

Abstract
The II-III loop of the skeletal muscle dihydropyridine receptor (DHPR) α1S subunit is responsible for bidirectional-signaling interactions with the ryanodine receptor (RyR1): transmitting an orthograde, excitation–contraction (EC) coupling signal to RyR1 and receiving a retrograde, current-enhancing signal from RyR1. Previously, several reports argued for the importance of two distinct regions of the skeletal II-III loop (residues R681–L690 and residues L720–Q765, respectively), claiming for each a key function in DHPR–RyR1 communication. To address whether residues 720–765 of the II-III loop are sufficient to enable skeletal-type (Ca2+ entry-independent) EC coupling and retrograde interaction with RyR1, we constructed a green fluorescent protein (GFP)-tagged chimera (GFP-SkLM) having rabbit skeletal (Sk) DHPR sequence except for a II-III loop (L) from the DHPR of the house fly, Musca domestica (M). The Musca II-III loop (75% dissimilarity to α1S) has no similarity to α1S in the regions R681–L690 and L720–Q765. GFP-SkLM expressed in dysgenic myotubes (which lack endogenous α1S subunits) was unable to restore EC coupling and displayed strongly reduced Ca2+ current densities despite normal surface expression levels and correct triad targeting (colocalization with RyR1). Introducing rabbit α1S residues L720–L764 into the Musca II-III loop of GFP-SkLM (substitution for Musca DHPR residues E724–T755) completely restored bidirectional coupling, indicating its dependence on α1S loop residues 720–764 but its independence from other regions of the loop. Thus, 45 α1S-residues embedded in a very dissimilar background are sufficient to restore bidirectional coupling, indicating that these residues may be a site of a protein–protein interaction required for bidirectional coupling.
Excitation–contraction (EC) coupling depends on the interaction of the voltage-gated L type Ca2+ channel or dihydropyridine receptor (DHPR) of the plasma membrane and the intracellular Ca2+-release channel or ryanodine receptor (RyR) of the sarcoplasmic reticulum (SR) (reviewed in refs. 1 and 2). Because EC coupling in skeletal muscle still occurs after blocking of the L type Ca2+ current, it is thought that membrane depolarization causes conformational changes of the DHPR (3, 4), which, in turn, trigger the opening of the RyR and the release of Ca2+ from SR stores (“mechanical hypothesis” of skeletal muscle EC coupling; ref. 3). In addition to this orthograde EC coupling signal, a retrograde signal exists, by which RyR1 (the skeletal isoform of the RyR) enhances L type current through the DHPR (5). Specifically, Ca2+ currents are small in dyspedic myotubes (which lack RyR1) despite normal densities of the DHPR, whereas expression of recombinant RyR1 restores the L type current density toward wild-type levels (5).
A fundamental goal for understanding the mechanism of bidirectional signaling is to identify the domains of the skeletal DHPR that directly participate in this process. One approach has been to analyze skeletal/cardiac chimeric DHPRs expressed in dysgenic myotubes, which lack the α1S subunit of the skeletal DHPR (6). Almost 10 years ago, this work demonstrated that a cardiac DHPR containing the skeletal II-III loop was able to restore Ca2+ entry-independent (skeletal-type) EC coupling (7). A subsequent study identified 46 aa (residues 720–765) of the skeletal II-III loop that are sufficient for transferring strong, skeletal-type EC coupling properties to an otherwise cardiac DHPR (8). More recently, it was shown that a skeletal DHPR with a cardiac II-III loop (SkLC) lacked both orthograde (skeletal EC-coupling) and retrograde (L current-enhancing) signaling (9). When α1S residues 720–765, which earlier had been shown to confer skeletal-type coupling on an otherwise cardiac DHPR (8), were introduced into SkLC, both skeletal-type EC coupling and wild-type Ca2+ current densities were restored (9). Thus, residues 720–765 of the skeletal DHPR II-III loop represent a “critical domain” for the bidirectional interaction between the skeletal DHPR and RyR1.
A limitation of chimeras is that they do not test the functional importance of regions that are conserved between the two parental proteins. This is a significant problem for the cardiac and skeletal DHPRs because the regions flanking the 46-residue critical domain are 56% identical between the cardiac and skeletal II-III loops. The potential importance of these flanking domains is emphasized by the results of experiments testing the effects of peptides on the function of RyR1 in vitro (ryanodine binding or Ca2+ release in SR vesicular preparations and open probability of RyRs reconstituted in artificial planar bilayers). In the earliest of these studies (10), recombinant peptides corresponding to either the skeletal or cardiac II-III loop were found to activate RyR1, which is difficult to reconcile with the results obtained with the chimeras (see above; refs. 7–9). Later, synthetic peptides (peptide A: α1S residues 671–690; peptide As10: residues 681–690), which corresponded to smaller portions of the skeletal loop and were upstream from the critical domain identified in the chimera studies, were found to activate RyR1 (11–14). In addition to the skeletal peptide (As10), the corresponding cardiac peptide (Ac10), which is homologous because of similar clusters of positively charged residues, also was found to cause activation of RyR1, although to a somewhat lower extent (15). Similar cardiac peptides also have been reported not to cause activation of RyR1 (16). Thus, the significance of the II-III loop-flanking domains remains uncertain.
Although the peptide experiments have the important advantage of providing a test of whether the skeletal DHPR and RyR1 interact directly, they have the disadvantage of lacking physiological context. To test the importance of the regions flanking the critical domain (L720–L764), including the As10 region, we created the chimera SkLM, a skeletal DHPR with a II-III loop from the house fly (Musca domestica) DHPR (17), which is highly divergent from both the skeletal and cardiac loops. SkLM was unable to support bidirectional coupling, but insertion of α1S residues L720–L764 into the Musca loop completely restored both orthograde and retrograde signaling. Thus, residues 720–764 represent or contain the sequence sufficient to mediate bidirectional coupling and, therefore, represent a potential site of protein–protein interaction necessary for this coupling. By contrast, the flanking domains are unlikely to be involved in such protein–protein interactions.
Materials and Methods
Construction of DHPR Chimeras.
DHPR II-III loop chimeras were constructed as follows, with nucleotide numbers given in parentheses and asterisks indicating restriction sites introduced by PCR.
Green fluorescent protein (GFP)-α1S.
The cDNA coding sequence of the rabbit skeletal muscle DHPR α1S subunit (18) was inserted in-frame 3′ to the coding region of GFP contained in a mammalian expression plasmid, as described previously (19).
GFP-SkLM.
The EcoRI-BalI fragment of the rabbit skeletal muscle DHPR α1S subunit (Sk) (nucleotides 1007–1973) was coligated with the BalI-NdeI fragment (nucleotides 1982–2296) from the II-III loop (L) from the body-wall muscle DHPR α1 subunit (M) of M. domestica (17) into plasmid pSP72 (Promega) by using the internal NdeI site (plasmid nucleotide 2379) and the EcoRI site of the polylinker. The NdeI/EcoRI restriction sites of pSP72 also were used to coligate two cDNA fragments, the NdeI*-XhoI fragment that was PCR-generated from the clone GFP-SkLC, GFP-α1S with the cardiac (α1C, C) II-III loop (nucleotides C2716–Sk2654) (9), plus the XhoI-BglII fragment of Sk (nucleotides 2654–4488). The PCR primer used to introduce the NdeI* site also mutated 2 aa of α1C (A907, S908) to the corresponding Musca residues (G767, T768: see Fig. 1). In a subsequent step, fragments EcoRI-NdeI (nucleotides Sk1007–M2297) and NdeI*-BglII (nucleotides C2716–Sk4488) were isolated from the two pSP72 subclones and coligated into the EcoRI/BglII-cleaved pSP72 vector. Finally, the SalI-EcoRI fragment of Sk (nucleotide 5′ polylinker-1007) was coligated with the EcoRI-BglII fragment (nucleotides Sk1007–Sk4488) from the last pSP72 subclone into the SalI/BglII sites of plasmid GFP-α1S.
Figure 1.

Schematic representation of the skeletal/Musca II-III loop DHPR chimeras GFP-SkLM and GFP-SkLMS45 and II-III loop sequence alignments. (A) DHPR chimeras were N-terminally fused to the green fluorescent protein GFP (19). Rabbit skeletal muscle (α1S) sequence is indicated in blue, and M. domestica (house fly) muscle (α1M) sequence is in black. I-IV, four homologous repeats of α1 subunits. (B) Alignment of cardiac (α1C), skeletal (α1S), and Musca (α1M) II-III loop sequences. The Musca sequence boxed in light gray (α1M residues E724–T755) was replaced by the portion of α1S sequence indicated by arrows (residues L720–L764) to yield GFP-SkLMS45. Sequences boxed in yellow indicate the so-called skeletal and cardiac “peptide A-10 region” or “activating domain” of EC coupling (12, 15). Asterisks indicate amino acids identical between α1S and α1C or α1S and α1M. Dots show corresponding residues with identical charge.
GFP-SkLMS45.
The MfeI-XbaI* fragment of M (nucleotides 2024–2177) was coligated with the XbaI*-XhoI fragment of GFP-SkLM (nucleotides M2258–Sk2654) into the MfeI/XhoI-cleaved plasmid GFP-SkLM. Together with the XbaI* (nucleotide M2177) site, the antisense PCR primer introduced an upstream AflII* site (nucleotide M2171). Similarly, the sense primer introduced an additional ClaI* site (nucleotide M2265) downstream of the XbaI* (nucleotide M2258)-cloning site. To yield plasmid GFP-SkLMS45, an AflII*-TaqI* fragment of Sk (nucleotides 2159–2292) was ligated into the AflII*/ClaI* sites of this subclone. All segments of cDNA generated and modified by PCR were checked by sequence analysis (MWG Biotec, Ebersberg, Germany).
Expression of cDNA.
The DHPR cDNAs were expressed in myotubes obtained as primary cultures from newborn dysgenic (mdg/mdg) mice (20) or myotubes produced by differentiation of the dysgenic cell line GLT (21). GLT cultures were transfected at the onset of myoblast fusion (2–4 days after addition of differentiation medium) by using the liposomal transfection reagent FuGene according to the manufacturer's protocol (Roche Diagnostics, Mannheim, Germany). Primary dysgenic myotubes were microinjected into a single nucleus (22) with solutions of DHPR cDNA (100–200 ng/μl) approximately 1 week after initial plating. Two to four days after transfection or injection, expressing myotubes were identified by GFP fluorescence and used in the experiments.
Electrophysiological Characterization.
Whole-cell patch clamp (23) recording of Ca2+
currents and charge movements (24) was used to obtain an estimate of
the ratio of maximum Ca2+ conductance to maximum
immobilization-resistant charge movement
(Gmax/Q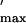 ),
which provides a quantitative assessment of the strength of retrograde
coupling. EC coupling was assayed in primary myotubes, as contractions
in response to pulses (100 ms, 100 V) applied via an extracellular
pipette (22), and in GLT myotubes, as fluorescence transients evoked in
cells loaded with Fluo-4AM and stimulated with pulses (1 ms, 20–30 V)
applied via electrodes placed on opposite sides of the culture dish
(25). To further assess EC coupling, depolarization-induced
intracellular Ca2+ transients were measured
microphotometrically during whole-cell recordings of primary myotubes
by including tetrapotassium-Fluo-3 (Molecular Probes) in the pipette
solution (26). All electrophysiological procedures, including test
protocols, equipment, solutions, and calculations essentially were the
same as described recently for primary myotubes (9) or GLT myotubes
(25), except that intracellular Ca2+ transients
were recorded in the present study from primary myotubes expressing
fluorescing GFP-tagged chimeras instead of coexpressing the CD8
reporter gene (9).
),
which provides a quantitative assessment of the strength of retrograde
coupling. EC coupling was assayed in primary myotubes, as contractions
in response to pulses (100 ms, 100 V) applied via an extracellular
pipette (22), and in GLT myotubes, as fluorescence transients evoked in
cells loaded with Fluo-4AM and stimulated with pulses (1 ms, 20–30 V)
applied via electrodes placed on opposite sides of the culture dish
(25). To further assess EC coupling, depolarization-induced
intracellular Ca2+ transients were measured
microphotometrically during whole-cell recordings of primary myotubes
by including tetrapotassium-Fluo-3 (Molecular Probes) in the pipette
solution (26). All electrophysiological procedures, including test
protocols, equipment, solutions, and calculations essentially were the
same as described recently for primary myotubes (9) or GLT myotubes
(25), except that intracellular Ca2+ transients
were recorded in the present study from primary myotubes expressing
fluorescing GFP-tagged chimeras instead of coexpressing the CD8
reporter gene (9).
Immunofluorescence Labeling.
Differentiated GLT cultures expressing GFP-SkLM were fixed and immunostained as described previously (27), using an affinity-purified anti-GFP antibody (Molecular Probes) at a dilution of 1:4,000 and the affinity-purified antibody 162 against RyR1 at a dilution of 1:5,000 (28). In double-labeling experiments, Alexa-conjugated secondary antibodies were used for GFP-SkLM so that the antibody label and the intrinsic GFP signal both were recorded in the green channel, and Texas red-conjugated secondary antibodies were used for RyR1. Controls, such as the omission of primary antibodies and incubation with inappropriate antibodies, were routinely performed. Images were recorded on a Zeiss Axiophot microscope by using a cooled charge-coupled device camera and metaview image-processing software (Universal Imaging, Media, PA). At least three different experiments were performed for comparison of the wild-type DHPR (GFP-α1S) with the DHPR chimera GFP-SkLM. Semiquantitative evaluation of the labeling patterns (25) revealed a clustering efficiency of greater than 50% in each experiment.
Results and Discussion
An Ancestral DHPR II-III Loop as a Tool to Test DHPR–RyR1 Interactions.
Previous analysis of chimeric DHPRs constructed from skeletal and cardiac sequence showed that a “critical domain” of the α1S II-III loop (residues L720–Q765) is required for both skeletal-type EC coupling (8, 9) and RyR1-mediated enhancement of Ca2+ currents (9). However, these experiments provided little or no information about the loop regions that flank the critical domain because these regions are well conserved between the cardiac and skeletal proteins (Fig. 1B). In the present work, we tested the importance of these flanking domains by replacing them with highly divergent sequences. To accomplish this, we created the chimera GFP-SkLM (Fig. 1A), in which the II-III loop of α1S was replaced by the highly divergent II-III loop of a DHPR cloned from the housefly (M. domestica) (17). Although we have not been able to express the Musca α1 subunit functionally in various heterologous systems (Xenopus oocytes, tsA-201 cells, or dysgenic myotubes), we did find that constructs containing parts of the Musca DHPR sequence were valuable for fine mapping of the DHP-binding domain (29). The Musca II-III loop has comparable length (126 residues) to the cardiac and skeletal loops, but only 19% overall identity, most of which is concentrated at the two ends. Importantly, there is absolutely no homology to the peptide A-10 (As10/Ac10) region (Fig. 1B); this region has been suggested to be important in EC coupling because the isolated peptide activates RyR1 (11–15). To test the role of the critical domain, we created the chimera GFP-SkLMS45 (Fig. 1A), in which 32 residues (E724–T755) from the Musca loop of GFP-SkLM were replaced by α1S residues L720–L764. In the absence of sequence homology that could serve as a guidepost for the insertion of the α1S critical domain into the Musca II-III loop, we chose to make this insertion within GFP-SkLMS45, such that the critical domain was separated from IIS6 and from IIIS1 by the same number of residues as in wild-type α1S. The final residue (Q765) of α1S sequence tested previously in α1S/α1C chimeras (8, 9) was omitted for cloning reasons.
The Presence of α1S Residues 720–764 in the Musca II-III Loop Supports Retrograde Coupling.
Expression of GFP-SkLM in dysgenic myotubes resulted in the presence of slowly activating (skeletal-type) Ca2+ currents, but these currents were much smaller than those for GFP-α1S (Fig. 2A). To determine whether a reduced density of surface expression could account for the small Ca2+ currents produced by SkLM, we measured immobilization-resistant charge movements (Fig. 2B). The charge vs. voltage relationship then was fitted to determine the maximal charge movement (Qmax) as an indirect measure of the expression density of DHPRs in the plasma membrane. Additionally, the current–voltage relationship for each cell was fitted (24) to yield a value of maximal Ca2+ conductance (Gmax). For each of the constructs, GFP-α1S and GFP-SkLM, neither Gmax nor Qmax was found to differ significantly between injected primary dysgenic myotubes and transfected GLT myotubes (Table 1). Thus, data from the two types of dysgenic myotubes were combined for all subsequent analyses. As shown in Fig. 2C, the average Gmax was significantly smaller for GFP-SkLM than for GFP-α1S, whereas Qmax was similar for the two constructs. Because the Qmax values are similar, it appears that the Musca loop does not alter surface expression and that the reduction in current amplitude occurs because the Musca loop does not support retrograde signaling with the RyR1. Unlike GFP-SkLM, GFP-SkLMS45 produced Ca2+ currents (Fig. 2A) similar in magnitude to those of GFP-α1S. This similarity between GFP-SkLMS45 and GFP-α1S also was evident in the average values of Gmax (Fig. 2C). Thus, the presence in the II-III loop of α1S residues L720–L764, even when surrounded by sequence very unlike α1S, was sufficient to restore the retrograde interaction whereby RyR1 increases the magnitude of slow L type Ca2+ current.
Figure 2.

Restoration of retrograde coupling after insertion of α1S
residues 720–764 into the Musca II-III loop (chimera
GFP-SkLMS45). (A) Representative whole-cell
Ca2+ currents recorded from dysgenic myotubes expressing
GFP-α1S (Left), GFP-SkLM
(Center), and GFP-SkLMS45
(Right). After a prepulse to inactivate T type currents
(24), macroscopic Ca2+ currents were elicited by 200-ms
step depolarizations from a holding potential of −80 mV to the
indicated test potentials. Current amplitudes were normalized by linear
cell capacitance and are expressed as pA/pF. (B)
Representative immobilization-resistant intramembrane charge movements
measured at +40 mV after blocking Ca2+ currents with 0.5 mM
Cd2+ and 0.1 mM La3+, recorded from cells
expressing the same three constructs shown above. (C)
Average maximal Ca2+ conductance
(Gmax, Left) and charge
movement (Qmax, Right) and
ratios of
Gmax/Q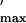 (Center) for GFP-α1S, GFP-SkLM, and
GFP-SkLMS45. The asterisk indicates a statistically
significant (P < 0. 001) difference in average
Gmax from the other two constructs after
comparison by an unpaired two-sample t test. No asterisk
indicates lack of statistically significant difference
(P > 0.05). (Bars = mean ± SEM of
12–20 recordings.)
(Center) for GFP-α1S, GFP-SkLM, and
GFP-SkLMS45. The asterisk indicates a statistically
significant (P < 0. 001) difference in average
Gmax from the other two constructs after
comparison by an unpaired two-sample t test. No asterisk
indicates lack of statistically significant difference
(P > 0.05). (Bars = mean ± SEM of
12–20 recordings.)
Table 1.
Ca2+ conductance and charge movement properties recorded from primary dysgenic myotubes and from the dysgenic cell line GLT are highly comparable
 |
Data are given as mean ± SEM (numbers in parentheses are the myotubes tested). Recordings from primary dysgenic myotubes (cDNA injected) are indicated in italic and from immortalized dysgenic GLT myotubes (cDNA transfected) in roman. Gmax is the maximal Ca2+ conductance (currents fitted according to ref. 24), Qmax is the maximum immobilization-resistant charge movement (Qon fitted according to ref. 24), and Q′max is the difference between Qmax and the average, endogenous charge movement Qdys(max) found in dysgenic myotubes (Qdys(max) = 2.5 nC/μF; ref. 24). Brackets indicate a lack of significant difference (P > 0.05) between data sets compared by an unpaired two-sample t test. Values for GFP-αIS recorded from primary dysgenic myotubes were listed for comparison and were published (9).
The Presence of α1S Residues 720–764 in the Musca II-III Loop Supports Orthograde Coupling.
In GLT myotubes expressing GFP-α1S, brief depolarizing pulses elicited transient elevations of intracellular Ca2+, which persisted even when Ca2+ influx was blocked by the addition of Cd2+ and La3+ to the bath (Fig. 3A). By contrast, the addition of Cd2+ and La3+ abolished the depolarization-evoked Ca2+ transients in GLT myotubes expressing GFP-α1C, although the SR was still capable of releasing Ca2+ in response to caffeine (Fig. 3B). Thus, GLT myotubes provide an appropriate system for distinguishing skeletal-type transients, which do not depend on entry of extracellular Ca2+, from cardiac-type transients, which do depend on such Ca2+ entry. Brief depolarizations failed entirely to evoke transients in GLT myotubes expressing GFP-SkLM, although the response to caffeine indicated that SR Ca2+ release was functional in these cells (Fig. 3C). By contrast, skeletal-type transients were present in GLT myotubes expressing GFP-SkLMS45 (Fig. 3D). As a more quantitative measurement of the strength of EC coupling, we determined for each chimeric construct the fraction of injected primary myotubes that contracted in response to electrical stimulation in Cd2+/La3+ solution. Contractions were never observed for GFP-SkLM, whereas the fraction of contracting myotubes was comparable for GFP-SkLMS45 and GFP-α1S (Fig. 3E). Like myotubes from normal mice (data not shown), electrical stimulation failed to cause contraction of a fraction of dysgenic myotubes transfected with GFP-α1S or GFP-SkLMS45. These noncontracting cells most likely represent myotubes in which components of the excitation–contraction coupling machinery are not fully developed (see ref. 25).
Figure 3.

Chimera GFP-SkLMS45 restores skeletal-type EC coupling on expression in dysgenic myotubes. Action-potential-induced Ca2+ transients recorded from dysgenic myotubes expressing DHPR constructs, loaded with the fluorescent Ca2+ indicator Fluo-4 AM. Tick marks on the horizontal axes indicate 2 s. The skeletal GFP-α1S (A) responded to 1-ms stimuli with Ca2+ transients that persisted after blocking currents with 0.5 mM Cd2+ and 0.1 mM La3+ (solid bar), whereas the cardiac GFP-α1C Ca2+ transients (B) were blocked by the Cd2+/La3+ solution. Myotubes expressing GFP-SkLM (C) failed to restore action-potential-induced Ca2+ transients (n = 10 dishes) even though Ca2+ release could be induced with 6 mM caffeine (shaded bar). GFP-SkLMS45 (D) fully restored action-potential-induced Ca2+ transients that were resistant to Cd2+/La3+ block of Ca2+ currents, indicating skeletal-type EC coupling. As for GFP-α1S, the application of Cd2+/La3+ sometimes caused a modest reduction in the amplitude of the transient in cells expressing GFP-SkLMS45. (E) Electrically evoked contractions (100 ms, 100 V) recorded in Cd2+/La3+ from dysgenic myotubes expressing either GFP-α1S, GFP-SkLM, or GFP-SkLMS45 indicated as percentage of myotubes stimulated.
In addition to examining EC coupling in intact myotubes, we also used whole-cell patch clamping to characterize the voltage dependence of Ca2+ release in primary dysgenic myotubes expressing GFP-α1S, GFP-SkLM, and GFP-SkLMS45. Very small Ca2+ transients were present for GFP-SkLM (Fig. 4A). Moreover, these transients appeared to depend on Ca2+ entry because the amplitude of the transient had a voltage dependence mirroring that of Ca2+ current (Fig. 4B). Additionally, the Ca2+ transients for GFP-SkLM were blocked by the addition of Cd2+ and La3+ to the bath (data not shown). In myotubes expressing GFP-SkLMS45, the Ca2+ transients were large (Fig. 4A) and did not differ significantly in either magnitude or voltage dependence from those of GFP-α1S (Fig. 4B). Thus, placing α1S residues L720–L764 into the very dissimilar background of the Musca II-III loop was able to restore both orthograde and retrograde coupling with RyR1.
Figure 4.

Restoration of bidirectional coupling by expression of chimera GFP-SkLMS45. (A) Whole-cell Ca2+ currents (Upper) and depolarization-induced Ca2+ transients (Lower) recorded simultaneously from dysgenic myotubes expressing GFP-SkLM or GFP-SkLMS45. Step depolarizations (200-ms pulses) were applied in 10-mV increments from a holding potential of −80 mV after a prepulse protocol (24). The vertical scale indicates ΔF/F, Ca2+-induced Fluo-3 fluorescence increments (ΔF) with respect to basal fluorescence (F). (B) Voltage dependence of depolarization-induced Ca2+ transients (ΔF/F, Upper) and of peak current densities (pA/pF, Lower) recorded from dysgenic myotubes expressing GFP-α1S (●), GFP-SkLM (♦), and GFP-SkLMS45 (○). Values represent the mean ± SEM of 11–20 recordings. The small Ca2+ transients for GFP-SkLM appeared to be a direct consequence of Ca2+ influx through the DHPR (see text).
A prerequisite for the bidirectional interaction between a DHPR construct and RyR1 is the colocalization of the two proteins in junctions between the plasma membrane and SR. If GFP-SkLM were not targeted into junctions, this could explain the absence of bidirectional signaling observed for this construct. Fig. 5 compares the subcellular distribution of GFP-SkLM (Top) with that of RyR1 (Middle). Both proteins are present in discrete clusters that overlap with one another, and clusters were observed in 54% of transfected myotubes (n = 200), which is close to the value observed in GFP-α1S-transfected myotubes (58%; n = 967). This colocalization, which indicates correct junctional targeting, is particularly evident in the pseudocolor overlay image (Fig. 5 Bottom), in which green and red indicate GFP-SkLM and RyR1, respectively, and yellow shows sites of colocalization. Because GFP-SkLM is able to target to junctions between the plasma membrane and SR, and because its targeting is not different from that of GFP-SkLMS45 [clusters found in 58% of transfected myotubes (n = 65); micrograph not shown], a failure to colocalize cannot account for the lack of bidirectional signaling by GFP-SkLM.
Figure 5.

Reduced Ca2+ currents and lack of EC coupling are not a result of failed junctional targeting of chimera GFP-SkLM. Subcellular localization of chimera GFP-SkLM in a transiently transfected dysgenic myotube (GLT). Double-immunofluorescence labeling was performed with antibodies against GFP, N-terminally fused to the DHPR chimera (Top) and against RyR1 (Middle). The “merged image” (Bottom) emphasizes the colocalization (yellow foci) of GFP-SkLM (green) and RyR1 (red) in clusters that represent junctions of the SR with transverse tubules or with the plasma membrane. Arrows indicate examples of GFP-SkLM/RyR1 colocalization. Inset shows a 2-fold-enlarged view of coclustered channels. N, nuclei. (Bar = 10 μm.)
EC coupling in skeletal muscle likely involves allosteric coupling between α1S and RyR1 either by direct contact between the two proteins or by way of intervening proteins. Using chimeras based on the II-III loop of the Musca DHPR, we have shown that residues L720–L764 of α1S contain a critical domain that is essential for both EC coupling and retrograde signaling. This result is in agreement with previous work using chimeras based on the II-III loop of α1C (8, 9). The new finding that bidirectional signaling survives a drastic change in the sequence of the As10 region makes it very unlikely that this region plays an important role in the activation of RyR1 during EC coupling as postulated previously (12, 13, 16). A similar conclusion also was reached on the basis of a skeletal DHPR in which only the As10 region was scrambled (30).
A model of allosteric coupling between α1S and RyR1 would appear to have two requirements. First, there must be anchoring interactions that maintain α1S and RyR1 in a precise spatial coordination with respect to one another. Second, the allosteric coupling between α1S and RyR1 during orthograde signaling must involve one or more cytoplasmic domains of α1S that undergo conformational changes in response to movement of the voltage-sensing domains. Thus, the critical domain of the II-III loop could be involved in protein–protein interactions that were either static (anchoring) or dynamic (undergoing conformational changes during orthograde signaling) or both. If the critical domain plays a dynamic role in EC coupling, then conformational changes are unlikely to be transmitted to it via the peptide backbone because signaling is normal after large changes in the sequence of the flanking regions. Whatever the role of the critical domain, the important result of the present work is that bidirectional signaling is not affected by dramatic changes in the primary sequence of the loop regions that flank the critical domain. Thus, these flanking regions are unlikely to be sites of protein–protein interaction necessary for bidirectional signaling.
Acknowledgments
We thank L. Grimes, K. Parsons, E. Emberger, and Dr. J. Hoflacher for their excellent experimental help and Dr. H. Glossmann for continuous support. This work was supported in part by Grants P13831- GEN (to M.G.) and P12653-MED (to B.E.F.) from the Fonds zur Förderung der wissenschaftlichen Forschung, Austria, by European Commissions Training and Mobility of Researchers Network Grant ERBFMRXCT960032 (to B.E.F.), and by National Institutes of Health Grant NS 24444 (to K.G.B.).
Abbreviations
- DHPR
dihydropyridine receptor
- RyR
ryanodine receptor
- EC
excitation–contraction
- GFP
green fluorescent protein
- SR
sarcoplasmic reticulum
- Sk
skeletal
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Flucher B E, Franzini-Armstrong C. Proc Natl Acad Sci USA. 1996;93:8101–8106. doi: 10.1073/pnas.93.15.8101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Franzini-Armstrong C, Protasi F. Physiol Rev. 1997;77:699–729. doi: 10.1152/physrev.1997.77.3.699. [DOI] [PubMed] [Google Scholar]
- 3.Schneider M F, Chandler W K. Nature (London) 1973;242:244–246. doi: 10.1038/242244a0. [DOI] [PubMed] [Google Scholar]
- 4.Rios E, Brum G. Nature (London) 1987;325:717–720. doi: 10.1038/325717a0. [DOI] [PubMed] [Google Scholar]
- 5.Nakai J, Dirksen R T, Nguyen H T, Pessah I N, Beam K G, Allen P D. Nature (London) 1996;380:72–75. doi: 10.1038/380072a0. [DOI] [PubMed] [Google Scholar]
- 6.Chaudhari N. J Biol Chem. 1992;267:25636–25639. [PubMed] [Google Scholar]
- 7.Tanabe T, Beam K G, Adams B A, Niidome T, Numa S. Nature (London) 1990;346:567–569. doi: 10.1038/346567a0. [DOI] [PubMed] [Google Scholar]
- 8.Nakai J, Tanabe T, Konno T, Adams B, Beam K G. J Biol Chem. 1998;273:24983–24986. doi: 10.1074/jbc.273.39.24983. [DOI] [PubMed] [Google Scholar]
- 9.Grabner M, Dirksen R T, Suda N, Beam K G. J Biol Chem. 1999;274:21913–21922. doi: 10.1074/jbc.274.31.21913. [DOI] [PubMed] [Google Scholar]
- 10.Lu X, Xu L, Meissner G. J Biol Chem. 1994;269:6511–6516. [PubMed] [Google Scholar]
- 11.El-Hayek R, Antoniu B, Wang J, Hamilton S L, Ikemoto N. J Biol Chem. 1995;270:22116–22118. doi: 10.1074/jbc.270.38.22116. [DOI] [PubMed] [Google Scholar]
- 12.El-Hayek R, Ikemoto N. Biochemistry. 1998;37:7015–7020. doi: 10.1021/bi972907o. [DOI] [PubMed] [Google Scholar]
- 13.Dulhunty A F, Laver D R, Gallant E M, Casarotto M G, Pace S M, Curtis S. Biophys J. 1999;77:189–203. doi: 10.1016/S0006-3495(99)76881-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Casarotto M G, Gibson F, Pace S M, Curtis S M, Mulcair M, Dulhunty A F. J Biol Chem. 2000;275:11631–11637. doi: 10.1074/jbc.275.16.11631. [DOI] [PubMed] [Google Scholar]
- 15.Saiki Y, El-Hayek R, Ikemoto N. J Biol Chem. 1999;274:7825–7832. doi: 10.1074/jbc.274.12.7825. [DOI] [PubMed] [Google Scholar]
- 16.Zhu X, Gurrola G, Jiang M T, Walker J W, Valdivia H H. FEBS Lett. 1999;450:221–226. doi: 10.1016/s0014-5793(99)00496-2. [DOI] [PubMed] [Google Scholar]
- 17.Grabner M, Bachmann A, Rosenthal F, Striessnig J, Schultz C, Tautz D, Glossmann H. FEBS Lett. 1994;339:189–194. doi: 10.1016/0014-5793(94)80413-3. [DOI] [PubMed] [Google Scholar]
- 18.Tanabe T, Takeshima H, Mikami A, Flockerzi V, Takahashi H, Kangawa K, Kojima M, Matsuo H, Hirose T, Numa S. Nature (London) 1987;328:313–318. doi: 10.1038/328313a0. [DOI] [PubMed] [Google Scholar]
- 19.Grabner M, Dirksen R T, Beam K G. Proc Natl Acad Sci USA. 1998;95:1903–1908. doi: 10.1073/pnas.95.4.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adams B A, Beam K G. J Gen Physiol. 1989;94:429–444. doi: 10.1085/jgp.94.3.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Powell J A, Petherbridge L, Flucher B E. J Cell Biol. 1996;134:375–387. doi: 10.1083/jcb.134.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tanabe T, Beam K G, Powell J A, Numa S. Nature (London) 1988;336:134–139. doi: 10.1038/336134a0. [DOI] [PubMed] [Google Scholar]
- 23.Hamill O P, Marty A, Neher E, Sakmann B, Sigworth F J. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 24.Adams B A, Tanabe T, Mikami A, Numa S, Beam K G. Nature (London) 1990;346:569–572. doi: 10.1038/346569a0. [DOI] [PubMed] [Google Scholar]
- 25.Flucher B E, Kasielke N, Grabner M. J Cell Biol. 2000;151:467–477. doi: 10.1083/jcb.151.2.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garcia J, Beam K G. J Gen Physiol. 1994;103:107–123. doi: 10.1085/jgp.103.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flucher B E, Andrews S B, Daniels M P. Mol Biol Cell. 1994;5:1105–1118. doi: 10.1091/mbc.5.10.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giannini G, Conti A, Mammarella S, Scrobogna M, Sorrentino V. J Cell Biol. 1995;128:893–904. doi: 10.1083/jcb.128.5.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sinnegger M J, Wang Z, Grabner M, Hering S, Striessnig J, Glossmann H, Mitterdorfer J. J Biol Chem. 1997;272:27686–27693. doi: 10.1074/jbc.272.44.27686. [DOI] [PubMed] [Google Scholar]
- 30.Proenza C, Wilkens C M, Beam K G. J Biol Chem. 2000;275:29935–29937. doi: 10.1074/jbc.C000464200. [DOI] [PubMed] [Google Scholar]