

Abstract
Human MinK and KCNQ1 subunits assemble to form IKs channels. When MinK position 55 is mutated to cysteine (MinK-55C), IKs channels can be blocked by external cadmium (Cd2+). We have supported a pore-associated location for MinK-55C because Cd2+ block is sensitive to voltage, permeant ions on the opposite side of the membrane (trans-ions), and external tetraethylammonium (TEA), an IKs pore-blocker. Two recent reports argue that MinK-55C is distant from the pore: one finds TEA does not affect Cd2+ block if channels are formed with a KCNQ1 mutant (K318I, V319Y) that increases TEA affinity; the second proposes that Cd2+ binds between MinK-55C and a cysteine in KCNQ1 that is posited to lie toward the channel periphery. Here, these discrepancies are considered. First, Cd2+ block of MinK-55C channels formed with wild-type KCNQ1 is shown to depend not only on voltage and trans-ions but state (showing decreased on-rate with increased open time and blocker trapping on channel closure). Conversely, MinK-55C channels with K318I, V319Y KCNQ1 are found to demonstrate Cd2+ block that is independent of voltage, trans-ions and state (and to have a lower unitary conductance): thus, the KCNQ1 mutations alter the process under study, yielding Cd2+ inhibition that is pore-independent and, perforce, TEA-insensitive. Second, MinK-55C channels are found to remain sensitive to Cd2+ despite mutation of any single native cysteine in KCNQ1 or all nine simultaneously; this suggests no KCNQ1 cysteine binds Cd2+ and can serve to localize MinK-55C. Despite many concerns that are enumerated, we remain obliged to conclude that Cd2+ enters and leaves the pore to reach MinK-55C, placing that residue in or near the pore.
INTRODUCTION
Classical voltage-gated potassium channels contain four pore-forming α-subunits that supply the domains that sense and respond to voltage and catalyze ion permeation. In native cells, potassium channels contain additional subunits such as the single-transmembrane domain MinK-related peptides (MiRPs; see Abbott and Goldstein, 1998). MiRPs establish the functional attributes of native complexes through their influence on surface expression, gating kinetics, unitary conductance, regulation/modulation, and pharmacology (Abbott and Goldstein, 2001, 2002). Thus, IKs channels are formed by KCNQ1 (a canonical α-subunit with 676 residues, one pore-forming P domain, and six transmembrane segments) and MinK (129 residues) (see Barhanin et al., 1996; Sanguinetti et al., 1996). Whereas channels formed only of KCNQ1 activate rapidly, exhibit current saturation, and have a small single-channel conductance, assembly with MinK yields IKs channels that activate slowly, do not saturate with prolonged depolarization, have a fourfold greater unitary conductance, exhibit altered discrimination among monovalent cations, and display modified responsiveness to a variety of activators and inhibitors (Busch et al., 1997; Kaczmarek and Blumenthal, 1997; Tai et al., 1997; Pusch, 1998; Sesti and Goldstein, 1998; Yang and Sigworth, 1998; Sesti et al., 2000b). Inherited missense mutations in KCNE1, the gene for MinK, are associated with cardiac arrhythmia and deafness and produce IKs channels that pass less potassium due to changes in these same functional attributes (Schulze-Bahr et al., 1997; Splawski et al., 1997; Tyson et al., 1997; Duggal et al., 1998; Sesti and Goldstein, 1998). Similarly, mutations in KCNE2 (Abbott et al., 1999; Sesti et al., 2000a) and KCNE3 (Abbott et al., 2001) are associated with cardiac arrhythmia and periodic paralysis, respectively, and decreased potassium flux through channels formed with those subunits.
The location of MinK relative to KCNQ1 in IKs channels remains a matter of controversy. Some posit that it crosses the membrane at the channel periphery (Romey et al., 1997; Wang et al., 1998; Tapper and George, 2001) or traverses the S4 canaliculus (Kurokawa et al., 2001). We have argued that MinK residues gain exposure in the outer pore vestibule (Wang et al., 1996), reside close to the ion conduction pathway near the selectivity filter (Tai and Goldstein, 1998), and influence the structure of the internal pore vestibule from an unknown distance (Sesti et al., 2000b). Most controversial have been our studies in which MinK residues 42–78 were sequentially altered to cysteine and IKs channels formed with wild-type KCNQ1 probed with sulfhydryl-reactive reagents and metals; some MinK sites were found to be accessible via a path whose attributes matched those of the IKs pore, suggesting they reside in or near the ion-conduction pathway (Tai and Goldstein, 1998). Thus, a cysteine at human MinK positions 54 or 55 allows blockade by external cadmium (Cd2+), but not Cd2+ applied from the inside; reciprocally, cysteine at positions 56 or 58 mediates inhibition by Cd2+ (or zinc) only from the inside (Tai and Goldstein, 1998). We reasoned that Cd2+ reaches these residues via the pore (rather than another transmembrane pathway) as follows: first, inhibition is voltage-dependent as if the blockers enter the transmembrane electric field to bind at 55 (from the outside) and 56 (from the inside). Second, permeant ions entering from the opposite side of the membrane alter inhibition in direct relationship to their relative permeability through the pore (a trans-ion effect that suggests ions traversing the pore interfere with blockade). Third, two adjacent residues (55 and 56) behave as if they are separated by the ion selectivity filter because transmembrane movement of sodium, Cd2+, and zinc are restricted at these residues; indeed, mutations at 55 alter permeation by cesium and ammonium, allow measurable sodium flux, and modify open-channel pore blockade by tetraethylammonium (TEA) (Goldstein and Miller, 1991; Wang et al., 1996; Tai and Goldstein, 1998). Finally, concurrent application of external TEA slows the timecourse of external Cd2+ blockade as if the two inhibitors compete for entry into the pore.
Recently, Kurokawa et al. (2001) sought to re-evaluate these findings through study of KCNQ1 mutants with enhanced affinity for the pore-blocker TEA. Failing to observe competition between external TEA and Cd2+ when channels contained MinK-55C and K318I, V319Y KCNQ1 subunits, they concluded that Cd2+ did not inhibit via the pore in mutant channels and, therefore, that MinK-55C was not pore-associated in channels with wild-type KCNQ1. However, we demonstrate here that changes attendant with these KCNQ1 mutations alter not only TEA affinity but the channel property under study—Cd2+ blockade. First, MinK-55C channels formed with wild-type KCNQ1 are confirmed to demonstrate Cd2+ inhibition that is voltage- and trans-ion-dependent when studied in the mammalian cells employed by Kurokawa et al. (2001), as previously found in oocytes (Tai and Goldstein, 1998). Next, MinK-55C channels formed with K318I, V319Y KCNQ1 are studied and found, in contrast, to be blocked by Cd2+ in a voltage- and trans-ion-insensitive fashion. Access and egress of Cd2+ is then observed to be state-dependent in channels with wild-type but not mutant KCNQ1 subunits. Finally, K318I, V319Y KCNQ1 subunits are judged to decrease single-channel potassium flux by noise-variance analysis. The results indicate that MinK-55C channels formed with K318I, V319Y KCNQ1 do not retain the conduction pathway attributes of channels with wild-type KCNQ1—specifically, those used to infer a pore locale for MinK-55C—and that failure to observe an effect of TEA on Cd2+ block should be expected, because Cd2+ no longer acts in a pore-dependent fashion.
Cadmium binding sites in proteins usually involve multiple coordinating side chains. To evaluate the contribution of native KCNQ1 cysteine residues to Cd2+ block of MinK-55C channels (and thereby identify potential sites of intersubunit contact), Tapper and George (2001) mutated the three cysteines predicted to lie in KCNQ1 transmembrane spans. Finding the C331A mutation to suppress Cd2+ blockade, they concluded that KCNQ1-331C and MinK-55C reside close together in IKs channels. In contrast, we studied all nine native cysteines altered individually or as a group (cysteine-free KCNQ1) and found no support for their role in Cd2+ blockade; we therefore conclude that these sites cannot serve to localize MinK-55C in IKs channels.
METHODS AND MATERIALS
Molecular biology
The K318I and V319Y KCNQ1 variant studied by Kurokawa and colleagues (2001) was made available to us immediately by those authors on our request. These and other mutants of human MinK and KCNQ1 were produced using the QuikChange mutagenesis kit (Stratagene, La Jolla, CA) followed by insertion of altered gene fragments into translationally silent restriction sites, as previously described (Sesti et al., 2000a). All products were confirmed by automated DNA sequencing. Human MinK (S38 isoform) and KCNQ1 cDNAs were, initially, gifts from R. Swanson (Merck) and M. T. Keating and M. Sanguinetti (University of Utah), respectively. cRNAs were synthesized using a mMessage mMachine kit (Ambion, Austin, TX) after constructs were moved into pRAT (Bockenhauer et al., 2001) and quantified by spectroscopy and comparison to control samples separated by electrophoresis and stained with ethidium bromide.
Expression protocols
Oocytes were isolated from Xenopus laevis frogs, defolliculated by collagenase treatment, injected the following day with 46 nl of sterile water containing 5 ng KCNQ1 and 1 ng MinK cRNA, and studied 2–4 days thereafter. Chinese hamster ovary (CHO) cells were transiently transfected by DEAE-Dextran, chloroquine, and DMSO shock, and were studied 20 h later, as before (Sesti et al., 2000a).
Electrophysiology
All experiments were performed at room temperature. Whole oocyte currents were measured by two electrode voltage clamp (Oocyte Clamp, Warner Instruments, Hamden, CT) with constant perfusion (∼1 ml/min, solution exchange <3 s). Data were sampled at 1 kHz and filtered at 0.25 kHz; if applied, leak correction was performed off-line. Standard bath solution was ND-96 (in mM): 96 NaCl, 2 KCl, 1 MgCl2, 0.3 CaCl2, and 5 HEPES/NaOH, pH 7.5.
Whole-CHO cell currents were recorded with the Axopatch 200B amplifier and Quadra 800 computer using Pulse software (HEKA Electronik, Lambrecht/Pfalz, Germany). For noise-variance analysis, data were stored filtered at 100 kHz on VHS tape (InstruTECH, Great Neck, NY) and analyzed with ACQUIRE and TAC software (Bruxton, Seattle, WA) and IGOR software packages (WaveMetrics, Lake Oswego, OR). Data are mean ± SE. The pipette contained (in mM): 100 KCl, 1 CaCl2, 1 MgCl2, 10 EGTA, and 10 HEPES/KOH, pH = 7.5. In trans-ion experiments, 100 KCl was replaced with 20 KCl and 80 NMDG. Bath solution was (in mM): 130 NaCl, 4 KCl, 2 CaCl2, 1.2 MgCl2, and 10 HEPES/NaOH, pH = 7.5. Chloride salts of Cd2+ and TEA were used without osmotic compensation.
Data analysis
The voltage-dependence of block was modeled assuming that occupying a single receptor was sufficient to block. The corresponding energy profile was composed of two barriers and one well following (Woodhull, 1973), with an internal barrier assumed to be infinitely high, so that blocked current was
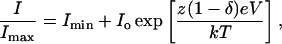 |
(1) |
where e, k, T, and z represent the electronic charge, the Boltzmann constant, absolute temperature and the charge on the blocker, respectively. Imin is the value of the blocked current for V → ∞ and Io is related to the part of the energy profile that is voltage-independent. δ is the apparent electrical distance and represents that fraction of the voltage drop experienced by the blocker.
Nonstationary noise-variance analysis was performed essentially as before (Sesti et al., 2000b). Currents and variances were obtained by fitting with a single Gaussian function all point histograms computed from 50- to 100-ms traces. IKs currents are characterized by slow development and failure to reach saturation. Currents elicited in the first 100 ms of each test pulse showed no time delay and were assumed to be non-channel-dependent; these leak currents and their variances were subtracted. Variance-current relationships were fitted to
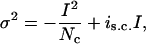 |
(2) |
where σ2 is the variance, I is the macroscopic current, Nc is the number of channels, and is.c. is the unitary current. Open probability, po, was obtained according to I/(Ncis.c.).
RESULTS
We have previously reported that IKs channels containing wild-type MinK and KCNQ1 subunits were insensitive to Cd2+, whereas mutation of MinK position 55 from glycine to cysteine (MinK-55C) allowed blockade by external (but not internal) Cd2+ (Tai and Goldstein, 1998). This work begins with consideration of channels formed by MinK-55C and a KCNQ1 variant with two point mutations, K318I and V319Y, as reported by Kurokawa and colleagues (Kurokawa et al., 2001). We find, as they did, that channels with MinK-55C and the KCNQ1 mutant are blocked by TEA with increased affinity compared to wild-type and that TEA does not alter the kinetics of Cd2+ blockade (not shown). Kurokawa and colleagues (2001) reasoned that loss of this evidence for Cd2+ block via the pore in mutant channels brought into question our conclusion that MinK-55C is accessed via the pore when assembled with wild-type KCNQ1 subunits. In this study, we sought to evaluate a simple explanation for the discordant conclusions: that K318I, V319Y KCNQ1 double mutation changed the attribute under study, that is, the effects of Cd2+, so that studies of the mutant were not germane (in this regard) to channels with wild-type KCNQ1. First, MinK-55C channels formed with wild-type KCNQ1 were compared to channels with the mutant for hallmarks of pore-dependent Cd2+ blockade.
Channels with wild-type, but not mutant, KCNQ1 show voltage-dependent Cd2+ block
Charged blockers that bind within a channel pore reveal the influence of the transmembrane electric field by changes in magnitude and/or kinetics of inhibition with applied voltage (Woodhull, 1973). Previously, we found Cd2+ inhibition of channels formed by MinK-55C and wild-type KCNQ1 in oocytes showed an effective electrical distance across the voltage drop (zδ) of ∼0.4 (Tai and Goldstein, 1998). Here, we compare the voltage-dependence of Cd2+ block of MinK-55C channels formed with wild-type or K318I, V319Y KCNQ1 subunits transiently expressed in Chinese hamster ovary (CHO) cells using whole-cell configuration in the fashion of Kurokawa and colleagues (2001). Although the application of 5 mM Cd2+ inhibits channels with wild-type KCNQ1 subunits (Fig. 1 A, WT) and those with mutant subunits (Fig. 1 B, Mutant), blockade of channels with wild-type KCNQ1 is sensitive to transmembrane voltage (Fig. 1 C, zδ = 0.36 ± 0.06, from −10 to 40 mV) while suppression of channels with mutant subunits is not (Fig. 1 D). Failure of voltage to alter block in the latter case suggests that the KCNQ1 mutations alter the pathway taken by Cd2+, and/or the location of the Cd2+ binding site, and/or the function of the Cd2+-occupied channel.
FIGURE 1.

Voltage alters cadmium block of channels with MinK-55C and wild-type (but not mutant) KCNQ1 subunits. Macroscopic current families in CHO cells. (A) MinK-55C and wild-type KCNQ1 (WT) channels in the absence (left) or presence of 5 mM cadmium (Cd2+) using 4 mM potassium bath solution and 100 mM potassium solution in the pipette. Voltage protocol: holding voltage −80 mV with 6-s steps from −60 mV to +40 mV in 10 mV increments. Interpulse interval, 2 s. Scale bars represent 0.5 nA and 1.5 s. (B) MinK-55C and K318I, V319Y KCNQ1 (Mutant) channels in the absence (left) or presence of 5 mM cadmium (Cd2+) as in A. Scale bars represent 0.5 nA and 1.5 s. (C) Cadmium block of outward current through channels formed with MinK-55C and wild-type KCNQ1 is voltage-dependent, studied as in A; fit to Eq. 1 gives zδ = 0.36 ± 0.06 (n = 5 cells). (D) Cadmium block of outward current through channels with MinK-55C and mutant KCNQ1 is not voltage-dependent; studied as in B (n = 5 cells).
Trans-ions alter Cd2+ block of channels with wild-type, but not mutant, KCNQ1
Pore blockers (especially those that are charged) are often affected by ions in the conduction pathway; thus, efflux of internal potassium, rubidium, and cesium alters external Cd2+ block of channels with MinK-55C and wild-type KCNQ1 expressed in oocytes (Tai and Goldstein, 1998). The same effect is observed here with CHO cells: when internal potassium concentration is lowered from 100 (Fig. 1 A) to 20 mM (Fig. 2 A) by isotonic substitution with NMDG, external Cd2+ blocks MinK-55C channels with wild-type KCNQ1 more effectively, decreasing the fraction of unblocked current at 40 mV from 0.50 ± 0.03 to 0.30 ± 0.02 (Fig. 2 C). Conversely, channels with mutant KCNQ1 subunits are insensitive to altered trans-potassium level (Figs. 1 B, 2 B, and 2 C; fu = 0.51 ± 0.03 and 0.53 ± 0.02). Further, the effective electrical distance for Cd2+ block of channels with wild-type KCNQ1 decreases with lowered internal permeant ion concentration, from zδ = 0.36 (Fig. 1 B) to 0.20 (Fig. 2 D), as expected for a pore-dependent process; in contrast, block of channels with mutant KCNQ1 subunits remains voltage-insensitive (Fig. 2 E).
FIGURE 2.

Trans-ions alter cadmium block of channels with MinK-55C and wild-type (but not mutant) KCNQ1 subunits. Macroscopic current families in CHO cells. (A) MinK-55C and wild-type KCNQ1 (WT) channels in the absence (left) or presence of 5 mM cadmium (Cd2+) using 4 mM potassium bath solution and 20 mM potassium solution in the pipette. Protocol: holding voltage −80 mV with 6-s steps from −60 mV to 40 mV in 10-mV increments; interpulse interval 2 s. Scale bars represent 0.1 nA and 1 s. (B) MinK-55C and K318I, V319Y KCNQ1 (Mutant) channels in the absence (left) or presence of 5 mM cadmium (Cd2+); conditions as in A. Scale bars represent 0.05 nA and 1 s. (C) Current inhibition by 5 mM cadmium at 40 mV with 100 mM or 20 mM potassium in the pipette for groups of six cells studied as in A and B; mean ± SE. (D) Cadmium block of outward current through channels formed with MinK-55C and wild-type KCNQ1 when internal potassium is lowered shows voltage-dependence, studied as in A; fit to Eq. 1 gives zδ = 0.20 ± 0.03 (n = 4 cells). (E) Cadmium block of outward current through channels with MinK-55C and mutant KCNQ1 when internal potassium is lowered is not voltage-dependent; as in B (n = 5 cells).
Cd2+ block is state-dependent in channels with wild-type, but not mutant, KCNQ1
Inhibition can be altered by channel state if the path taken by a blocker (or its binding site) change with gating transitions; in extreme cases, agents bind significantly in only one state (Armstrong, 1971; Yellen et al., 1994) or can be trapped in the channel by a state change (Miller et al., 1987). When the timecourse for current decline is assessed, channels with wild-type KCNQ1 and MinK-55C show state-dependent Cd2+ block kinetics whereas those with mutant KCNQ1 do not (Fig. 3, Table 1). Thus, acute Cd2+ application (in oocytes to facilitate long recording sessions) with repetitive depolarizing test pulses of 2 or 8 s and an interpulse interval of 11 s reveals channels with MinK-55C and wild-type KCNQ1 to have a slower apparent Cd2+ on-rate and diminished steady-state block with longer test pulses (Fig. 3 A, Table 1). This suggests that Cd2+ blocks channels with wild-type KCNQ1 less readily when they are open. Conversely, block proceeds in a state-insensitive manner for channels with mutant KCNQ1 subunits (Fig. 3 B, Table 1).
FIGURE 3.

Cadmium block kinetics are state-dependent with MinK-55C and wild-type (but not mutant) KCNQ1 subunits. Macroscopic current in oocytes (mean ± SE for 8–15 cells). The data are fitted to a single exponential function, and values are reported in Table 1. Protocol: repeated steps from −80 mV to +20 mV for 2 s (circle) or 8 s (square) with 11-s interpulse interval and 2 mM potassium bath solution; the bar indicates application of 5 mM Cd2+. (A) MinK-55C and wild-type KCNQ1 (WT) channels. (B) MinK-55C and K318I, V319Y KCNQ1 (Mutant) channels.
TABLE 1.
Kinetics and steady-state cadmium block with varied open time
| KCNQ1 type | OT/CT (s) | I∞ | Iα | τ (cycles) |
|---|---|---|---|---|
| WT | 2/11 | 0.34 ± 0.02 | 0.63 ± 0.04 | 2.2 ± 0.3 |
| WT | 8/11 | 0.52 ± 0.01 | 0.46 ± 0.02 | 3.0 ± 0.3 |
| Mutant | 2/11 | 0.49 ± 0.01 | 0.49 ± 0.03 | 2.2 ± 0.4 |
| Mutant | 8/11 | 0.47 ± 0.02 | 0.52 ± 0.04 | 2.0 ± 0.2 |
The timecourse of cadmium blockade (5 mM) was measured in 8–15 oocytes expressing MinK-55C and wild-type (WT) KCNQ1 or K318I, V319Y KCNQ1. Currents were elicited with cycles of 2- or 8-s steps from −80 mV to +20 mV with 11-s interpulse intervals. OT and CT indicate duration, in seconds, of the depolarizing and interpulse intervals. Data were fitted to a single exponential function: I∞ + Iα EXP(−n/τ) where I∞ represents the asymptotic fractional current, n is the cycle number, and τ and Iα are constants.
Closure sustains block of channels with wild-type, but not mutant, KCNQ1
Cadmium unblock was assessed first by cyclical stimulation in the presence of Cd2+ to achieve steady-state blockade, hyperpolarization to hold channels closed for 3 min after bath Cd2+ was removed, and cyclical restimulation in the absence of blocker: channels with MinK-55C and wild-type KCNQ1 remained inhibited (Fig. 4 A, arrow) on average by 50% (Fig. 4 B) whereas those with mutant subunits were fully unblocked (Fig. 4, B and C). Thus, Cd2+ appears to exit closed channels containing wild-type KCNQ1 subunits slowly compared to those formed with mutant subunits.
FIGURE 4.

Cadmium-unblock kinetics from the closed state are slow with MinK-55C and wild-type (but not mutant) KCNQ1 subunits. Macroscopic current in oocytes with 2 mM potassium bath solution. Cells are repeatedly stepped from −80 mV to +20 mV for 8 s with an 11-s interpulse interval; 5 mM Cd2+ was added (bar) until equilibrium blockade was achieved; cells were then held for 3 min at −80 mV and Cd2+ washed away; cyclical stimulation was then resumed. (A) MinK-55C and wild-type KCNQ1 (WT) channels unblock slowly when held closed. (B) MinK-55C and K318I, V319Y KCNQ1 (Mutant) channels unblock readily when held closed. (C) Fractional recovery from block during 3-min closed period after cadmium washout; as in A and B (arrow) for groups of six cells.
State-dependent unblock of channels with wild-type (but not mutant) KCNQ1 is also apparent when block is achieved by sustained application of Cd2+ to hyperpolarized channels (Fig. 5). Resumption of cyclical stimulation in the continued presence of Cd2+ reveals slow relaxation to a higher current level for channels with wild-type KCNQ1 (Fig. 5, A and B); changes with mutant KCNQ1 were either too rapid to discern, or minimal (Fig. 5, B and C). This may reflect diminished block with increasing open time, as observed above (Fig. 3).
FIGURE 5.

Cadmium-unblock kinetics from the open state are slow with MinK-55C and wild-type (but not mutant) KCNQ1 subunits. Macroscopic current in oocytes with 2 mM potassium bath solution. Cells are repeatedly stepped from −80 mV to +20 mV for 8 s with an 11-s interpulse interval, then held at −80 mV for 3 min with 5 mM added Cd2+ (bar) to achieve steady-state closed state block; cyclical stimulation is then resumed in the continued presence of Cd2+ and then on Cd2+ washout. (A) MinK-55C and wild type KCNQ1 (WT) channels unblock slowly. (B) MinK-55C and K318I, V319Y KCNQ1 (Mutant) channels unblock readily. (C) Fractional change in block after steady-state closed state block is achieved; and cyclical stimulation resumed; as in A and B for groups of 4–5 cells.
Finally, removal of Cd2+ during continuous stimulation shows unblock to be slow for channels with wild-type KCNQ1 (Fig. 5 A, τ = 69 ± 15 s, n = 4 cells) compared to those with the mutant (Fig. 5 B, τ < 10 s, n = 5 cells). Of note, channels with wild-type KCNQ1 that are closed before removal of Cd2+ (Fig. 4 A) show somewhat faster unblock kinetics (Fig. 4 A, τ = 27 ± 4 s, n = 5 cells) suggesting that the two blocked states are not equivalent.
Taken together, these findings suggest that Cd2+ enters and leaves MinK-55C channels containing wild-type KCNQ1 in a state-dependent manner; Cd2+ enters open channels less readily (perhaps revealing a destabilizing influence of ions traversing the open pore, in keeping with trans-ion effects, Fig. 2 C), and acts as if “locked” inside closed channels. Conversely, Cd2+ appears to enter and exit channels with mutant KCNQ1 subunits in a state-independent manner.
K318I, V319Y KCNQ1 double mutation decreases unitary current
Since the KCNQ1 mutations alter pore block by TEA and the effects of Cd2+ on the channels, other pore-associated attributes were evaluated. The mutations produce no apparent change in selectivity of the channel for potassium over sodium based on bi-ionic reversal potential measurements; CHO cells expressing MinK-55C with wild-type or mutant KCNQ1 show shifts in reversal potential of 53 ± 0.2 and 51 ± 0.1 mV, respectively, with a change in bath potassium concentration from 4 to 40 mM (n = 3–8 cells). Conversely, the mutations do alter unitary current as judged by noise-variance analysis. This approach is helpful because IKs channels open only briefly to pass small currents, but it is limited by failure of the currents to saturate despite prolonged depolarizing pulses (Sesti and Goldstein, 1998; Yang and Sigworth, 1998; Sesti et al., 2000b). Channels formed with MinK-55C and mutant KCNQ1 subunits exhibit a threefold decrease in the unitary current compared to those with wild type KCNQ1, is.c. = 0.10 ± 0.03 and 0.30 ± 0.08 pA, respectively, without significant differences in open probability (Fig. 6). This suggests the KCNQ1 mutations change the structure and function of the pore not only to increase TEA affinity and alter the character of Cd2+ block, but to decrease single-channel conductance.
FIGURE 6.

Channels with MinK-55C and mutant KCNQ1 have a decreased unitary conductance compared to those with wild-type KCNQ1 subunits. Macroscopic current families studied in CHO cells. (A) Representative variance-current relationships for cells expressing MinK-55C with wild-type KCNQ1 (WT, solid squares) or K318I, V319Y KCNQ1 (Mutant, open squares) at 40 mV. Curves are best fit of the data to Eq. 2 with is.c. = 0.27 pA, Nc = 5,000 channels, po = 0.37 for WT; and is.c. = 0.08 pA, Nc = 14,000 channels, and po = 0.44 for mutant channels. (B) Unitary current at 40 mV by noise-variance for MinK-55C channels with wild-type (WT) or mutant KCNQ1; each bar represents the average ± SE for five cells, and corresponded to po = 0.38 ± 0.08 and 0.45 ± 0.06, respectively.
No cysteine in KCNQ1 is required for Cd2+ block
As Cd2+ binding sites often involve the collaboration of multiple sulfhydryl groups, we interrogated each of the nine cysteine residues in KCNQ1 through their individual mutation to serine or alanine, and replacement en toto to produce a cysteine-free subunit. Changing each or all the native cysteines in KCNQ1 did not relieve MinK-55C channels from blockade after 3 min exposure to 5 mM external Cd2+ (Fig. 7). This screen included two mutants of KCNQ1 residue C331 (see Fig. 7 B, C331S or C331A) and was performed in oocytes, the experimental cells used by Tapper and George (2001); nonetheless, we were unable to reproduce their finding of loss of Cd2+ sensitivity with mutation of KCNQ1 C331. This discrepancy was not due to the level of Cd2+ employed, as when oocytes were studied first with 0.5 mM Cd2+ and then 5 mM Cd2+, the unblocked fractional current at steady state (5–8 min Cd2+ application) for MinK-55C channels with wild-type KCNQ1 was 0.89 ± 0.02 and 0.34 ± 0.02 (n = 7 cells); with KCNQ1-331A it was 0.79 ± 0.03 and 0.30 ± 0.04 (n = 11 cells); and with cysteine-free KCNQ1 it was 0.88 ± 0.01 and 0.33 ± 0.03 (n = 9 cells).
FIGURE 7.

Cadmium blocks channels with MinK-55C and wild-type KCNQ1 like those with KCNQ1 subunits with altered native cysteine residues. Macroscopic currents measured in oocytes with and without 5 mM cadmium. (A) Cadmium block of channels formed with MinK-55C and KCNQ1-C331A in oocytes. Protocol: 5 s pulse to 20 mV from −80 mV every 20 s; current is shown normalized to the value before cadmium application; inset shows indicated current traces. (B) No native cysteine in KCNQ1 is required for cadmium blockade of channels with MinK-55C subunits in oocytes; protocol as in A, mean ± SE, n = 6–13 cells. Oocytes expressed MinK or MinK-55C and wild-type KCNQ1 or KCNQ1 with the indicated cysteine altered to serine (S) or alanine (A). Fraction unblocked current after 3 min cadmium application, normalized to unblocked current level. Values plotted are: MinK/KCNQ1, 0.97 ± 0.03; MinK-55C/KCNQ1, 0.42 ± 0.05; MinK-55C/KCNQ1-Δ34, 0.42 ± 0.06; MinK-55C/KCNQ1-C122S, 0.45 ± 0.03; MinK-55C/KCNQ1-C136S, 0.37 ± 0.05; MinK-55C/KCNQ1-C180S, 0.40 ± 0.03; MinK-55C/KCNQ1-C214S, 0.31 ± 0.04; MinK-55C/KCNQ1-C331S, 0.48 ± 0.03; MinK-55C/KCNQ1-C331A, 0.44 ± 0.03; MinK-55C/KCNQ1-C381S, 0.26 ± 0.02; MinK-55C/KCNQ1-C445S, 0.40 ± 0.03; MinK-55C/KCNQ1-C642A, 0.43 ± 0.02; and MinK-55C/cysteine-free KCNQ1, 0.45 ± 0.02. KCNQ1-Δ34 is a native splice variant of KCNQ1 (Sanguinetti et al., 1996) and lacks C34 that is found in the longer KCNQ1 variant used otherwise in this work.
Cd2+ effects on channels with wild-type MinK in CHO cells
As previously reported (Tai and Goldstein, 1998), 5 mM external Cd2+ has no significant effect on channels formed with wild-type MinK and KCNQ1 subunits studied in oocytes (Fig. 7 B). Conversely, as noted by Kurokawa and colleagues (2001), the same channels in CHO cells show ∼11% suppression after just 75 s of cyclical stimulation and inhibition is not readily reversed on Cd2+ removal (Fig. 8 A). This effect is observed whether the KCNQ1 subunits employed are wild-type or cysteine-free (Fig. 8 B). The effect is slow to reverse and insensitive to voltage with both wild-type KCNQ1 and K318I, V319Y KCNQ1 subunits (Fig. 8 C); it follows that the effect is not a confounding variable in studies of voltage or trans-ions performed in CHO cells with MinK-55C channels (Figs. 1 and 2). As in oocytes, inhibition of MinK-55C channels in CHO cells was readily recognized by its magnitude and reversibility whether KCNQ1 subunit were wild-type, cysteine-free, C331S (Fig. 8 B) or C331A (Fig. 8, B and D).
FIGURE 8.

Studies in CHO cells. Macroscopic currents with and without 5 mM cadmium with a holding voltage of −80 mV, a test pulse of 5 s to 20 mV and a 10-s interpulse interval. Macroscopic current families studied in CHO cells. (A) Cadmium effect on channels with wild-type MinK in CHO cells: weak and poorly reversible inhibition whether KCNQ1 is cysteine-free (shown) or wild-type, B. Plot is current from a cell at the end of a test pulse; inset shows indicated current traces. (B) Removal of native cysteines in KCNQ1 does not alter the effect of cadmium on channels with wild-type or MinK-55C subunits. Plot is fraction of unblocked current after 75 s cadmium application, normalized to unblocked current level (Mean ± SE), n = 8 cells; MinK/KCNQ1, 0.89 ± 0.05; MinK/cysteine-free KCNQ1, 0.88 ± 0.02; MinK-55C/KCNQ1, 0.59 ± 0.04; MinK-55C/cysteine-free KCNQ1, 0.61 ± 0.05; MinK-55C/KCNQ1-C331S, 0.59 ± 0.04; and MinK-55C/KCNQ1-C331A, 0.51 ± 0.05. (C) Cadmium effects on channels with wild-type MinK are insensitive to voltage whether channels carry wild-type KCNQ1 (solid square) or K318I, V319Y KCNQ1 subunits (open square); protocol and plot are as in Fig. 1 C (n = 4–8 cells). (D) Cadmium block of channels with MinK-55C and KCNQ1-C331A; plot as in A.
DISCUSSION
A KCNQ1 mutant that alters TEA affinity, Cd2+ block, and potassium flux
This study supports the case that external Cd2+ blocks IKs channels formed with MinK-55C and wild-type KCNQ1 in a pore-dependent fashion. Previously, we demonstrated that Cd2+ block is altered by transmembrane voltage, trans-ions, and concurrent application of the pore-blocker TEA; we also found that mutation of position 55 influenced open-channel pore blockade and selectivity among monovalent cations consistent with its proximity to the pore (Goldstein and Miller, 1991; Tai and Goldstein, 1998). Here, we show that Cd2+ appears to enter and exit the channels in a state-dependent manner, and that no native KCNQ1 cysteine residue is required to achieve Cd2+ blockade. Conversely, we observe none of these pore-associated attributes for Cd2+ block of channels containing MinK-55C and a KCNQ1 mutant (K318I, V319Y) that binds TEA with high affinity. This indicates that the K318I, V319Y mutations alter the effects of Cd2+ on the channels. A decrease in unitary conductance with K318I, V319Y KCNQ1 subunits supports the idea that the structure and function of the pore are changed by the mutations. Because these channels do not show pore-dependent Cd2+ blockade, pore occlusion by TEA cannot be expected to influence Cd2+ inhibition. It follows that failure to observe an affect of TEA on Cd2+ inhibition of channels with K318I, V319Y KCNQ1 subunits neither supports nor contradicts our prior assertion that block of channels with wild-type KCNQ1 is pore-dependent, or the inference that MinK-55C is in, or near, the ion-conduction pathway.
One possible explanation for differences in Cd2+ blockade with wild-type and mutant KCNQ1 subunits is that binding occurs at the same location in the two channel types but that Cd2+ travels via different pathways to its blocking site. Our findings suggest that Cd2+ enters channels with wild-type KCNQ1 via the pore, doing so more rapidly when channels are closed (perhaps due to the absence of trans-ion flux), and that changes associated with opening expedite unblock. Conversely, channels with mutant KCNQ1 show no significant dependence of Cd2+ inhibition on voltage, trans-ions, or state suggesting pore-independent access and egress of the blocker. Another possibility is that KCNQ1 mutation produces an additional Cd2+ blocking site outside the electric field; however, equilibrium inhibition of channels with wild-type or mutant KCNQ1 subunits is roughly similar and multiple blocking sites are not suggested by studies of the mutant. A third possibility is that mutation alters one site that influences TEA affinity, Cd2+ block, and unitary conductance—a plausible scenario in light of evidence that TEA and magnesium bind at interacting sites inside the pore of an inward rectifier potassium channel and that both agents manifest voltage-dependence due to transmembrane movement of permeant ions rather than significant entry of the blockers into the electric field (Spassova and Lu, 1998, 1999). While we favor the notion that Cd2+ reaches its blocking site via the pore when channels contain wild-type KCNQ1 and an alternative pathway with mutant subunits, our findings do not eliminate the other possibilities, nor stipulate the location where Cd2+ binds.
A puzzling model stands
These results continue to support a pore-associated location for MinK-55C in IKs channels; this is an unsettling notion for at least three reasons. First, the microbial potassium-selective channels visualized at high resolution (KcsA and MthK) offer little guidance as to how MinK subunits might be accommodated in close proximity to those pores (Zhou et al., 2001; Jiang et al., 2002). On the other hand, these subunits have just two transmembrane segments and significant sequence variations compared to the pore-forming α-subunits of voltage-gated potassium channels that may rationalize assembly with MinK and its relatives. Indeed, structural differences are suggested by functional studies of α-subunits from eukaryotes with two-transmembrane segments (Minor et al., 1999) or six-spans (del Camino et al., 2000), as predicted for KCNQ1.
A second issue is that our ideas about location rest on the assumption that Cd2+ binds directly to the substituted cysteine (MinK-55C) rather than some other site. The assumption seems reasonable, first, because Cd2+ does not block channels with wild-type MinK and, second, because cysteine substitution across MinK yields Cd2+ blocking sites with unique attributes; for example, some substitutions mediate block only by external Cd2+ while others yield block only from inside solution (Tai and Goldstein, 1998). Direct Cd2+-cysteine interaction is also supported by observation that zinc blocks reversibly at the same sites whereas covalent modification by sulfhydryl reagents produces irreversible effects (Wang and Goldstein, 1995; Tai and Goldstein, 1998).
A third concern is that Cd2+ inhibits channels with MinK-55C slowly and weakly compared to cysteine-substituted Shaker channel α-subunits, some of which demonstrate on-rates approaching free diffusion (Yellen, 1998). Slow block kinetics of a pore-associated MinK-55C site might result from infrequent exposure of MinK-55C in the pore or slow movement of Cd2+ from the pore to a location adjacent to the ion conduction pathway. It seems unlikely that Cd2+ moves extensively within the IKs channel complex, because it inhibits channels containing MinK-54C or MinK-55C only by entering from the outside, and must be applied from the inside to block those with MinK-56C or MinK-58C. Weak blockade might result from interaction with just one or two MinK-55C residues; indeed, we have suggested that just two MinK monomers are present in each IKs complex (Wang and Goldstein, 1995), although others argue MinK number is variable and can be greater (Wang et al., 1998).
A fourth concern is that these studies are performed with a MinK mutant (55C) and just as K318I, V319Y KCNQ1 mutations alter Cd2+ blockade from that observed with wild-type KCNQ1, so, too, MinK mutation could alter the channel in a difficult-to-discern fashion.
If MinK-55C sites do not coordinate Cd2+ on their own, identification of non-sulfhydryl KCNQ1 residues that collaborate in binding may offer some answers to these concerns. Ultimately, direct visualization may be required to address outstanding conundra such as the trajectory of MinK through the IKs channel complex, the usual number of MinK subunits in wild-type channels, and the location where Cd2+ binds.
Acknowledgments
We are grateful to G. W. Abbott and N. Goldstein for critical feedback.
This work was supported by a grant from the National Institutes of Health to S.A.N.G., who is a recipient of the Doris Duke Charitable Foundation Distinguished Clinical Scientist Award.
Federico Sesti's present address is UMDNJ-Robert Wood Johnson Medical School, Dept. of Physiology and Biophysics, Piscataway, NJ 08854.
References
- Abbott, G. W., M. H. Butler, S. Bendahhou, M. C. Dalakas, L. J. Ptacek, and S. A. N. Goldstein. 2001. MiRP2 forms potassium channels in skeletal muscle with Kv3.4 and is associated with periodic paralysis. Cell. 104:217–231. [DOI] [PubMed] [Google Scholar]
- Abbott, G. W., and S. A. N. Goldstein. 1998. A superfamily of small potassium channel subunits: form and function of the MinK-related peptides (MiRPs). Q. Rev. Biophys. 31:357–398. [DOI] [PubMed] [Google Scholar]
- Abbott, G. W., and S. A. N. Goldstein. 2002. Disease-associated mutations in KCNE potassium channel subunits (MiRPs) reveal promiscuous disruption of multiple currents and conservation of mechanism. FASEB J. 16:390–400. [DOI] [PubMed] [Google Scholar]
- Abbott, G. W., and S. A. N. Goldstein. 2001. Potassium channel subunits encoded by the KCNE gene family: physiology and pathophysiology of the MinK-related peptides (MiRPs). Mol. Intervent. 1:95–107. [PubMed] [Google Scholar]
- Abbott, G. W., F. Sesti, I. Splawski, M. Buck, M. H. Lehmann, K. W. Timothy, M. T. Keating, and S. A. N. Goldstein. 1999. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell. 97:175–187. [DOI] [PubMed] [Google Scholar]
- Armstrong, C. M. 1971. Interaction of tetraethylammonium ion derivatives with the potassium channels of giant axons. J. Gen. Physiol. 58:413–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barhanin, J., F. Lesage, E. Guillemare, M. Fink, M. Lazdunski, and G. Romey. 1996. K(V)LQT1 and IsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature. 384:78–80. [DOI] [PubMed] [Google Scholar]
- Bockenhauer, D., N. Zilberberg, and S. A. N. Goldstein. 2001. KCNK2: reversible conversion of a hippocampal potassium leak into a voltage-dependent channel. Nat. Neurosci. 4:486–491. [DOI] [PubMed] [Google Scholar]
- Busch, A. E., G. L. Busch, E. Ford, H. Suessbrich, H. J. Lang, R. Greger, K. Kunzelmann, B. Attali, and W. Stuhmer. 1997. The role of the IsK protein in the specific pharmacological properties of the IKs channel complex. Br. J. Pharmacol. 122:187–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Camino, D., M. Holmgren, Y. Liu, and G. Yellen. 2000. Blocker protection in the pore of a voltage-gated K+ channel and its structural implications. Nature. 403:321–325. [DOI] [PubMed] [Google Scholar]
- Duggal, P., M. R. Vesely, D. Wattanasirichaigoon, J. Villafane, V. Kaushik, and A. H. Beggs. 1998. Mutation of the gene for IsK associated with both Jervell and Lange-Nielsen and Romano-Ward forms of long-QT syndrome. Circulation. 97:142–146. [DOI] [PubMed] [Google Scholar]
- Goldstein, S. A., and C. Miller. 1991. Site-specific mutations in a minimal voltage-dependent K+ channel alter ion selectivity and open-channel block. Neuron. 7:403–408. [DOI] [PubMed] [Google Scholar]
- Jiang, Y. X., A. Lee, J. Chen, M. Cadene, B. T. Chait, and R. MacKinnon. 2002. Crystal structure and mechanism of a calcium-gated potassium channel. Nature. 417:515–522. [DOI] [PubMed] [Google Scholar]
- Kaczmarek, L. K., and E. M. Blumenthal. 1997. Properties and regulation of the minK potassium channel protein. Physiol. Rev. 77:627–641. [DOI] [PubMed] [Google Scholar]
- Kurokawa, J., H. K. Motoike, and R. S. Kass. 2001. TEA(+)-sensitive KCNQ1 constructs reveal pore-independent access to KCNE1 in assembled I-Ks channels. J. Gen. Physiol. 117:43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, C., R. Latorre, and I. Reisin. 1987. Coupling of voltage-dependent gating and Ba2+ block in the high-conductance, Ca2+-activated K+ channel. J. Gen. Physiol. 90:427–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor, D. L., Jr., S. J. Masseling, Y. N. Jan, and L. Y. Jan. 1999. Transmembrane structure of an inwardly rectifying potassium channel. Cell. 96:879–891. [DOI] [PubMed] [Google Scholar]
- Pusch, M. 1998. Increase of the single-channel conductance of KvLQT1 potassium channels induced by the association with MinK. Pflugers Arch. 437:172–174. [DOI] [PubMed] [Google Scholar]
- Romey, G., B. Attali, C. Chouabe, I. Abitbol, E. Guilemare, J. Barhanin, and M. Lazdunski. 1997. Molecular mechanism and functional significance of the MinK control of the KvLQT1 channel activity. J. Biol. Chem. 272:16713–16716. [DOI] [PubMed] [Google Scholar]
- Sanguinetti, M. C., M. E. Curran, A. Zou, J. Shen, P. S. Spector, D. L. Atkinson, and M. T. Keating. 1996. Coassembly of K(V)Lqt1 and MinK (IsK) proteins to form cardiac IKs potassium channel. Nature. 384:80–83. [DOI] [PubMed] [Google Scholar]
- Schulze-Bahr, E., Q. Wang, H. Wedekind, W. Haverkamp, Q. Chen, and Y. Sun. 1997. KCNE1 mutations cause Jervell and Lange-Nielsen syndrome. Nat. Genet. 17:267–268. [DOI] [PubMed] [Google Scholar]
- Sesti, F., G. W. Abbott, J. Wei, K. T. Murray, S. Saksena, P. J. Schwartz, S. G. Priori, D. M. Roden, A. L. J. George, and S. A. N. Goldstein. 2000a. A common polymorphism associated with antibiotic-induced cardiac arrhythmia. Proc. Natl. Acad. Sci. USA. 97:10613–10618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesti, F., and S. A. N. Goldstein. 1998. Single-channel characteristics of wildtype IKs channels and channels formed with two MinK mutants that cause long-QT syndrome. J. Gen. Phys. 112:651–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesti, F., K. K. Tai, and S. A. N. Goldstein. 2000b. MinK endows the IKs potassium channel with sensitivity to internal TEA. Biophys. J. 79:1369–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spassova, M., and Z. Lu. 1998. Coupled ion movement underlies rectification in an inward-rectifier K+ channel. J. Gen. Physiol. 112:211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spassova, M., and Z. Lu. 1999. Tuning the voltage dependence of tetraethylammonium block with permeant ions in an inward-rectifier K+ channel. J. Gen. Physiol. 114:415–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splawski, I., M. Tristani-Firouzi, M. H. Lehmann, M. C. Sanguinetti, and M. T. Keating. 1997. Mutations in the hminK gene cause long-QT syndrome and suppress IKs function. Nat. Genet. 17:338–340. [DOI] [PubMed] [Google Scholar]
- Tai, K.-K., K.-W. Wang, and S. A. N. Goldstein. 1997. MinK potassium channels are heteromultimeric complexes. J. Biol. Chem. 272:1654–1658. [DOI] [PubMed] [Google Scholar]
- Tai, K. K., and S. A. N. Goldstein. 1998. The conduction pore of a cardiac potassium channel. Nature. 391:605–608. [DOI] [PubMed] [Google Scholar]
- Tapper, A. R., and A. L. J. George. 2001. Location and orientation of MinK within the IKs potassium channel complex. J. Biol. Chem. 276:38249–38254. [DOI] [PubMed] [Google Scholar]
- Tyson, J., L. Tranebjaerg, S. Bellman, C. Wren, J. F. Taylor, J. Bathen, B. Aslaksen, S. J. Sorland, O. Lund, S. Malcolm, M. Pembrey, S. Bhattacharya, and M. Bitner-Glindzicz. 1997. IsK and KvLQT1: mutation in either of the two subunits of the slow component of the delayed rectifier potassium channel can cause Jervell and Lange-Nielsen syndrome. Hum. Mol. Genet. 6:2179–2185. [DOI] [PubMed] [Google Scholar]
- Wang, K.-W., K.-K. Tai, and S. A. N. Goldstein. 1996. MinK residues line a potassium channel pore. Neuron. 16:571–577. [DOI] [PubMed] [Google Scholar]
- Wang, K. W., and S. A. N. Goldstein. 1995. Subunit composition of MinK potassium channels. Neuron. 14:1303–1309. [DOI] [PubMed] [Google Scholar]
- Wang, W., J. Xia, and R. S. Kass. 1998. MinK-KvLQT1 fusion proteins, evidence for multiple stoichiometries of the assembled IsK channel. J. Biol. Chem. 273:34069–34074. [DOI] [PubMed] [Google Scholar]
- Woodhull, A. M. 1973. Ionic blockage of sodium channels in nerve. J. Gen. Physiol. 61:687–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y., and F. Sigworth. 1998. Single-channel properties of IKs potassium channels. J. Gen. Physiol. 112:665–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellen, G. 1998. The moving parts of voltage-gated ion channels. Q. Rev. Biophys. 31:239–295. [DOI] [PubMed] [Google Scholar]
- Yellen, G., D. Sodickson, T. Y. Chen, and M. E. Jurman. 1994. An engineered cysteine in the external mouth of a K+ channel allows inactivation to be modulated by metal binding. Biophys. J. 66:1068–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Y., J. H. Morais-Cabral, A. Kaufman, and R. MacKinnon. 2001. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 Å resolution. Nature. 414:43–48. [DOI] [PubMed] [Google Scholar]