

Abstract
Ca2+-induced Ca2+-release (CICR)—the mechanism of cardiac excitation-contraction (EC) coupling—also contributes to skeletal muscle contraction; however, its properties are still poorly understood. CICR in skeletal muscle can be induced independently of direct, calcium-independent activation of sarcoplasmic reticulum Ca2+ release, by reconstituting dysgenic myotubes with the cardiac Ca2+ channel α1C (CaV1.2) subunit. Ca2+ influx through α1C provides the trigger for opening the sarcoplasmic reticulum Ca2+ release channels. Here we show that also the Ca2+ channel α1D isoform (CaV1.3) can restore cardiac-type EC-coupling. GFP-α1D expressed in dysgenic myotubes is correctly targeted into the triad junctions and generates action potential-induced Ca2+ transients with the same efficiency as GFP-α1C despite threefold smaller Ca2+ currents. In contrast, GFP-α1A, which generates large currents but is not targeted into triads, rarely restores action potential-induced Ca2+ transients. Thus, cardiac-type EC-coupling in skeletal myotubes depends primarily on the correct targeting of the voltage-gated Ca2+ channels and less on their current size. Combined patch-clamp/fluo-4 Ca2+ recordings revealed that the induction of Ca2+ transients and their maximal amplitudes are independent of the different current densities of GFP-α1C and GFP-α1D. These properties of cardiac-type EC-coupling in dysgenic myotubes are consistent with a CICR mechanism under the control of local Ca2+ gradients in the triad junctions.
INTRODUCTION
Excitation-contraction (EC) coupling in muscle depends on the close interaction of a voltage-gated Ca2+ channel in the t-tubule or the plasma membrane with a Ca2+ release channel (ryanodine receptor, RyR) in the sarcoplasmic reticulum (SR). In cardiac EC-coupling this interaction is mediated by Ca2+ entering the cell through a voltage-gated Ca2+ channel that in turn activates Ca2+ release from the SR. In skeletal muscle, SR Ca2+ release is activated in the absence of Ca2+ influx, presumably by a physical interaction of the two Ca2+ channels. Under experimental conditions, the skeletal muscle Ca2+ release channel (RyR1) can also be activated by μM concentrations of cytoplasmic free Ca2+ (Nagasaki and Kasai, 1983; Smith et al., 1986). Whether Ca2+-induced Ca2+-release (CICR) in skeletal muscle occurs under physiological conditions, and if so, to what extent it contributes to skeletal muscle EC-coupling is still not resolved.
Reconstitution of Ca2+ channel null-mutant skeletal myotubes with wild-type and mutant heterologous Ca2+ channels has been a potent tool for analyzing the mechanism of EC-coupling in an intact muscle system. Normal function can be restored in dysgenic myotubes, which lack the skeletal muscle Ca2+ channel α1S subunit, by heterologous expression of α1S (Tanabe et al., 1988). When dysgenic myotubes are reconstituted with the cardiac Ca2+ channel α1C subunit, “cardiac-type” EC-coupling is restored, which is characterized by its dependence on Ca2+ influx (Tanabe et al., 1990). Presumably the skeletal muscle Ca2+ release channel is activated by trigger Ca2+ entering the myotube through α1C. However, not all examined Ca2+ channel isoforms that produced Ca2+ currents when expressed in dysgenic myotubes also triggered CICR from the SR. The neuronal isoform α1A, which is not targeted into triads of dysgenic myotubes, only very rarely displayed evoked contractions (Adams et al., 1994) or Ca2+ transients in response to electrically induced action potentials (action potential-induced Ca2+ transients) (Flucher et al., 2000). Apparently the correct subcellular distribution of the channel is important for the activation of cardiac-type EC-coupling in skeletal myotubes. Skeletal-type EC-coupling properties could be conferred onto the cardiac α1C subunit by replacing a short sequence of the cytoplasmic loop connecting the homologous repeats II and III with that of α1S (Nakai et al., 1998). Thus, the chimera CSk53 was created, which combines skeletal-type activation of EC-coupling with cardiac current properties.
If CICR participates in normal skeletal muscle EC-coupling, it is unlikely that the trigger Ca2+ comes from the Ca2+ currents across the sarcolemma, because the slow activation of the skeletal Ca2+ current lags behind activation of SR calcium release (Garcia et al., 1989; Feldmeyer et al., 1990). Instead, trigger Ca2+ must be provided by Ca2+ released from the SR by the population of the skeletal muscle RyR1 that is under the direct control of the voltage-sensor α1S subunit (reviewed in Rios and Stern, 1997). In other words, Ca2+ released through the RyR1 provides a positive feedback resulting in release of more Ca2+ from the same or adjacent RyRs. A recent report by O'Brien et al. (2002) indicates that, whereas CICR appears not to be necessary for activation of EC-coupling in skeletal muscle, SR Ca2+ release is reduced by fivefold when Ca2+ activation of RyR1 is impeded. This indicates a significant contribution of CICR to skeletal muscle EC-coupling.
Here we studied the properties of CICR in skeletal myotubes separate from skeletal-type activation of Ca2+ release by expressing nonskeletal α1 subunits with distinct current characteristics and with distinct targeting characteristics in dysgenic myotubes. Analyzing the capability of different types of Ca2+ channels to generate action potential-induced and voltage-clamp-induced Ca2+ transients indicates that in the absence of direct skeletal coupling, Ca2+ currents of similar size as those of the skeletal α1S are sufficient to trigger CICR. Moreover, the results indicate that activation of SR Ca2+ release seems to depend less on the magnitude of the whole-cell current than on the correct localization of the channels in the triads.
MATERIALS AND METHODS
Cell culture and transfections
Myotubes of the homozygous dysgenic (mdg/mdg) cell line GLT were cultured as described in Powell et al. (1996). At the onset of myoblast fusion (2 days after addition of differentiation medium), GLT cultures were transfected using FuGene transfection reagent (Roche, Basel, Switzerland). Cultures were analyzed 3–5 days after transfection.
α1 subunit constructs
The following GFP-tagged Ca2+ channel constructs have been used: α1S, α1A, α1C (Grabner et al., 1998), α1D (Koschak et al., 2001), and α1G (Monteil et al., 2000). GFP-CSk53: The BamHI-EcoRV fragment (nt 1265–4351; α1C numbering) of clone CSk53 (Nakai et al., 1998), where part of the II-III loop coding α1C cDNA sequence (nt 2549–2690) was replaced by α1S sequence (nt 2156–2297), was ligated into the corresponding restriction enzyme sites of clone GFP-α1C (Grabner et al., 1998) resulting in an N-terminally GFP-tagged CSk53.
GFP and immunofluorescence labeling
Differentiated GLT cultures were fixed and immunostained as previously described (Flucher et al., 1994; Flucher et al., 2000). For double-immunofluorescence labeling we used an affinity-purified anti-GFP antibody (Molecular Probes, Eugene, OR) at a final dilution of 1:4000 and the affinity-purified antibody 162 (Giannini et al., 1995) against RyR1 at a dilution of 1:5000. Alexa-488- and Alexa-594-conjugated secondary antibodies (Molecular Probes) at dilutions of 1:4000 were used to achieve a wide separation of the fluorescence signals. Alexa-488 was usually used with the anti-GFP antibody so that the antibody label and the intrinsic GFP signal were both recorded in the green channel. Controls, for example multiple combinations of primary and secondary antibodies and the omission of primary antibodies, were routinely performed.
Images of the double labeling experiments were recorded on an Axiophot microscope (Carl Zeiss, Jena, Germany) using a cooled CCD camera and Meta View image processing software (Universal Imaging Corporation, West Chester, PA). Quantitative analysis of the labeling patterns was performed by systematically screening the cover glasses for transfected myotubes using a 63× objective. The labeling pattern in transfected myotubes with more than two nuclei was classified as “clustered” when punctuate α1 subunit fluorescence was co-localized with a similar RyR1 label (Fig. 1). The counts were obtained from several samples of at least three different experiments.
FIGURE 1.
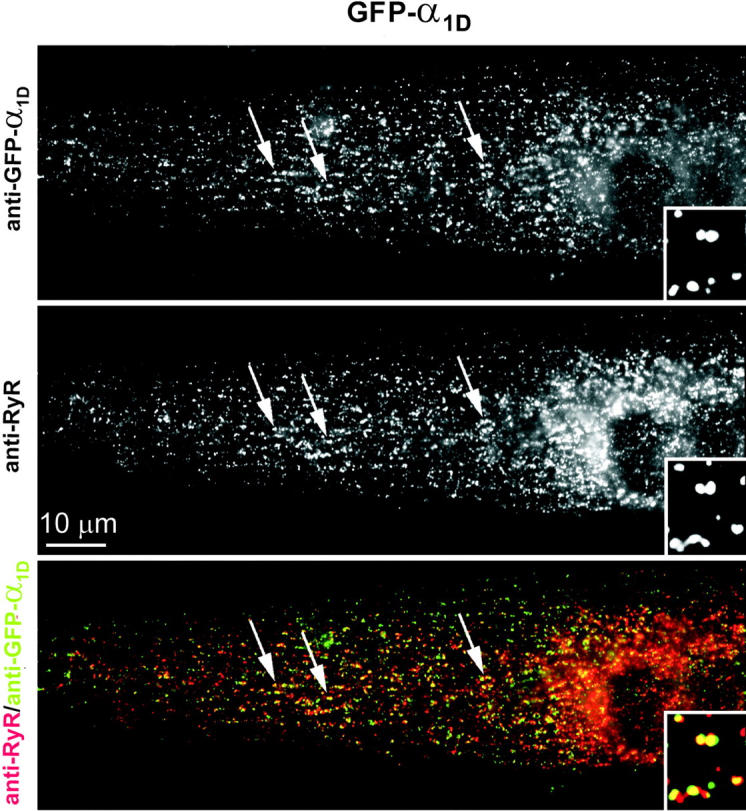
The L-type Ca2+ channel subunit GFP-α1D is targeted into skeletal muscle triads of dysgenic myotubes. Dysgenic myotubes transfected with GFP-α1D were double immunofluorescence labeled with antibodies against GFP (top) and against the skeletal muscle RyR1 (middle). GFP-α1D is distributed in clusters co-localized with RyR1 (examples indicated by arrows), which indicates a localization of GFP-α1D in junctions of the SR with the plasma membrane or the t-tubules (triads). The color overlay (bottom) shows the co-localization of GFP-α1D clusters (green) and RyR1 clusters (red) as yellow foci. The inset shows a fourfold enlarged area. Bar, 10 μm.
Patch-clamp and intracellular Ca2+ recording
Whole-cell patch-clamp recordings were performed with an Axopatch 200A amplifier controlled by pClamp 8.0 software (Axon Instruments Inc., Union City, CA, USA). The bath solution contained (mM): 10 CaCl2, 145 tetraethylammonium chloride, and 10 HEPES (pH 7.4 with TEA-OH). Patch pipettes, pulled from borosilicate glass (Harvard Apparatus, Kent, UK) had resistances of 1.8 to 2.5 MΩ when filled with 145 Cs-aspartate, 2 MgCl2, 10 HEPES, 0.1 Cs-EGTA, 2 Mg-ATP (pH 7.4 with Cs-OH). For simultaneous recording of whole-cell currents and Ca2+ transients the pipette solution additionally contained 0.2 mM fluo-4-K5 (Molecular Probes). The Ca2+ fluorescence signal was recorded by a photometer system (PTI, S. Brunswick, NJ, USA) adjusted to the Zeiss Axiovert epifluorescence microscope. Ca2+ transients were normalized by the resting fluorescence (ΔF/F). To attempt block of SR Ca2+ release, ryanodine and ruthenium red (both Sigma-Aldrich, Vienna, Austria) were dissolved in absolute ethanol or water, respectively, and added to the pipette solution at final concentrations of 10–40 μM for ryanodine and 10–160 μM for ruthenium red.
Leak currents were digitally subtracted by a P/4 prepulse protocol. Recordings were low-pass Bessel filtered at 1 kHz and sampled at 5 kHz. Currents were determined with 200 ms depolarizing steps from a holding potential of −80 mV to test potentials between −40 and +80 mV in 10 mV increments. Test pulses were preceded by a 1-s prepulse to −30 mV to inactivate endogenous T-type Ca2+ currents (Adams et al., 1990). Current recordings of cells transfected with the T-type Ca2+ channel α1G subunit were measured with 200 ms depolarizing steps from a holding potential of −90 mV to test potentials between −80 mV and +80 mV in 10 mV increments. Ca2+ current densities were normalized by linear cell capacitance and expressed in pA/pF.
Action potential–induced Ca2+ transients were recorded in cultures loaded for 45–60 min at room temperature with 5 μM fluo-4-AM plus 0.1% Pluronic F-127 (Molecular Probes) in HEPES and bicarbonate-buffered DME, as previously described (Flucher et al., 1993; Powell et al., 1996). Action potentials were elicited by passing 1 ms pulses of 30 V across the 19-mm incubation chamber. 0.5 mM Cd2+ and 0.1 mM La3+ were added to block Ca2+ influx and therefore allow discrimination between CICR and skeletal-type EC coupling. Application of 6 mM caffeine to the bath solution resulted in rapid Ca2+ release and thus proved the capacity of SR Ca2+ release.
Statistics
Data were expressed as mean ± SE where n is the number of cells examined. Data analysis, unpaired Student's t-test, and one-way-ANOVA analysis followed by the Tukey post test were performed with Clampfit 8.0 (Axon instruments, Union City, CA, USA) and GraphPad Prism software (GraphPad Inc., San Diego, CA, USA). P < 0.05 was considered statistically significant.
RESULTS
It has previously been demonstrated that heterologous expression of different muscle α1 subunit isoforms in dysgenic myotubes restores EC-coupling by two distinct mechanisms. Whereas the skeletal muscle α1S triggers SR Ca2+ release independently of Ca2+ influx through the L-type Ca2+ channel, the cardiac α1C depends on Ca2+ influx for activation of EC-coupling, presumably by CICR (Garcia et al., 1994). In contrast to the two muscle isoforms α1S and α1C, the neuronal non-L-type α1A subunit was not capable of restoring EC-coupling in dysgenic myotubes, although α1A generated sizable Ca2+ currents (Adams et al., 1994; Flucher et al., 2000). Here we expressed α1S, α1C, α1A, and for the first time the L-type Ca2+ channel α1 subunit isoform α1D in dysgenic myotubes to compare their current properties and targeting characteristics with their ability to restore EC-coupling.
GFP-α1D is targeted into triads of dysgenic myotubes
Immunofluorescence analysis (Fig. 1) shows that a fusion protein of GFP attached to the N-terminus of α1D (GFP-α1D) is efficiently expressed in myotubes of the dysgenic cell line GLT and is distributed in a clustered pattern. Double labeling of GFP-α1D and RyR1 demonstrated that GFP-α1D clusters were co-localized with clusters of RyR1, thus identifying the clusters as t-tubule/SR or plasma membrane/SR junctions, all of which will from here on be called triads. GFP-α1D/RyR1 coclustering was found in 62% of 454 analyzed GFP-α1D-expressing myotubes, which was similar to the degree of clustering obtained with the native α1S or with α1C (see also Fig. 4 C). Myotubes in which GFP-α1D was expressed in a nonclustered pattern typically showed ER/SR labeling (not shown), presumably because these myotubes were still very immature or due to unbalanced expression of the heterologous channel and endogenous muscle proteins (Flucher et al., 1994). Localizing GFP-α1D (Fig. 1, green) in clusters coincident with the RyR (Fig. 1, red), makes α1D the third member of the L-type Ca2+ channel family that, when expressed in skeletal muscle cells, is targeted into triads.
FIGURE 4.
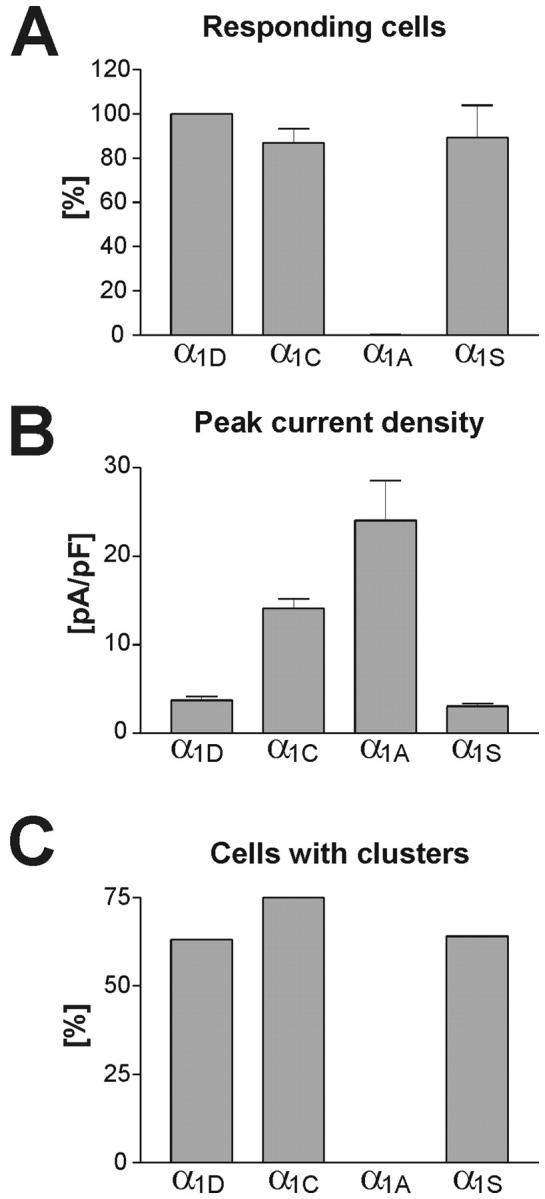
The ability of Ca2+ channel isoforms to restore EC-coupling compared to their current densities and targeting properties. (A) Myotubes responding to field stimulation with Ca2+ transients (see Fig. 3) were counted in cultures transfected with GFP-α1D, GFP-α1C, GFP-α1A, and GFP-α1S. Counts from seven experiments are expressed in percent (mean ± SE) with the values of GFP-α1D normalized to 100%. All three L-type channels (GFP-α1D, GFP-α1C, GFP-α1S) restored EC-coupling with similar efficiency, whereas with GFP-α1A only a single responding cell was observed. (B) Average peak current densities shown for the same channel isoforms as above (see also Table 1). Although the nonskeletal α1 isoforms activate action potential-induced Ca2+ transients by CICR, no correlation of current density and restoration of EC-coupling exists (cf. A and B). (C) Fraction of myotubes in which the channel isoforms were targeted into the triads (see Fig. 1). The triad targeting properties of the Ca2+ channel isoforms exactly correspond to their ability to restore EC-coupling (cf. A and C).
GFP-α1D restores Ca2+ currents in dysgenic myotubes
Whole-cell patch-clamp measurements of dysgenic myotubes expressing GFP-α1D revealed Ca2+ currents with properties distinct from those of GFP-tagged skeletal and cardiac α1 subunits (Fig. 2). Myotubes were selected for electrophysiological analysis based on the GFP fluorescence. Ca2+ currents in response to depolarizing steps from a holding potential of −80 mV to voltages between −40 and +80 mV in 10 mM extracellular Ca2+ were recorded and used to calculate the current-to-voltage (I/V) relationship of GFP-α1D. Representative current traces for the three α1 subunit isoforms are shown in Fig. 2 C. Comparing the average I/V curves of GFP-α1D currents to those of GFP-α1S and GFP-α1C expressed in dysgenic myotubes (Fig. 2 A) shows that the average maximal current density of GFP-α1D is 3.7 ± 0.4 pA/pF, which is close to the value obtained with GFP-α1S but significantly lower (P < 0.01) than the average current density of GFP-α1C (Table 1). Furthermore, GFP-α1D activates at more negative potentials than the two other α1 subunits. The point of half-maximal activation for GFP-α1D is ∼13 mV more negative than that of GFP-α1C and 37 mV more negative than that of GFP-α1S (Table 1). The distinct activation characteristics can be best appreciated when the I/V curves are normalized for maximal amplitudes (Fig. 2 B). This analysis demonstrates that GFP-α1D is functionally expressed in dysgenic myotubes and that its activation characteristics resemble those reported for α1D in native cells and for GFP-α1D in a nonmuscle expression system (Zidanic and Fuchs, 1995; Koschak et al., 2001).
FIGURE 2.

Current properties of GFP-α1D expressed in dysgenic myotubes compared to those of GFP-α1S and GFP-α1C. (A) Voltage dependence of peak Ca2+ current densities (pA/pF; mean ± SE) of GFP-α1D (▪; n = 42), GFP-α1S (□; n = 33), and GFP-α1C (•; n = 28). Maximal current density of GFP-α1D is similar to that of GFP-α1S and about four times lower than that of GFP-α1C. (B) Normalized I/V curves show that GFP-α1D activates at 13 mV more negative potentials than GFP-α1C and at 37 mV more negative potentials than GFP-α1S (for values of half maximal activation, see Table 1). (C) Representative whole-cell current recordings of GFP-α1D, GFP-α1S, and GFP-α1C expressed in dysgenic myotubes. Cells were held at −80 mV and after a prepulse to inactivate low-voltage activated currents test pulses to increasing voltages from −40 to +80 mV were applied; currents were recorded in 10 mM Ca2+. GFP-α1D activates faster than GFP-α1S.
TABLE 1.
Properties of Ca2+ currents and Ca2+ transients recorded in reconstituted dysgenic myotubes with combined patch-clamp and fluo-4 Ca2+ measurements
| α1-subunit | Current density (pA/pF) | Gmax (nS/nF) | V1/2 (mV) | k (mV) | (ΔF/F)max |
|---|---|---|---|---|---|
| α1D | 3.7 ± 0.4 (42) | 51.3 ± 5.3 | −0.6 ± 1.2 | 6.7 ± 0.3 | 0.27 ± 0.03 *** (9) |
| α1S | 3.1 ± 0.3 (33) | 112.9 ± 6.8 | 36.0 ± 1.4 | 7.5 ± 0.3 | 0.96 ± 0.09 (34) |
| α1C | 14.1 ± 1.1** (28) | 236.2 ± 20.2 | 12.2 ± 1.6 | 6.2 ± 0.4 | 0.30 ± 0.03 *** (10) |
| CSk53 | 22.3 ± 2.2*** (35) | 177.7 ± 18.0 | 5.9 ± 1.7 | 5.8 ± 0.5 | 0.41 ± 0.03 *** (18) |
| α1A | 24.1 ± 4.5*** (18) | 264.7 ± 55.9 | 6.0 ± 1.2 | 1.0 ± 0.2 | 0.50 ± 0.12 ** (12) |
Entries correspond to mean ± SE; numbers of cells are given in parentheses.
Significance (*** P < 0.001; ** P < 0.01) is given in relation to α1S.
(ΔF/F)max values other than that of α1S are not significantly different from one another.
GFP-α1D restores cardiac-type EC-coupling in dysgenic myotubes
Next it was of interest to examine whether the small Ca2+ currents generated by GFP-α1D would be sufficient to stimulate EC-coupling in dysgenic myotubes. This was first analyzed by recording intracellular Ca2+ signals in response to 1-ms current pulses passed through the recording chamber, in myotubes loaded with the fluorescent Ca2+ indicator fluo-4-AM. In normal myotubes and in dysgenic myotubes transfected with GFP-α1S or GFP-α1C, this field stimulation protocol has previously been shown to elicit Ca2+ transients with an all-or-none response to increasing voltages. This characteristic identifies the transients as action potential-induced Ca2+ transients (Flucher et al., 1993), which represent the most physiological parameter for assessing the restoration of EC-coupling. However, even in cultures transfected with the native GFP-α1S, action potential-induced Ca2+ transients are observed only in a subset of expressing myotubes, presumably because excitability is achieved only in well differentiated cultured myotubes.
Cultures transfected with GFP-α1D responded to field stimulation with brief Ca2+ transients (Fig. 3). The transients were characterized by a rapid upstroke that terminated abruptly, indicative of a strong voltage-dependence of both, the activation and the deactivation of the EC-coupling mechanism. The average amplitudes of the transients reached a (ΔF/F)max of 0.46 ± 0.04, which was not significantly different from the amplitudes recorded with GFP-α1C or GFP-α1S (Table 2). The speed of the upstroke and the decay of the transients were similar between GFP-α1D and GFP-α1C; however, the average values obtained with these channel isoforms were significantly slower than those of GFP-α1S. Unless the expression of nonskeletal channels adversely affects the overall differentiation of the myotubes, for which there is no independent evidence based on the morphology of the myotubes and on the expression of proteins seen with immunocytochemistry, this difference in the time course of the transients probably reflects the different modes of EC-coupling (see below). But most important, despite the differences observed in the whole-cell current properties (Fig. 2), time-to-peak, amplitudes, and time constant of the decline of the Ca2+ transients were not significantly different (P>0.05) between GFP-α1D and GFP-α1C (Table 2).
FIGURE 3.

Restoration of action potential-induced Ca2+ transients by GFP-α1D in dysgenic myotubes. Field stimulation at low frequency (30 V, 1 ms, 0.3 Hz/0.5 Hz) evoked rapid Ca2+ transients in myotubes expressing GFP-α1D, GFP-α1C, or GFP-α1S. Bath application of 0.5 mM Cd2+/0.1 mM La3+ immediately stopped the activation of transients in myotubes expressing GFP-α1D (A) and GFP-α1C (B), indicating that the transients were dependent on Ca2+ influx through the L-type channels. Addition of 6 mM caffeine (A, right trace) triggered a strong Ca2+ transient, proving that SR Ca2+ release was still functional during the Cd2+/La3+ block. (C) Persistent Ca2+ transients after addition of Cd2+/La3+ in myotubes expressing GFP-α1S characterizes skeletal-type EC-coupling, which is independent of Ca2+ influx. In contrast, GFP-α1D restored action potential-induced Ca2+ transients with cardiac characteristics. Marks underneath the traces indicate the approximate times of stimulation; bars indicate periods of Cd2+/La3+ and caffeine application.
TABLE 2.
Properties of action potential-induced Ca2+ transients recorded in reconstituted dysgenic myotubes with fluo-4 Ca2+ measurements
| α1-subunit | Time to peak (s) | (ΔF/F)max | Decay time constant τ (s) | N |
|---|---|---|---|---|
| α1S | 0.068 ± 0.010 | 0.65 ± 0.09 | 0.255 ± 0.019 | 26 |
| α1D | 0.370 ± 0.057 *** | 0.46 ± 0.04 | 1.408 ± 0.170 *** | 9 |
| α1C | 0.257 ± 0.030 *** | 0.55 ± 0.10 | 1.113 ± 0.103 *** | 18 |
Entries correspond to mean ± SE. Significance (*** P < 0.001) is given in relation to α1S; values of α1D and α1C are not significantly different from each other (P>0.05).
When Ca2+ currents were blocked by the addition of 0.5 mM Cd2+/0.1 mM La3+ to the bath solution while continuously stimulating, the action potential-induced Ca2+ transients in GFP-α1D-transfected myotubes ceased immediately (Fig. 3 A). This dependence of the Ca2+ transients on Ca2+ influx was similarly observed with GFP-α1C (Fig. 3 B) and is the hallmark of cardiac-type EC-coupling. In contrast, because skeletal EC-coupling is independent of Ca2+ influx, blocking Ca2+ currents with Cd2+/La3+ in myotubes transfected with GFP-α1S did not stop action potential-induced Ca2+ transients (Fig. 3 C). To make sure that the inhibition of Ca2+ transients in myotubes expressing GFP-α1D was indeed due to blocking the activation mechanism, SR Ca2+ release was tested by the application of 6 mM caffeine immediately after a blocking experiment. The strong Ca2+ release induced by caffeine (Fig. 3 A, right trace) showed that the failing response to field stimulation in Cd2+/La3+ was not due to depletion of the SR or other effects on the release apparatus. Thus, GFP-α1D restored cardiac-type EC-coupling in dysgenic myotubes.
Restoration of cardiac-type EC-coupling depends on triad targeting but not on the magnitude of the current density
In the field stimulation experiments GFP-α1D was as efficient as GFP-α1C in restoring action potential-induced Ca2+ transients in dysgenic myotubes. In Fig. 4 A we compare the frequencies at which action potential-induced Ca2+ transients were observed in cultures transfected with GFP-α1D, GFP-α1C, GFP-α1A, and GFP-α1S. The number of responding cells in cultures transfected with GFP-α1D was not significantly different from those of GFP-α1C and GFP-α1S. As previously shown (Flucher et al., 2000), GFP-α1A very rarely produced action potential-induced Ca2+ transients, probably due to the fact that this channel is not targeted into the triads. Out of six culture dishes transfected with GFP-α1A, only a single myotube in one dish responded with an action potential-induced Ca2+ transient; therefore, the bar is not visible at this scale (Fig. 4 A). Comparing the capability of each of these channel isoforms to restore EC-coupling with the average densities of their Ca2+ currents shows no correlation of the two parameters (cf. Fig. 4, A and B). For GFP-α1S, which directly activates SR Ca2+ release, such a correlation would not have been expected. However, for the constructs which elicit cardiac-type EC-coupling—presumably by CICR—a correlation of current density and their ability to activate action potential-induced Ca2+ transients was to be expected. Most interestingly, GFP-α1D, with a current density of less than a third of that of GFP-α1C, activated EC-coupling just as efficiently as the cardiac channel. In contrast to current density, restoration of EC-coupling correlated very well with the triad targeting properties of the individual channel isoforms (cf. Fig. 4, A and C). Those constructs which are targeted into the triads (GFP-α1S, GFP-α1C, GFP-α1D) all activated EC-coupling with similar efficiency. However GFP-α1A, which generated the largest current density of all examined channel isoforms (see also Table 1) but is not targeted into triads, hardly ever activated action potential-induced Ca2+ transients in dysgenic myotubes. Thus, for the activation of cardiac-type EC-coupling, the spatial arrangement of the Ca2+ channels opposite the RyR appears to be more important than the pure size of the Ca2+ currents.
Ca2+ transients activated by GFP-α1C and GFP-α1D are qualitatively and quantitatively different from those activated by GFP-α1S
To further characterize the dependence of cardiac-type EC-coupling on Ca2+ currents we performed combined patch-clamp and fluo-4 Ca2+ recording experiments in dysgenic myotubes expressing different α1 subunit isoforms. This approach enables us to simultaneously monitor the whole-cell Ca2+ influx and intracellular Ca2+ transients in the same myotubes, and it allows us to analyze the voltage-dependence of Ca2+ transients. But these experiments differ from the analysis of action potential-induced Ca2+ transients in several important aspects: 1), Sampling—whereas with field stimulation we see and record only those myotubes that have achieved excitability and a good degree of EC-coupling (probably the peak of the population of expressing cells), with the patch-clamp approach we get a random sample of differently well developed and expressing myotubes; 2), Stimulation—instead of a 1-ms extracellular current pulse, which stimulates an action potential lasting only a few milliseconds, in the patch-clamp experiments we depolarize the cells for 200 ms giving rise to unnaturally long periods of Ca2+ influx; and 3), Intracellular milieu—while the myotube is being loaded with the Ca2+ indicator through the patch pipette, the cells are dialyzed and Ca2+ buffers and the entire intracellular milieu are altered. Thus, in this set of experiments we trade in better control and quantitative analysis for less physiological conditions. This is reflected in the shape of the Ca2+ transients in response to the 200-ms depolarization (Fig. 5 C), which does not resemble that of action potential-induced Ca2+ transients (Fig. 3).
FIGURE 5.

Simultaneous recording of whole-cell Ca2+ currents and Ca2+ transients in dysgenic myotubes transfected with skeletal and nonskeletal α1 subunit isoforms. (A, B) Voltage-dependence of Ca2+ transients (A) and of Ca2+ currents (B). Ca2+ transients of GFP-α1S (▪; n = 34) have a sigmoidal voltage relationship that does not follow the I/V curve. Transient-to-voltage curves for GFP-α1C (•; n = 10), GFP-α1D (□; n = 9), and GFP-α1A (○; n = 11) are bell-shaped and follow their I/V curves with respect to activation properties but not with respect to amplitudes. Whereas current densities for GFP-α1D are significantly smaller than those of GFP-α1C (P = 0.0011) or GFP-α1A (P = 0.014), their maximal amplitudes of Ca2+ transients are not significantly different from each other (P > 0.05). The dotted line shows the shift in the GFP-α1A I/V curve after removing one exceptionally high recording from the analysis. But transients of all nonskeletal isoforms are significantly smaller (P < 0.001) than that of GFP-α1S. (C) Representative examples of Ca2+ transients recorded with fluo-4 in parallel to whole-cell currents show the difference between skeletal and nonskeletal α1 isoforms. (D) A scatter plot of the maximal peak current densities versus peak ΔF/F values for GFP-α1S (▪), GFP-α1C (•), and GFP-α1D (□) shows that skeletal and nonskeletal Ca2+ signals clearly separate into two distinct groups. Superthreshold Ca2+ transients of GFP-α1C and GFP-α1D are confined to a narrow window of amplitudes over a wide range of current densities. (E) I/V curves of GFP-α1C (•) and GFP-α1D (□) (both the same as in B) compared to the average I/V curves of those currents for each group, which did not evoke Ca2+ transients (▴, GFP-α1C, n = 5; ▵ GFP-α1D, n = 16). For both channels, currents without transients were relatively smaller than those that evoked transients, but the absolute values differed significantly.
Nevertheless, important functional characteristics of the transients, like the skeletal- and cardiac-type activation of EC-coupling, can be distinguished in the voltage-dependence curves of Ca2+ transients (Garcia et al., 1994). The amplitudes of Ca2+ transients stimulated by GFP-α1C mirror those of the Ca2+ currents over a wide voltage range (Figs. 5, A and B). In contrast, with GFP-α1S Ca2+ transients activate at more negative potentials as the Ca2+ currents and stay fully activated at voltages near the reversal potential where currents decline. As expected from the dependence of action potential-induced Ca2+ transients on Ca2+ influx shown in Fig. 3, the voltage-dependence curves of Ca2+ transients in myotubes expressing GFP-α1D showed the cardiac characteristics (Fig. 5 A). With this construct the Ca2+ transients activated in parallel with the currents at more negative potentials than those of GFP-α1C and declined at voltages above +20 mV. Interestingly, the maximal amplitudes of the transients for GFP-α1D and GFP-α1C were not significantly different (P > 0.05) despite the more than threefold difference in current densities (Fig. 5; Table 1). This suggests that currents of both channels reach the activation threshold of CICR and trigger equal amounts of Ca2+ release. However, the maximal amplitudes of the Ca2+ transients activated by GFP-α1D or GFP-α1C were much lower than that activated by GFP-α1S. This indicates that the native channel either activates a larger Ca2+ pool or activates SR Ca2+ release more efficiently than the two nonskeletal channel isoforms.
Unexpectedly, also GFP-α1A, which in the field stimulation experiments restored action potential-induced Ca2+ transients very rarely (Fig.4 A), frequently produced cardiac-type Ca2+ transients in the combined patch-clamp/fluo-4 Ca2+ recording experiments. Apparently the mechanism that limited activation of cardiac-type EC-coupling in response to action potential-induced Ca2+ transients to the correctly targeted channel isoforms broke down under the conditions of the patch-clamp experiments. But even in this case, the average maximal amplitudes of Ca2+ transients were similar to those of GFP-α1C and GFP-α1D (Fig. 5). Whereas the transient-to-voltage relationship for GFP-α1A is somewhat higher than that of GFP-α1C and GFP-α1D (Table 1), this difference is not statistically significant (P > 0.05) and mainly reflects a single recording with an exceptionally high current and transient. If this recording is excluded from the analysis, the transients of GFP-α1A are also in the exact same range as GFP-α1D and GFP-α1C (dotted curve in Fig. 5 A). At this point we have no explanation for the occurrence of such exceptionally strong responders. However, one needs to remember that also in the field stimulation experiments occasional responders were observed with GFP-α1A (see above; Adams et al., 1994; Flucher et al., 2000).
Analysis of the combined patch-clamp/fluo-4 Ca2+ recording experiments revealed striking differences between the skeletal and nonskeletal α1 subunits expressed in dysgenic myotubes. Plotting the amplitudes of the Ca2+ transients versus the peak current densities for all individual experiments (Fig. 5 D) shows that dysgenic myotubes reconstituted with GFP-α1S produce Ca2+ transients of greatly variable size (ΔF/F values up to 2.60) with no correlation to current densities. Ca2+ transients are observed even in myotubes with no detectable currents, and some of the myotubes with the highest current densities showed very small transients. In contrast, GFP-α1C and GFP-α1D display a completely different picture. Data points of both channels spread out over a wide range of current densities but within a relatively narrow window of Ca2+ transient amplitudes (ΔF/F values between 0.15 and 0.42). The maximal transient amplitudes of both nonskeletal channel isoforms do not reach those of the skeletal GFP-α1S.
Furthermore, a fraction of GFP-α1C- and GFP-α1D-expressing myotubes showed Ca2+ currents but no detectable Ca2+ transient (33% and 64% of myotubes expressing GFP-α1C and GFP-α1D, respectively). This was never the case with GFP-α1S. In Fig. 5 E a second set of I/V curves is shown for GFP-α1C and GFP-α1D. In addition to the I/V curve of those myotubes responding with both currents and transients (same as in Fig. 5 B), I/V curves were calculated from those recordings, in which Ca2+ currents were not accompanied with Ca2+ transients. For both channel isoforms the currents not associated with Ca2+ transients were significantly smaller (α1D: P = 0.0001; α1C: P = 0.0180) than those associated with transients. Thus, in both cases the larger currents activated transients whereas the smaller currents did not. This could be interpreted as the existence of a threshold for triggering the Ca2+ signal. However, the whole-cell current densities necessary to activate Ca2+ transients differed greatly between the two channel isoforms. The amplitudes of Ca2+ currents of GFP-α1D that were associated with Ca2+ transients were almost identical to those of GFP-α1C that were not associated with transients (Fig. 5 E). Thus, whereas current amplitudes matter for the activation of Ca2+ release in skeletal myotubes, the absolute values of whole-cell current densities do not reliably describe the activation threshold in the triad.
The two distinct groups of Ca2+ transient amplitudes do not depend on the skeletal versus cardiac EC-coupling mechanisms
The data presented above show that GFP-α1C and GFP-α1D expressed in dysgenic myotubes give rise to Ca2+ transients with an average maximal amplitude at about one third of that activated by GFP-α1S (Table 1). Therefore it was reasonable to assume that the difference in skeletal versus cardiac EC-coupling mechanisms (direct or Ca2+-dependent) is responsible for this striking difference. This hypothesis was tested with a channel chimera, GFP-CSk53, which consists of GFP-α1C with a 46-amino-acid sequence in the II-III cytoplasmic loop replaced by the corresponding sequence of α1S (Nakai et al., 1998). This sequence is sufficient to confer skeletal-type EC-coupling properties onto the cardiac channel. Action potential-induced Ca2+ transients persist after application of Cd2+/La3+ to block Ca2+ currents (Fig. 6 E). This observation is in agreement with earlier reports showing that GFP-CSk53 activated EC-coupling by the skeletal Ca2+-independent mechanism (Nakai et al., 1998). Fig. 6, A–D shows the voltage-dependence of Ca2+ transients and currents of GFP-CSk53 compared to those of GFP-α1S and GFP-α1C. Peak current densities similar to those of GFP-α1C (Fig. 6 B; Table 1) demonstrate the normal functional expression of the channel chimera. But surprisingly, on first sight the voltage relationship of the transients of GFP-CSk53 resembled that of the cardiac and not of the skeletal isoforms in both amplitude and shape (Fig. 6 A). However, the analysis of individual recordings from the combined patch-clamp/fluo-4 Ca2+ experiments showed that the cardiac-like decline of the transients at voltages above +20 mV was preferentially seen in those recordings where the baseline shifted upward during the duration of the experiment. Dividing the recordings into those with baseline shifts below and those above 10% (Fig. 6 C) revealed that the records with little or no shift displayed the typical skeletal characteristics of the voltage-dependence of the Ca2+ transients, i.e., no decline at higher test potentials. The same analysis performed with the recordings of GFP-α1C showed typical cardiac characteristics in both groups (Fig. 6 D). Therefore, the apparent decline of the amplitudes of Ca2+ transients at positive potentials was an experimental artifact that could be avoided by excluding the data of recordings with a baseline shift greater than 10%.
FIGURE 6.

Comparison of the cardiac-skeletal chimera CSk53 with α1C and α1S. The voltage-dependences of Ca2+ transients (A) and of peak current densities (B) of CSk53 (□, n = 18) resemble in size and shape those of GFP-α1C (•) but not those of GFP-α1S (▪). The bell-shaped (cardiac-type) Ca2+ transient-to-voltage curve of CSk53 is in disagreement with skeletal-type EC-coupling properties as indicated by the insensitivity of action potential-induced Ca2+ transients to Cd2+/La3+ block (E). (C) Dividing the CSk53 records into two groups based on the occurrence and magnitude of a fluorescence baseline shift (above and below 10%) shows that the stable recordings (baseline shift < 10%; □, n =7; > 10%, ♦, n = 11) display a sigmoidal transient-to-voltage relationship. (D) In contrast, the transient-to-voltage curves of α1C are bell-shaped regardless of the size of a baseline shift (baseline shift < 10%, C, n = 5; >10%, •, n = 5). Therefore, the bell-shaped appearance of the curve for CSk53 in A is due to this artifact, and the true character of the CSk53 transients is skeletal. (F) A scatter plot of maximal peak current densities versus ΔF/F values emphasizes the difference between Ca2+ transients of CSk53 and GFP-α1S. Whereas current densities for CSk53 vary between 1.5 and 40 pA/pF, the amplitudes of the transients do not exceed ΔF/F values of 0.6.
GFP-CSk53 activates Ca2+ transients by the skeletal mechanism
Nevertheless, its maximal Ca2+ transient amplitudes were similar to those of the nonskeletal α1 subunits (Fig. 6, A and F). The average maximal amplitude of Ca2+ transients for GFP-CSk53 was 0.41 ± 0.03, which is not significantly different from those of GFP-α1C and GFP-α1D but significantly lower than that of GFP-α1S (P < 0.001; Table 1). Thus, the difference in amplitudes of Ca2+ transients observed in myotubes transfected with GFP-α1C and GFP-α1D on one hand, and with GFP-α1S on the other, cannot be solely explained by the difference between the cardiac and skeletal activation mechanisms.
Cardiac-type and skeletal-type Ca2+ transients can be individually activated in the same myotube
Finally, we wanted to exclude the possibility that the relatively low Ca2+ transients in myotubes transfected with nonskeletal muscle α1 subunit isoforms and the chimera CSk53 were simply due to a reduced Ca2+ release capacity of those myotubes. Therefore we examined whether the same myotube responded with different size Ca2+ transients when activated by the native GFP-α1S or by a nonskeletal channel. The low-voltage-activated Ca2+ channel GFP-α1G (Monteil et al., 2000) also gave rise to Ca2+ transients in dysgenic myotubes. These activated at potentials of −60 mV and peaked at about −40 mV, both at ∼40 mV more negative potentials than transients in GFP-α1S-expressing myotubes (Fig. 7 A). Similar to the Ca2+ transients of all other examined nonskeletal α1 subunits, GFP-α1G transients reached maximal amplitudes that were substantially smaller than those of GFP-α1S. When coexpressed with GFP-α1S, Ca2+ transients activated by GFP-α1G could be well separated from transients activated by the high-voltage-activated GFP-α1S (Fig. 7 C). The transient-to-voltage curve of cotransfected myotubes displayed a shoulder at −30 mV and continued to rise at −10 mV (Fig. 7 A). Subtracting the transient-to-voltage curve of GFP-α1G alone from that of the cotransfection experiments eliminated the shoulder, resulting in a curve with typical skeletal characteristics (Fig. 7 B). Thus, we conclude that the shoulder represents the contribution of GFP-α1G. The height of this shoulder is near ΔF/F values of 0.3, which corresponds to transients observed with GFP-α1C, GFP-α1D, GFP-α1A, and GFP-CSk53, and it is only about one third of the maximal Ca2+ signal activated by GFP-α1S in the same cells at more positive voltages. Thus, in our experiments Ca2+ transients activated by channels other than the native GFP-α1S peak at amplitudes significantly below that activated by GFP-α1S, even in the same myotube.
FIGURE 7.

Cardiac-type and skeletal-type EC-coupling activated independently in the same myotube. (A) Voltage-dependence of Ca2+ transients in dysgenic myotubes transfected with GFP-α1G alone (♦; n = 9) and in combination with GFP-α1S (▪; n = 9). The low-voltage activated Ca2+ channel GFP-α1G generates cardiac-type Ca2+ transients activated at negative voltages with maximal amplitudes (ΔF/F) not significantly different from those of GFP-α1C and GFP-α1D (cf. Fig. 5). The I/V curve of cotransfected myotubes displays a shoulder at negative potentials and continues to rise at positive potentials to levels similar to those achieved with GFP-α1S alone (□; data from Fig. 5 A). Maintained transients at +80 V near the reversal potential characterize this component as skeletal. (B) Subtraction of the I/V curve of GFP-α1G from that of the GFP-α1G/GFP-α1S-cotransfected myotubes results in a curve (•) resembling that of GFP-α1S (□). (C) Examples of Ca2+ transients and currents from a cotransfected cell depolarized to −10 mV (left traces) and to +80 mV (right traces). Ca2+-dependent (cardiac-type) and Ca2+-independent (skeletal-type) transients can be activated independently from each other in the same cell.
DISCUSSION
In this study we investigated the properties of “cardiac-type” EC-coupling activated in skeletal muscle cells by reconstituting dysgenic myotubes with nonskeletal α1 subunit isoforms. GFP-α1D—an L-type Ca2+ channel isoform found in pancreatic β cells, hair cells of the cochlea, and in the sinus node of the heart (Hell et al., 1993; Iwashima et al., 1993; Kollmar et al., 1997; Takimoto et al., 1997)—was targeted into the triads and restored action potential-induced Ca2+ transients as efficiently as GFP-α1C, even though its whole-cell current density was much smaller. In combined patch-clamp/fluo-4 measurements, Ca2+ transients activated by GFP-α1C and GFP-α1D peaked at amplitudes significantly below those of the skeletal GFP-α1S. However, this difference in the size of Ca2+ transients was not due to the different EC-coupling activation mechanisms, because CSk53, a cardiac channel with skeletal EC-coupling characteristics, also gave rise to transients of reduced magnitude.
GFP-α1D, together with the native α1S and the cardiac α1C, is now the third member of the voltage-gated Ca2+ channel family that has been demonstrated to become normally targeted into the skeletal muscle triads. Apparently all the members of the L-type subclass so far examined possess the triad targeting signals, whereas the neuronal, non-L-type α1A does not (Flucher et al., 2000). We have previously shown that an important triad targeting signal resides in the C-terminus of α1S and can be conferred onto GFP-α1A by replacing a short C-terminal sequence with the corresponding sequence of α1S (Flucher et al., 2000). Similarly the C-terminus of GFP-α1D was able to confer triad targeting properties onto GFP-α1A (Flucher and Grabner, unpublished results). Thus, triad targeting signals could be a conserved property of the L-type Ca2+ channel family. Alternatively, since α1S, α1C, and α1D isoforms are all expressed in various muscle tissues whereas α1A is not, the triad targeting property could be a muscle-specific feature.
Ca2+ currents generated by GFP-α1D expressed in dysgenic myotubes showed the typical characteristics previously reported for α1D in native cells (Zidanic and Fuchs, 1995) and of GFP-α1D expressed in mammalian heterologous expression systems (Koschak et al., 2001). GFP-α1D currents activated at 13 mV more negative potentials compared to GFP-α1C. The current density of GFP-α1D in dysgenic myotubes was less than one third of that of GFP-α1C. Nevertheless, both channels restored action potential-induced Ca2+ transients in dysgenic myotubes equally well and as efficiently as the native GFP-α1S. The fact that these Ca2+ transients were blocked by Cd2+/La3+ demonstrated that the mechanism by which GFP-α1D activated action potential-induced Ca2+ transients was CICR, with the trigger Ca2+ provided by the L-type Ca2+ current. Therefore it was surprising that the considerable difference in the size of the currents of GFP-α1D and GFP-α1C was not reflected in the size of the transients or in the rate at which action potential-induced Ca2+ transients were observed. Thus, activation of CICR is not strongly dependent on the total influx of Ca2+ manifested in the whole-cell current densities. This conclusion is further supported by data on α1A collected by us and others (Adams et al., 1994; Flucher et al., 2000; and present study). While α1A expressed in dysgenic myotubes produces high current densities the size of GFP-α1C or larger, it restores contractions or action potential-induced Ca2+ transients very rarely.
Whereas the ability of the examined α1 subunits to trigger Ca2+ transients does not correlate with the magnitude of their Ca2+ currents, it correlates well with their triad targeting properties. All channel isoforms that are efficiently targeted into the triads (GFP-α1S, GFP-α1C, GFP-α1D) also restore action potential-induced Ca2+ transients. On the other hand, GFP-α1A fails to do either. This suggests that activation of EC-coupling by CICR is dependent on local Ca2+ concentrations achieved in the restricted space of the triad in the moment of depolarization by an action potential. In the triad, even the small GFP-α1D currents—which are of similar size as those of α1S—attain the concentration necessary for activating SR Ca2+ release. However, the Ca2+ activation threshold of Ca2+ release is not reached during fivefold larger Ca2+ currents generated by the nontargeted GFP-α1A channel isoform. A local activation mechanism for CICR is also consistent with the observation that the termination of the Ca2+ release during action potential-induced Ca2+ transients is under the tight control of the membrane potential. At least in the dysgenic system, CICR is not a regenerative process that is self-sustained by Ca2+ released through the RyRs alone. In that case transients would be expected to continue to rise after repolarization and the deactivation of voltage-gated Ca2+ channels. On the contrary, the abrupt termination of the transients upon repolarization argues that the sustenance of the CICR requires the continued influx of Ca2+ through a voltage-gated channel. This tight control of Ca2+ activation of the Ca2+ release channels by voltage-gated Ca2+ currents in dysgenic myotubes reconstituted with GFP-α1C or GFP-α1D is reminiscent of EC coupling in cardiac myocytes (Cannell et al., 1987; Stern et al., 1999; Bers, 2000) and can only be envisioned in the restricted space of the triad junction, where α1C or α1D come within nanometers of the Ca2+ release channel (Rios and Stern, 1997).
A role of local Ca2+ gradients in the activation of CICR in skeletal myotubes is further corroborated by the observation that the lower limits of current densities required for triggering Ca2+ transients differ between GFP-α1C and GFP-α1D. In both cases failure of activation of Ca2+ transients was preferentially observed in myotubes with low current densities, whereas the myotubes with higher current densities usually produced Ca2+ transients. However, the current density required for the activation of Ca2+ transients was not constant. It varied between individual myotubes, and it was clearly different between GFP-α1C and GFP-α1D. The existence of a threshold of current density below which no Ca2+ release would be stimulated is expected for CICR. But this threshold should be the same in myotubes expressing GFP-α1C and GFP-α1D. Thus, we have to conclude that the whole-cell current densities only incompletely reflected the actual Ca2+ concentration at the activation site. Either the geometry of the channels in the triad is such that the spatiotemporal Ca2+ gradients required for Ca2+ activation of the RyR are similar for GFP-α1C and GFP-α1D even though their channel properties differ or, alternatively, the fractions of channels inside and outside the triad might differ for the two α1 isoforms, so that there is no linear relationship between whole-cell current densities and current densities in the triads.
GFP-α1A, which is not targeted into triads and usually fails to trigger action potential-induced Ca2+ transients, generated Ca2+ transients in the combined patch-clamp and fluo-4 Ca2+ recording experiments. On first sight, this result appears to contradict the hypothesis that triggering cardiac-type EC-coupling by CICR depends on the co-localization of the t-tubular and SR Ca2+ channels in the triads. However, if activation of CICR in the triads depends on local Ca2+ gradients, these could easily break down during the 200-ms depolarization of the patch-clamp experiments. During a brief action potential locally high Ca2+ concentrations near the mouth of a channel may activate SR Ca2+ release in the case of channels concentrated in the triad junction, whereas similarly high Ca2+ concentrations generated by α1A channels diffusely located outside the triads may dissipate in the highly buffered cytoplasm before reaching the RyRs in the triad. However, during continued activation over the period of 200 ms, Ca2+ entering the myotube through α1A may flood the cell to a degree that spatial gradients can no longer be maintained and CICR is triggered even by a channel positioned outside the triad. Interfering with the Ca2+ buffering milieu in the patched myotubes may further contribute to this inconsistent behavior under the different experimental conditions. The different shapes of Ca2+ transients produced by the field stimulation and in the combined patch-clamp recordings support this explanation (see also Garcia et al., 1994). The fact that in these experiments Ca2+ transients do not decline immediately after repolarization suggests that in contrast to action potential-induced Ca2+ transients, CICR stimulated by long depolarization contains a regenerative component, which sustains Ca2+ release even in the absence of influx. Also the slow decline of the Ca2+ transients indicates that the cytoplasmic Ca2+ buffering capacity has been exceeded under these experimental conditions. Applying shorter test pulses (10 ms) partially reversed this problem, and Ca2+ transients looked more like those of the action potential-induced Ca2+ transients (not shown). However, analyzing the voltage-dependence of current and transient activation and a reliable comparison of current densities of channels with different activation kinetics necessitated long depolarization. Therefore, even though analysis of Ca2+ signals under voltage-clamp control is common practice and has contributed significantly to our current understanding of EC-coupling (e.g., Baylor et al., 1983; Brum et al., 1987; Melzer et al., 1984; Beurg et al., 1997; Dietze et al., 1998; Jurkat-Rott et al., 1998; Grabner et al., 1999; Wilkens et al., 2001), it has to be kept in mind that spatiotemporal Ca2+ gradients, which appear to be critical for activation of CICR under physiological conditions, probably break down during prolonged patch-clamp recordings.
A trivial alternative explanation for the different size, shape, and behavior of Ca2+ transients obtained with patch-clamp stimulation would be that the transients recorded from nonskeletal Ca2+ channels in the patch-clamp mode are not CICR at all, but simply reflect the Ca2+ influx through these channels. This would explain the reduced amplitude compared to that of the skeletal transients as well as the fact that GFP-α1A, which failed to restore action potential-induced Ca2+ transients, generates a Ca2+ signal under patch-clamp conditions. Attempts to test this possibility by pharmacologically isolating a possible Ca2+ influx signal from that of CICR did not resolve the issue either, because even concentrations between 10 μM and 40 μM ryanodine (Lipp et al., 2002) or 10 μM and 160 μM ruthenium red (Xu et al., 1999) in the patch pipette did not completely block Ca2+ release in control myotubes transfected with GFP-α1S. However, important evidence from this and previous studies (Tanabe et al., 1990; Garcia et al., 1994; Garcia and Beam, 1994) argues against the possibility that these Ca2+ transients arise exclusively from Ca2+ influx: first, the poor correlation of current densities with the occurrence of associated Ca2+ transients in individual myotubes; i.e., myotubes with current densities as small as 3 pA/pF showed transients whereas other myotubes with much larger currents showed no measurable transients (Fig. 5 D) (see also Garcia et al., 1994); second, the poor correlation of current densities with the amplitudes of Ca2+ transient for GFP-α1C and GFP-α1D, i.e., whereas their current densities differed by almost threefold, GFP-α1C and GFP-α1D produced Ca2+ transients of similar magnitude (Fig. 5, A and B); and finally, Ca2+ transients triggered by the cardiac-skeletal chimera GFP-CSk53 were not significantly larger than those of GFP-α1C, as would be expected from a construct that triggers skeletal-type Ca2+ release in addition to a fluo-4 signal reflecting pure Ca2+ influx. If we had merely recorded the influx of Ca2+, the magnitude of the Ca2+ transients should be highly correlated to the underlying currents. On the contrary, the behavior of the observed Ca2+ transients is consistent with a CICR mechanism that depends not only on the size of the Ca2+ influx but also on its local density.
Comparing the voltage-dependence of the Ca2+ currents with that of the Ca2+ transients revealed important properties of CICR in skeletal myotubes. For the nonskeletal channels, the voltage-dependence of the Ca2+ transients faithfully followed that of the current density, resulting in the bell-shaped curve typical for cardiac-type EC-coupling (Tanabe et al., 1990). Moreover, differences in the voltage-dependence of the currents, like the left-shifted activation of GFP-α1D, can similarly be observed for the transients. This strict correlation of current amplitudes and transient amplitudes at different voltages clearly indicates that CICR is not simply an all-or-none mechanism. Smaller currents give rise to smaller transients and larger currents give rise to larger transients. Thus, CICR in individual skeletal myotubes functions as a graded amplification system in which Ca2+ entering the myotube through the voltage-gated channels is proportionally amplified by Ca2+ released from the SR. Nevertheless, considerable differences in current densities between individual cells and the differences in the average current densities of GFP-α1C and GFP-α1D are not reflected in the maximal amplitudes of the transients. These are very similar in all myotubes expressing nonskeletal α1 subunits, and they peak at a level way below the maximal capacity of SR Ca2+ release. Thus, it seems that irrespective of their different current properties, GFP-α1C, GFP-α1D, GFP-α1A, and GFP-α1G activate the same maximal amount of Ca2+ release in the combined patch-clamp/fluo-4 recordings.
This reduced maximum of Ca2+ release achieved by the nonskeletal channels is, however, not due to a limited release capacity of the EC-coupling apparatus. Ca2+ transients activated by GFP-α1S were on average three times larger than those of nonskeletal channels. This observation is consistent with similar differences seen in an earlier report comparing α1S and α1C expressed in myotubes of dysgenic primary cultures (Garcia et al., 1994). But perhaps myotubes reconstituted with the skeletal GFP-α1S channel differentiate better than myotubes expressing the heterologous channel isoforms and therefore generate more robust Ca2+ transients? However, even on a single myotube level we observed that nonskeletal Ca2+ channels released only a fraction of the Ca2+ releasable by GFP-α1S. Myotubes cotransfected with a low-voltage-activated channel GFP-α1G and the high-voltage-activated GFP-α1S showed the typical small cardiac-type Ca2+ transients at negative voltages followed by up to fivefold larger skeletal-type transients at positive voltages. This indicates that nonskeletal channels only partially activate the Ca2+ release capacity of dysgenic myotubes.
Interestingly, the cause of this differential ability to activate Ca2+ release is not, as one might think, the difference between skeletal and cardiac-type activation of Ca2+ release. GFP-CSk53, a cardiac-based channel chimera with skeletal EC-coupling properties (Nakai et al., 1998), also induced small transients like those of GFP-α1C and GFP-α1D. Apparently channel isoforms and chimeras other than α1S activate SR Ca2+ release less efficiently than the native channel, and transferring skeletal EC-coupling properties onto a cardiac-type channel is not sufficient to overcome this deficiency. It may well be that other properties specific to the skeletal α1S subunit enable α1S to activate more Ca2+ release channels or the same number more efficiently than nonskeletal channels. Thus, the body of the channel—skeletal or nonskeletal—as well as the region in the II-III loop important for skeletal-type activation of EC-coupling seems to be important for the magnitude of Ca2+ release achieved in reconstituted dysgenic myotubes.
The combined evidence from this and previous studies allows drafting of the following model of the role of CICR in skeletal muscle contraction. CICR appears to be a significant component of skeletal muscle EC-coupling (O'Brien et al., 2002). To activate CICR in skeletal muscle, a threshold concentration of Ca2+ in the triads needs to be exceeded. During the brief depolarization of an action potential, this is achieved only by channels located in close range of the RyRs within the triad junction, but not by Ca2+ influx outside the triads. Interestingly, Ca2+ currents the size of the skeletal α1S are capable of activating CICR, whereas SR Ca2+ release through the RyRs alone cannot sustain its own further release after repolarization. Thus, in normal EC-coupling CICR is not regenerative but amplifies the trigger Ca2+ signal while remaining under the strict control of the voltage-activated channels. Upon long depolarization, and possibly also during tetanic stimulation or certain pathologic conditions like malignant hyperthermia, the strict local control of CICR breaks down and regenerative activation of CICR in skeletal muscle may occur. Thus, the spatial arrangement of voltage-activated channels and SR Ca2+ release channels in the triad plays an essential part in the control of Ca2+ release in skeletal muscle EC-coupling.
Acknowledgments
We thank Drs. P. Lory and S.J. Dubel (Institut de Genetique Humaine, CNRS UPR 1142, Montpellier) for generously supplying the expression plasmid for GFP-α1G, Dr. W. Melzer and his team in Ulm for fruitful exchange of know-how, Dr. J. Hoflacher and Ms. D. Kandler for their experimental help, and Dr. H. Glossmann for generously providing support for the pursuit of this project.
This work was supported in part by the European Commissions Training and Mobility of Researchers Network Grant ERBFMRXCT960032 (to B.E.F.) and by the Fonds zur Förderung der wissenschaftlichen Forschung, Austria, Grants P12653-MED and P15338-MED (to B.E.F.), and P13831-GEN and Austrian National Bank (to M.G.). This work is part of the Ph.D. thesis of N.K.
References
- Adams, B. A., T. Tanabe, A. Mikami, S. Numa, and K. G. Beam. 1990. Intramembrane charge movement restored in dysgenic skeletal muscle by injection of dihydropyridine receptor cDNAs. Nature. 346:569–572. [DOI] [PubMed] [Google Scholar]
- Adams, B. A., Y. Mori, M. S. Kim, T. Tanabe, and K. G. Beam. 1994. Heterologous expression of BI Ca2+ channels in dysgenic skeletal muscle. J. Gen. Physiol. 104:985–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylor, S. M., W. K. Chandler, and M. W. Marshall. 1983. Sarcoplasmic reticulum calcium release in frog skeletal muscle fibres estimated from Arsenazo III calcium transients. J. Physiol. 344:625–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers, D. M. 2000. Calcium fluxes involved in control of cardiac myocyte contraction. Circ. Res. 87:275–281. [DOI] [PubMed] [Google Scholar]
- Beurg, M., M. Sukhareva, C. Strube, P. A. Powers, R. G. Gregg, and R. Coronado. 1997. Recovery of Ca2+ current, charge movements, and Ca2+ transients in myotubes deficient in dihydropyridine receptor beta 1 subunit transfected with beta 1 cDNA. Biophys. J. 73:807–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brum, G., E. Stefani, and E. Rios. 1987. Simultaneous measurements of Ca2+ currents and intracellular Ca2+ concentrations in single skeletal muscle fibers of the frog. Can. J. Physiol. Pharmacol. 65:681–685. [DOI] [PubMed] [Google Scholar]
- Cannell, M. B., J. R. Berlin, and W. J. Lederer. 1987. Effect of membrane potential changes on the calcium transient in single rat cardiac muscle cells. Science. 238:1419–1423. [DOI] [PubMed] [Google Scholar]
- Dietze, B., F. Bertocchini, V. Barone, A. Struk, V. Sorrentino, and W. Melzer. 1998. Voltage-controlled Ca2+ release in normal and ryanodine receptor type 3 (RyR3)-deficient mouse myotubes. J. Physiol. 513:3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmeyer, D., W. Melzer, B. Pohl, and P. Zollner. 1990. Fast gating kinetics of the slow Ca2+ current in cut skeletal muscle fibres of the frog. J. Physiol. 425:347–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flucher, B. E., S. B. Andrews, S. Fleischer, A. R. Marks, A. Caswell, and J. A. Powell. 1993. Triad formation: organization and function of the sarcoplasmic reticulum calcium release channel and triadin in normal and dysgenic muscle in vitro. J. Cell Biol. 123:1161–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flucher, B. E., S. B. Andrews, and M. P. Daniels. 1994. Molecular organization of transverse tubule/sarcoplasmic reticulum junctions during development of excitation-contraction coupling in skeletal muscle. Mol. Biol. Cell. 5:1105–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flucher, B. E., N. Kasielke, and M. Grabner. 2000. The triad targeting signal of the skeletal muscle calcium channel is localized in the COOH terminus of the α1S subunit. J. Cell Biol. 151:467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia, J., M. Amador, and E. Stefani. 1989. Relationship between myoplasmic calcium transients and calcium currents in frog skeletal muscle. J. Gen. Physiol. 94:973–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia, J., and K. G. Beam. 1994. Calcium transients associated with the T type calcium current in myotubes. J. Gen. Physiol. 104:1113–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia, J., T. Tanabe, and K. G. Beam. 1994. Relationship of calcium transients to calcium currents and charge movements in myotubes expressing skeletal and cardiac dihydropyridine receptors. J. Gen. Physiol. 103:125–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannini, G., A. Conti, S. Mammarella, M. Scrobogna, and V. Sorrentino. 1995. The ryanodine receptor/calcium channel genes are widely and differentially expressed in murine brain and peripheral tissues. J. Cell Biol. 128:893–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabner, M., R. T. Dirksen, and K. G. Beam. 1998. Tagging with green fluorescent protein reveals a distinct subcellular distribution of L-type and non-L-type Ca2+ channels expressed in dysgenic myotubes. Proc. Natl. Acad. Sci. USA. 95:1903–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabner, M., R. T. Dirksen, N. Suda, and K. G. Beam. 1999. The II–III loop of the skeletal muscle dihydropyridine receptor is responsible for the bidirectional coupling with the ryanodine receptor. J. Biol. Chem. 274:21913–21919. [DOI] [PubMed] [Google Scholar]
- Hell, J. W., R. E. Westenbroek, C. Warner, M. K. Ahlijanian, W. Prystay, M. M. Gilbert, T. P. Snutch, and W. A. Catterall. 1993. Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel alpha 1 subunits. J. Cell Biol. 123:949–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwashima, Y., W. Pugh, A. M. Depaoli, J. Takeda, S. Seino, G. I. Bell, and K. S. Polonsky. 1993. Expression of calcium channel mRNAs in rat pancreatic islets and downregulation after glucose infusion. Diabetes. 42:948–955. [DOI] [PubMed] [Google Scholar]
- Jurkat-Rott, K., U. Uetz, U. Pika-Hartlaub, J. Powell, B. Fontaine, W. Melzer, and F. Lehmann-Horn. 1998. Calcium currents and transients of native and heterologously expressed mutant skeletal muscle DHP receptor alpha1 subunits (R528H). FEBS Lett. 423:198–204. [DOI] [PubMed] [Google Scholar]
- Kollmar, R., L. G. Montgomery, J. Fak, L. J. Henry, and A. J. Hudspeth. 1997. Predominance of the alpha1D subunit in L-type voltage-gated Ca2+ channels of hair cells in the chicken's cochlea. Proc. Natl. Acad. Sci. USA. 94:14883–14888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koschak, A., D. Reimer, I. Huber, M. Grabner, H. Glossmann, J. Engel, and J. Striessnig. 2001. alpha 1D (Cav1.3) subunits can form L-type Ca2+ channels activating at negative voltages. J. Biol. Chem. 276:22100–22106. [DOI] [PubMed] [Google Scholar]
- Lipp, P., M. Egger, and E. Niggli. 2002. Spatial characteristics of sarcoplasmic reticulum Ca2+ release events triggered by L-type Ca2+ current and Na+ current in guinea-pig cardiac myocytes. J. Physiol. 542:383–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melzer, W., E. Rios, and M. F. Schneider. 1984. Time course of calcium release and removal in skeletal muscle fibers. Biophys. J. 45:637–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteil, A., J. Chemin, E. Bourinet, G. Mennessier, P. Lory, and J. Nargeot. 2000. Molecular and functional properties of the human alpha(1G) subunit that forms T-type calcium channels. J. Biol. Chem. 275:6090–6100. [DOI] [PubMed] [Google Scholar]
- Nagasaki, K., and M. Kasai. 1983. Fast release of calcium from sarcoplasmic reticulum vesicles monitored by chlortetracycline fluorescence. J. Biochem. (Tokyo). 94:1101–1109. [DOI] [PubMed] [Google Scholar]
- Nakai, J., T. Tanabe, T. Konno, B. Adams, and K. G. Beam. 1998. Localization in the II–III loop of the dihydropyridine receptor of a sequence critical for excitation-contraction coupling. J. Biol. Chem. 273:24983–24986. [DOI] [PubMed] [Google Scholar]
- O'Brien, J. J., W. Feng, P. D. Allen, S. R. Chen, I. N. Pessah, and K. G. Beam.2002. Ca2+ activation of RyR1 is not necessary for the initiation of skeletal-type excitation-contraction coupling. Biophys. J. 82:2428–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell, J. A., L. Petherbridge, and B. E. Flucher. 1996. Formation of triads without the dihydropyridine receptor α subunits in cell lines from dysgenic skeletal muscle. J. Cell Biol. 134:375–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios, E., and M. D. Stern. 1997. Calcium in close quarters: microdomain feedback in excitation-contraction coupling and other cell biological phenomena. Annu. Rev. Biophys. Biomol. Struct. 26:47–82. [DOI] [PubMed] [Google Scholar]
- Smith, J. S., R. Coronado, and G. Meissner. 1986. Single channel measurements of the calcium release channel from skeletal muscle sarcoplasmic reticulum. Activation by Ca2+ and ATP and modulation by Mg2+. J. Gen. Physiol. 88:573–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern, M. D., L. S. Song, H. Cheng, J. S. Sham, H. T. Yang, K. R. Boheler, and E. Rios. 1999. Local control models of cardiac excitation-contraction coupling. A possible role for allosteric interactions between ryanodine receptors. J. Gen. Physiol. 113:469–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takimoto, K., D. Li, J. M. Nerbonne, and E. S. Levitan. 1997. Distribution, splicing and glucocorticoid-induced expression of cardiac alpha 1C and alpha 1D voltage-gated Ca2+ channel mRNAs. J. Mol. Cell. Cardiol. 29:3035–3042. [DOI] [PubMed] [Google Scholar]
- Tanabe, T., K. G. Beam, J. A. Powell, and S. Numa. 1988. Restoration of excitation-contraction coupling and slow calcium current in dysgenic muscle by dihydropyridine receptor complementary DNA. Nature. 336:134–139. [DOI] [PubMed] [Google Scholar]
- Tanabe, T., A. Mikami, S. Numa, and K. G. Beam. 1990. Cardiac-type excitation-contraction coupling in dysgenic skeletal muscle injected with cardiac dihydropyridine receptor cDNA. Nature. 344:451–453. [DOI] [PubMed] [Google Scholar]
- Wilkens, C. M., N. Kasielke, B. E. Flucher, K. G. Beam, and M. Grabner. 2001. Excitation-contraction coupling is unaffected by drastic alteration of the sequence surrounding residues L720–L764 of the alpha 1S II–III loop. Proc. Natl. Acad. Sci. USA. 98:5892–5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, L., A. Tripathy, D. A. Pasek, and G. Meissner. 1999. Ruthenium red modifies the cardiac and skeletal muscle Ca(2+) release channels (ryanodine receptors) by multiple mechanisms. J. Biol. Chem. 274:32680–32691. [DOI] [PubMed] [Google Scholar]
- Zidanic, M., and P. A. Fuchs. 1995. Kinetic analysis of barium currents in chick cochlear hair cells. Biophys. J. 68:1323–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]