure the initial LDAO to egg PC ratio was 1.6. We do not expect that at this detergent-to-lipid ratio, a complete disruption of the molecular structure of the egg PC liposomes will occur, but rather the formation of holes in the bilayer structure.
FIGURE 2.

Turbidity (optical absorbance at 524 nm) of egg PC liposomes at various LDAO to egg PC ratios. The effect of the detergent on the physical state of the liposomes can be followed. The three indicated stages are discussed in the text.
Reconstitution experiments at lower relative amounts of LDAO were unsuccessful. It seems that detergent-mediated liposomes are more suitable for transmembrane protein reconstitution than pure liposomes, most likely, the formation of holes in the bilayer structure facilitates the insertion of transmembrane proteins.
AFM imaging of two-dimensional aggregates of LH2
Mica was used as a substrate. It is atomically flat, hydrophilic, and, in water, negatively charged (Israelachvili and Adams, 1977; Pashley, 1981). When a solution of proteoliposomes is exposed to the mica substrate, proteoliposomes first adsorb on the mica substrate. Theoretical and experimental studies have shown that if liposomes/proteoliposomes are big enough they rupture on a hydrophilic surface, leading to the formation of single (proteo) bilayer patches, exposing the interior of the liposome to the air/water interface. (Vikholm et al., 1995; Sackmann, 1996; Seifert, 1997; Keller and Kasemo, 1998; Reviakine and Brisson, 2000; Leonenko et al., 2000; Kumar and Hoh, 2000).
Negatively charged biomolecules can be immobilized on mica, by use of ions in the buffer solution. The counter ions act as a bridge between the negatively charged mica and the negatively charged biomolecules (Müller et al., 1997a). At a pH ∼7.2, the headgroups of egg PC are neutral, the periplasmic side of LH2 is almost neutral, and the cytoplasmic side is negatively charged. LH2 liposomes were adsorbed on the mica substrate using an electrolyte solution consisting of a mixture of monovalent and divalent ions, buffer C. The ruptured LH2 liposomes on the mica substrate were imaged in tapping mode AFM in ambient conditions and in contact mode AFM in a buffer solution.
TMAFM of two-dimensional aggregates of LH2
Using TMAFM in ambient conditions, round patches were observed with a typical height of ∼10 nm and a typical diameter of ∼400 nm (Fig. 3 A). Several patches had part of the edges elevated to a height of typically 14 nm. Within the patches, domains can be distinguished which show protrusions assembled in a lattice with unit lattice dimensions of α = 13.4 nm, β = 16.4 nm, γ = 63°. The ellipsoidal-shaped blobs have lateral dimensions of 6.5 nm and 9 nm and protrude by ∼0.4 nm from the lipid bilayer, as seen in a high magnification image, Fig. 3 B. Two types of domains were distinguished differing by their height as measured from the mica surface (shown in Fig. 4), a thin domain with a height of ∼9.5 nm and a thicker one of ∼12 nm. (Due to high aggregation of LH2 in the bilayer and the small height of the tip apex, the actual dimensions of the part of the LH2 extending from the lipid bilayer could only be measured at the edges of the domains.)
FIGURE 3.

(A) TMAFM topograph of reconstituted light-harvesting 2 complex in a lipid bilayer imaged under ambient conditions. The gray scale represents a height range of 15 nm. (B) The higher magnification image shows periodic structures, with an ellipsoidal-like shape. The gray scale represents a height range of 5 nm.
FIGURE 4.

TMAFM topograph of reconstituted light-harvesting 2 complexes in a lipid bilayer. The gray scale represents a height range of 15 nm. Thinner and thicker domains are observed in the imaged patch. (Inset) Section analysis along the white line in image showing the protrusions of the thinner and thicker domain. The height difference is 2 nm.
CMAFM of two-dimensional aggregates of LH2
TMAFM imaging in ambient conditions can be considered effortless. However so far, high-resolution imaging of biomolecules has been performed successfully only in a liquid environment. The mode of operation commonly used in liquid is CMAFM. High-resolution imaging in liquid is limited by two main factors, the sharpness of the tip and the deformation of the biomolecule under the probe. Commercially available Si3N4 probes can be sharp enough (Sheng et al., 1999). The second limiting factor is the deformation of the biomolecule under the tip pressure. To avoid deformation of the biomolecule, one has to neutralize the electrostatic and van der Waals forces between the tip and the biomolecule, and at the same time retain a contact between the tip and the sample. To achieve this condition for the LH2-lipid samples, we use a buffer-containing electrolyte solution, buffer D.
At low magnification, CMAFM topographs in the above mentioned buffer (Fig. 5 A) revealed similar patches as the TMAFM topographs in ambient conditions (Fig. 3 A). The height of the patch (measured from the mica surface) was on average 11 nm. Also here two domains were evident, as in TMAFM (Fig. 4), with a height difference of ∼2 nm (Fig. 5 A). The big, mostly round objects (bright color) in the image are presumably small liposomes or contaminants adsorbed on the patch.
FIGURE 5.

(A) CMAFM topograph of reconstituted light-harvesting 2 complex in a lipid bilayer imaged in buffer D. The gray scale represents a height range of 20 nm. (B) High-resolution images on the thinner layer, revealing the ring structure of the LH2 complex. The gray scale represents a height range of 1.5 nm. (Inset) Single LH2 complex. (C) High-resolution image on the thicker layer. The gray scale represents a height range of 2 nm.
Higher magnification images demonstrated the difference between TMAFM in ambient and CMAFM in liquid conditions. Higher magnification images on the thinner domain (Fig. 5 B) revealed roughly doughnut-shaped structures, the ring structure of the LH2 complex. An inner diameter of 3.6 nm and an outer diameter of 7.3 nm were measured. The rings extended 0.21 nm from the lipid bilayer. On the thicker domain, higher magnification images (Fig. 5 C) did not reveal ring structures, but protrusions with a diameter of 8 nm extending slightly higher from the underlying layer, by 0.3 nm.
Submolecular resolution was obtained in some of the imaged LH2 complexes. We were able to identify nine subunits, corresponding to the nine αβ-heterodimers of the LH2 complex (Fig. 5 B, inset). In Fig. 5 B, some of the LH2 complexes seem to miss some subunits. Unfortunately we cannot make a definitive statement for the existence of open rings in our aggregates with the current resolution. The observation could be due to a local deformation of the imaged biomolecule, as a result of height variations and the low degree of aggregation. Future, higher-resolution images could verify the validity of this observation.
Orientation of LH2 in lipid bilayer
Structure of LH2 from Rps. acidophila strain 10050 in lipid membrane
The polypeptides of the LH2 complex of Rps. acidophila strain 10050 are quite small (the α-apoprotein contains 53 amino acids, and the β-apoprotein 41 amino acids). Both polypeptides consist of polar N- and C-termini and a central hydrophobic region. The N-termini lie on the cytoplasmic side of the membrane and the C-termini on the periplasmic side. The hydrophobic regions are single transmembrane α-helices. The amino acid sequence and corresponding x-ray structure of the α- and β-polypeptides of LH2 is shown in Table 1.
TABLE 1.
Amino acid sequence of LH2s α and β polypeptides
| Polypeptide | Sequence | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| α-apoprotein | * | * | * | M | N | Q | G | K | I | W | T | V | V | N | P | A | I | G | I | P | A | L | L | G | S | V | T | V | I | A | I | L | V | H | L | A | I | L | S | H | T | T | W | F | P | A | Y | W | Q | G | G | V | K | K | A | A | |
| 1 | 5 | 10 | 15 | 20 | 25 | 30 | 35 | 40 | 45 | 50 | |||||||||||||||||||||||||||||||||||||||||||||||
| * | * | * | C | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | H | H | H | H | H | H | H | H | H | H | H | H | H | H | H | H | H | H | H | H | H | H | H | H | H | H | C | C | C | H | H | H | H | H | H | H | ? | ? | ? | ? | ? | ? | ? | ||
| β-apoprotein | * | * | * | * | * | A | T | L | T | A | E | Q | S | E | E | L | H | K | Y | V | I | D | G | T | R | V | F | L | G | L | A | L | V | A | H | F | L | A | F | S | A | T | P | W | L | H | |||||||||||
| 1 | 5 | 10 | 15 | 20 | 25 | 30 | 35 | 40 | |||||||||||||||||||||||||||||||||||||||||||||||||
| * | * | * | * | * | C | C | C | C | C | C | H | H | H | H | H | H | H | H | H | H | H | H | H | H | H | H | H | H | H | H | H | H | H | H | H | H | H | H | H | H | H | C | C | C | C | ||||||||||||
The corresponding x-ray secondary structure is assigned: * - unknown; 3 - 310 Helix; H - α Helix; ? - disordered; C - coil.
The amino acid sequence of the polypeptides of various purple bacteria antenna complexes has been published before (Zuber, 1990). The main feature is that all α- and β-polypeptides contain a hystidine residue. This conserved residue is ligated to the bacteriochlorophyls in the periplasmic side of the membrane bilayer. A sequence alignment for these amino acid sequences was performed, and for each α- and β-polypeptide the amino acid sequence corresponding to the membrane spanning, N- and C-terminal portions were assigned.
Alignment of the amino acid sequence of the polypeptides of the LH2 complex from Rps. acidophila strain 10050 with the published sequence alignment was done using as a reference point the most conserved residue, hystidine H (the residue is highlighted at position 30 in the amino acid sequence in Table 1). We found that residues 14-34 for the α-apoprotein, and 14-36 for the β-apoprotein are transmembrane (the presumed position of the hydrophobic layer thickness membrane is shown by the gray boxed areas in Table 1).
According to the x-ray structure the total length of the αβ-heterodimer is 5.6 nm, but is asymmetric. From the cytoplasmic side it extends by 1.0 nm from the lipid bilayer and only by 0.2 nm from the periplasmic side (including a diameter of 5.5 Å per lipid head (Parsegian et al., 1979)).
We also used several transmembrane prediction algorithms available on the Internet: TMHMM (http://www.cbs.dtu.dk/services/TMHMM/); DAS (http://www.sbc.su.se/∼miklos/DAS/); TMPRED (http://www.ch.embnet.org/cgi-bin/TMPRED_form_parser); HMMTOP (http://www.enzim.hu/hmmtop1.1/server/hmmtop.cgi); SOSUI (http://sosui.proteome.bio.tuat.ac.jp/cgi-bin/sosui.cgi?/); SWISS-PROT (http://www.expasy.ch/sprot/). All gave different results, however, all the algorithms predicted that the polypeptide portion of the cytoplasmic side of the LH2 complex would extend further from the lipid bilayer, with respect to the periplasmic side.
Based on the above observations, we believe that the observed thinner domains contain LH2s oriented into the lipid bilayer with the periplasmic side exposed to the AFM tip, because the periplasmic side of the LH2 complex extends the least from the lipid bilayer. The previously mentioned thicker domains consist of LH2 aggregates but with opposite orientation in the lipid bilayer.
Insertion of LH2 in lipid liposome
For reconstitution of transmembrane proteins in preformed liposomes, it has been argued (Eytan, 1982; Jain and Zakim, 1987) that if the membrane protein structure is asymmetric, the protein will preferentially insert in the lipid bilayer with its more hydrophobic moiety first, thus exposing its more hydrophilic part to the exterior of the lipid bilayer.
Unidirectional reconstitution of the lactose protein of Escherichia coli in egg PC preformed liposomes mediated with Triton X-100 (Knol et al., 1998), and in E. coli phospholipid extracted preformed liposomes mediated with α-d-octylglucoside and Triton X-100 (Jung et al., 1998) have been reported. Unidirectional reconstitution of reaction centers in a mixture of dipalmitoyl-l-α-phosphatidydylcholine and dipalmitoyl-l-α-phosphatidylglycerol liposomes was also achieved by the freeze-thaw method (Miyake and Hara, 1997); 90% of the RCs were incorporated with the H-subunit in the outer surface of the liposomes.
The x-ray structure of LH2 reveals that the cytoplasmic side is more polar than the periplasmic side. With the rupture of the LH2 liposomes on the hydrophilic mica surface the interior of the liposome (which would be the periplasmic side) is exposed to the AFM tip. A preferred orientation of LH2s in the lipid bilayer was indeed observed. The periplasmic side was observed in 83% of the imaged LH2s, therefore we can conclude that the periplasmic side, which is more hydrophobic preferentially inserts first.
LH2 dimensions measured with the AFM
Table 2 shows an overview of the dimensions of the reconstituted samples measured with AFM. The lateral dimensions are in agreement with the measurements performed with x-ray crystallography on cryogenic 3D LH2 crystals (McDermott et al., 1995). But the dimensions perpendicular to the bilayer, namely the thickness of the LH2 bilayer, the distance between the top of the LH2 ring, and the lipid bilayer, deviate from the expected values based on crystal parameters.
TABLE 2.
Single LH2 and LH2-bilayer dimensions measured with the AFM under physiological conditions
| Dimension | Average ± SD (nm) |
|---|---|
| Thickness | |
| Domain I | 9.5 ± 1.0 |
| Domain II | 12.2 ±1.2 |
| LH2 dimensions | |
| Inner diameter | 3.6 ± 0.2 |
| Outer diameter | 7.3 ± 0.5 |
| Ring diameter | 5.5 ± 0.2 |
| Height (lipid-top) | 0.21 ± 0.06 |
| Height (ring center-top) | 0.27 ± 0.07 |
Absolute normal dimensions measured with AFM have to be treated with caution. Height deviations of imaged biomolecules have been reported previously. Height differences in AFM topographs between different materials not only have a structural origin, but also depend on the specific tip-sample interactions (Butt et al., 1990; Müller and Engel, 1997b; Heinz and Hoh, 1999; Müller et al., 1999). For example, by varying the salt concentration of the imaging buffer, the thickness of the purple membrane could be varied with a factor 2 (Müller and Engel, 1997b). In our system, the underlying mica substrate, the headgroups of egg PC, the cytoplasmic side, and the periplasmic side of the LH2 complex all have quite different surface charge densities, likely leading to the observed discrepancies in the experimentally determined heights of the corresponding domains.
The height of an LH2 complex, as determined from the length of the αβ-heterodimer, is 5.6 nm. As observed, the LH2 complexes are not all oriented in the same direction in the lipid bilayer. Treating the LH2 bilayer as rigid, for patches containing LH2s facing in both directions a patch thickness of 6.5 nm is expected. It is well established that the headgroups of phospholipids interact with the mica surface mediated by a thin layer of water (Ragier et al., 1995). Therefore, an extra 1–2 nm should be added, resulting in a LH2-bilayer thickness of ∼8.5 nm. We measured an average patch thickness of 10 nm in TMAFM and 11 nm in CMAFM. Measurements in aqueous solutions with different salt concentrations may make it possible to investigate the remaining difference of 1.5–2.5 nm, and deduce the actual perpendicular dimensions.
Small height variations of ∼0.3 nm can be observed in the AFM images. Egg PC is a mixture of phosphatidylcholines lipids with hydrocarbon chains of different length. According to the manufacturer's specifications, egg PC is a mixture of C16 and C18 saturated and unsaturated alkyl chains with a small content of unsaturated C20 phosphatidylcholines. The difference between the thickness of the hydrophobic part of a lipid bilayer for a C16 and a C18 alkyl chain is of the order of 0.3 nm. This length difference will result in a hydrophobic mismatch between the hydrophobic part of the LH2 complex and the hydrophobic core of the lipids. There are however, various possible effects (Killian, 1998) of hydrophobic mismatch between α-helices proteins and lipids making it difficult to draw any conclusions on how this particular complex system will adapt to the mismatch.
Energy transfer in two-dimensional aggregates of LH2
Various LH2 complexes of purple bacteria have been reconstituted and crystallized in lipid bilayers (Montoya, et al., 1995; Oling, et al., 1996; Savage, et al., 1996; Walz, et al. 1998; Scheuring, et al., 2001; Ranck, et al., 2001). In Table 3 an overview of the unit cell configurations is given. The area occupied by a LH2 complex varies from ∼5000 to 25,000 Å. This diversity is not surprising because the systems are quite different (the size and composition of light-harvesting complexes vary from species to species, as do the lipids used for reconstitution and the conditions of the reconstitution itself, such as salt concentration and pH). It is interesting, however, to attempt to compare the degree of aggregation observed in our system with that of native photosynthetic membranes. One way is to estimate the distance dependent dipole strength for interLH2 excitation transfer on the basis of our observed unit crystal, using the Föster theory.
TABLE 3.
Summary of light-harvesting 2 ordering in two-dimensional crystals
| LH2 | Unit cell vectors | Area/LH2 (Å2) | Reference |
|---|---|---|---|
| Rhodovulum sulfidophilum | α = β = 157 Å, γ = 90° | 24,649 | Montoya, et al., 1995 |
| Rhodovulum sulfidophilum | α = 160 Å, β = 140 Å, γ = 90° | 22,400 | Savage, et al., 1996 |
| Ectothiorhodospira sp. | α = β = 99 Å, γ = 90° | 9801 | Oling, et al., 1996 |
| Rhodobacter capsulatus | α = β = 81 Å, γ = 60° | 6561 | Oling, et al., 1996 |
| Rhodobacter sphaeroides | α = β = 149 Å, γ = 90° | 22,201 | Walz, et al. 1998 |
| Rubrivivax gelatiosus | α = β = 66 Å, γ = 60° | 4356 | Ranck, et al., 2001 |
| Rubrivivax gelatiosus | α = 75 Å, β = 175, Å, γ = 92° | 13,125 | Ranck, et al., 2001 |
| Rubrivivax gelatiosus | α = 90 Å, β = 120 Å, γ = 92°, | 10,800 | Ranck, et al., 2001 |
| Rubrivivax gelatiosus | α = β = 76 Å, γ = 60° | 5776 | Scheuring, et al., 2001 |
| Rubrivivax gelatiosus | α = 82 Å, β = 133 Å, γ = 90° | 10,906 | Scheuring, et al., 2001 |
| Rhodopseudomonas acidophila | α = 134 Å, β = 164 Å, γ = 63° | 22,000 | this study |
The in vivo architecture of the photosynthetic unit of purple bacteria consists also of LH1-RC complexes, which may alter the nature and degree of aggregation. Our description of the energy transfer between two LH2s would hold if the photosynthetic unit arranges itself in a strip architecture, as has been previously proposed (Jungas et al., 1999; Ritz et al., 2001). In the strip configuration, LH2 complexes form arrays within the middle of such an array a strip of LH1-RC complexes is located.
If we assume that the excitation transfer process between a donor (LH2) and an acceptor (LH2) pigment is an incoherent hopping process, the transfer rate can be calculated according to the expression
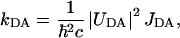 |
where ℏ is the Planck constant (divided by 2π), c is the speed of light, UDA is the coupling energy between the donor and acceptor electronic levels, and JDA represents the overlap on the frequency scale between the donor emission and acceptor absorption spectra.
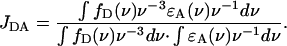 |
A value of 9.4 × 10−4 cm was obtained for solubilized LH2 complexes at room temperature.
The dipole-dipole interaction can be written as
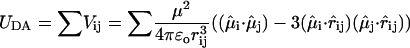 |
here, the subscripts ij denote Bchl's belonging to two different LH2 complexes. rij is the distance between two Bchls and μ is the dipole strength. For the above formula, the distances and angles between adjacent Bchls of two neighboring LH2s were determined for our unit cell configuration. Pairwise summation for the determination of the coupling energy was performed between adjacent LH2 rings.
Employing a value of 1/10 ps−1 (Ritz et al., 2001) for the transfer rate we obtained a dipole strength, μ, of 1.6 ± 0.2 (in units of monomeric dipole strength), for the closest unit distance [13.4 nm] and 4.3 ± 0.5 (in units of monomeric dipole strength), for the largest unit distance [16.4 nm]. The uncertainty arises from the value of the monomeric dipole strength; values in the literature range from 6.1 D to 7.7 D (Ritz et al., 2001).
A value close to 3 has been reported in literature. Superradiance measurements (Monshouwer et al. 1997) on the light-harvesting LH2 complex of Rhodobacter sphaeroides gave an emitting dipole strength equal to 2.8.
CONCLUSIONS
In this work, aggregated LH2s reconstituted in lipid bilayers were produced by detergent removal, using freeze-thaw and dialysis techniques. The produced 2D aggregates were ideal for AFM imaging, because the solution contained mainly LH2 liposomes that opened up on the surface. They were imaged with atomic force microscopy in ambient conditions with tapping mode, and under physiological conditions with contact mode. The AFM images revealed that the LH2s were aggregated in 2D quasicrystalline domains. The ring structure could only be resolved in an aqueous solution. In ambient conditions the AFM images revealed protrusions of the size of an LH2 complex. Useful information, however, could be extracted from the dry AFM images, such as the orientation and 2D organization in the lipid bilayer. A realistic value for the dipole strength for our LH2 spatial configuration in the bilayer was calculated using a simple theory. The organization observed in the AFM images may be relevant to the assembly of light-harvesting complexes in native photosynthetic membranes.
In the last few years, structural details of many individual pigment-protein complexes have emerged, adding to our understanding of the function of the photosynthetic apparatus. One of the next challenges will be to understand the organization of the photosynthetic apparatus in native membranes, in which the complexes are also believed to be in an aggregated configuration. Our results clearly demonstrate that also these types of aggregates can be successfully imaged with the AFM technique.
Acknowledgments
The authors thank A. Hoff for his valuable contribution in the beginning stages of this project and S. Oellerich for obtaining an emission spectrum of a solubilized LH2.
This work is part of the research program of the Stichting voor Fundamenteel Onderzoek der Materie with financial aid from the Nederlandse Organisatie voor Wetenschappelijk Onderzoek. R. Cogdell was funded by the Biotechnology and Biological Sciences Research Council agency.
References
- Butt, H. J., K. H. Downing, and P. K. Hansma. 1990. Imaging the membrane protein bacteriorhodopsin with the atomic force microscope. Biophys. J. 58:1473–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eytan, G. D. 1982. Use of liposomes for reconstitution of biological functions. Biochim. Biophys. Acta. 694:185–202. [DOI] [PubMed] [Google Scholar]
- Heinz, W. F., and J. H. Hoh. 1999. Relative surface charge density mapping with the atomic force microscope. Biophys. J. 76:528–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda-Yamasaki, I., T. Odahara, K. Mitsuoka, Y. Fujiyoshi, and K. Murata. 1998. Projection map of the reaction center-light harvesting 1 complex from Rhodopseudomonas viridis at 10 angstrom resolution. FEBS Lett. 425:505–508. [DOI] [PubMed] [Google Scholar]
- Israelachvili, J., and G. Adams. 1977. Measurement of forces between two mica surfaces in aqueous electrolyte solutions in the range 0–100 nm. J. Chem. Soc. Faraday Trans. I. 74:975–1000. [Google Scholar]
- Jain, M. K., and D. Zakim. 1987. The spontaneous incorporation of proteins into preformed bilayers. Biochim. Biophys. Acta. 906:33–68. [DOI] [PubMed] [Google Scholar]
- Jap, B. K., M. Zulauf, T. Scheybani, A. Hefti, W. Baumeister, U. Aebi, and A. Engel. 1992. 2D crystallization: from art to science. Ultramicroscopy. 46:45–84. [DOI] [PubMed] [Google Scholar]
- Jung, H., S. Tebbe, R. Schmid, and K. Jung. 1998. Unidirectional reconstitution and characterization of purified Na + /proline transporter of Escherichia coli. Biochemistry. 37:11083–11088. [DOI] [PubMed] [Google Scholar]
- Jungas, C., J. L. Ranck, J. L. Rigaud, P. Joliot, and A. Verméglio. 1999. Supramolecular organization of the photosynthetic apparatus of Rhodobacter sphaeroides. EMBO J. 18:534–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller, C. A., and B. Kasemo. 1998. Surface specific kinetics of lipid vesicle adsorption measured with a quartz crystal microbalance. Biophys. J. 75:1397–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killian, J. A. 1998. Hydrophobic mismatch between proteins and lipids in membranes. Biochim. Biophys. Acta. 1376:401–416. [DOI] [PubMed] [Google Scholar]
- Knol, J., K. Sjollema, and B. Poolman. 1998. Detergent-mediated reconstitution of membrane proteins. Biochemistry. 37:16410–16415. [DOI] [PubMed] [Google Scholar]
- Koepke, J., X. C. Hu, C. Muenke, K. Schulten, and H. Michel. 1996. The crystal structure of the light-harvesting complex II (B800–850) from Rhodospirillum molischianum. Structure. 4:581–597. [DOI] [PubMed] [Google Scholar]
- Kumar, S., and J. H. Hoh. 2000. Direct visualization of vesicle-bilayer complexes by atomic force microscopy. Langmuir. 16:9936–9940. [Google Scholar]
- Kühlbrandt, W. 1992. Two-dimensional crystallization of membrane proteins. Q. Rev. Biophys. 25:1–49. [DOI] [PubMed] [Google Scholar]
- Lacapere, J. J., D. L. Stokes, A. Olofsson, and J. L. Rigaud. 1998. Two-dimensional crystallization of Ca-ATPase by detergent removal. Biophys. J. 75:1319–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasch, J. 1995. Interaction of detergents with lipid vescicles. Biochim. Biophys. Acta. 1241:269–292. [DOI] [PubMed] [Google Scholar]
- Leonenko, Z. V., A. Carnini, and D. T. Cramb. 2000. Supported planar bilayer formation by vesicle fusion: the interaction of phospholipid vesicles with surfaces and the effect of gramicidin on bilayer properties using atomic force microscopy. Biochim. Biophys. Acta. 1509:131–147. [DOI] [PubMed] [Google Scholar]
- McDermott, G., S. M. Prince, A. A. Freer, A. M. Hawthornwaite-Lawless, M. Z. Paziz, R. J. Cogdell, and N. W. Isaacs. 1995. Crystal structure of an integral membrane light-harvesting complex from photosynthetic bacteria. Nature. 374:517–521. [Google Scholar]
- Miyake, J., and M. Hara. 1997. Protein-based nanotechnology: molecular construction of proteins. Materials Science and Engineering. C4:213–219. [Google Scholar]
- Monshouwer, R., M. Abrahamsson, F. van Mourik, and R. van Grondelle. 1997. Superradiance and exciton delocalization in bacterial photosynthetic light-harvesting systems. J. Phys. Chem. B. 101:7241–7248. [Google Scholar]
- Montoya, G., M. Cyrklaff, and I. Sinning. 1995. Two-dimensional crystallization and preliminary structure analysis of light harvesting II (B800–850) complex from the purple bacterium. Rhodovulum Sulfidophilum. J. Mol. Biol. 250:1–10. [DOI] [PubMed] [Google Scholar]
- Mosser, G. 2001. Two-dimensional crystallogenesis of transmembrane proteins. Micron. 32:517–540. [DOI] [PubMed] [Google Scholar]
- Müller, D. J., A. Engel, and M. Amrein. 1997a. Preparation techniques for the observation of native biological systems with the atomic force microscope. Biosens. Bioelectron. 8:867–877. [Google Scholar]
- Müller, D. J., and A. Engel. 1997b. The height of biomolecules measured with the atomic force microscope depends on electrostatic interactions. Biophys. J. 73:1633–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller, D. J., D. Fotiadis, S. Scheuring, S. A. Müller, and A. Engel. 1999. Electrostatically balanced subnanometer imaging of biological specimens by atomic force microscope. Biophys. J. 76:1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissen, J., S. Gritsch, G. Wiegand, and J. O. Radler. 1999. Wetting of phospholipid membranes on hydrophilic surfaces. Concepts towards self-healing membranes. Eur. Phys. J. B. 10:335–344. [Google Scholar]
- Oling, F., E. J. Boekema, I. O. de Zarate, R. Visschers, R. van Grondelle, W. Keegstra, A. Brisson, and R. Picorel. 1996. Two-dimensional crystals of LH2 light-harvesting complexes from Ectothiorhodospira sp. and Rhodobacter Capsulatus investigated with electron microscopy. Biochim. Biophys. Acta. 1273:44–50. [Google Scholar]
- Parsegian, V. A., N. L. Fuller, and R. P. Rand. 1979. Measured work of deformation and repulsion of lecithin bilayers. Proc. Natl. Acad. Sci. USA. 76:2750–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pashley, R. M. 1981. DLVO and hydration forces between mica surfaces in Li, Na, K and Sc electrolyte solutions: a correlation of double-layer and hydration forces with the surface cation exchange properties. J. Colloidal Interface Sci. 83:531–546. [Google Scholar]
- Pick, U. 1981. Liposomes with a large trapping capacity prepared by freezing and thawing of sonicated phospholipid mixtures. Arch. Biochem. Biophys. 212:186–194. [DOI] [PubMed] [Google Scholar]
- Ranck, J. L., T. Ruiz, G. Pehau-Arnaudet, B. Arnoux, and F. Reiss-Husson. 2001. Two-dimensional structure of the native light-harvesting complex LH2 from Rubrivivax gelatinosus and a truncated form. Biochim. Biophys. Acta. 1506:67–78. [DOI] [PubMed] [Google Scholar]
- Reviakine, I., and A. Brisson. 2000. Formation of supported phospholipid bilayers from unilamellar vesicles investigated by atomic force microscopy. Langmuir. 16:1806–1815. [Google Scholar]
- Rigaud, J. L., B. Pitard, and D. Levy. 1995. Reconstitution of membrane proteins into liposomes: application to energy-transducing membrane proteins. Biochim. Biophys. Acta. 1231:223–246. [DOI] [PubMed] [Google Scholar]
- Rigaud, J. L., G. Mosser, J. J. Lacapere, A. Olofsson, D. Levy, and J. L. Ranck. 1997. Bio-beads: an efficient strategy for two-dimensional crystallization of membrane proteins. J. Struct. Biol. 118:226–235. [DOI] [PubMed] [Google Scholar]
- Ritz, T., S. Park, and K. Schulten. 2001. Kinetics of excitation migration and trapping in the photosynthetic unit of purple bacteria. J. Phys. Chem. B. 105:8259–8267. [Google Scholar]
- Sackmann, E. 1996. Supported membranes: scientific and practical applications. Science. 271:43–48. [DOI] [PubMed] [Google Scholar]
- Savage, H., M. Cyrklaff, G. Montoya, W. Kuhlbrandt, and I. Sinning. 1996. Two-dimensional structure of light harvesting complex II (LHII) from the purple bacterium Rhodovulum sulfidophilum and compared with LHII from Rhodopseudomonas acidophila. Structure. 4:243–252. [DOI] [PubMed] [Google Scholar]
- Scheuring, S., F. Reiss-Husson, A. Engel, J. L. Rigaud, and J. L. Ranck. 2001. High-resolution AFM topographs of the Rubrivivax gelatinosus light harvesting complex 2. EMBO J. 20:3029–3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert, U. 1997. Configurations of fluid membranes and vesicles. Adv. Phys. 46:13–137. [Google Scholar]
- Shao, Z. 1999. Probing nanometer structures with atomic force microscopy. News in Physiological Sciences. 14:142–149. [DOI] [PubMed] [Google Scholar]
- Sheng, S., D. M. Czajkowsky, and Z. Shao. 1999. AFM tips: how sharp are they? Journal of Microscopy-Oxford. 196:1–5. [DOI] [PubMed] [Google Scholar]
- Szoka, F., and D. Papahadjopoulos. 1978. Procedure for preparation of liposomes with large internal aqueous space and high capture by reverse phase evaporation. Proc. Natl. Acad. Sci. USA. 75:4194–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vikholm, I., J. Peltonen, and O. Teleman. 1995. Atomic force microscope images of lipid layers spread from vesicle suspensions. Biochim. Biophys. Acta. 1233:111–117. [DOI] [PubMed] [Google Scholar]
- Walz, T., S. J. Jamieson, C. M. Bower, P. A. Bullough, and C. N. Hunter. 1998. Projection structures of three photosynthetic complexes from Rhodobacter sphaeroides: LH2 at 6 Å and RC-LH1 at 25 Å. J. Mol. Biol. 282:833–845. [DOI] [PubMed] [Google Scholar]
- Zuber, H. 1985. Structure and function of light-harvesting complexes and their polypeptides. Photochem. Photobiol. 42:821–844. [Google Scholar]
- Zuber, H. 1990. Considerations on the structural principles of the antenna complexes pf phototropic bacteria. In Molecular Biology of Membrane-Bound Complexes in Phototropic Bacteria. G. Drews and E. A. Dawes, editors. Plenum Press, New York. 161–180.