

Abstract
Membrane fusion mediated by influenza virus hemagglutinin (HA) yields different phenotypes depending on the surface density of activated HAs. A key question is whether different phenotypes arise from different fusion machines or whether different numbers of identical fusion machines yield different probabilistic outcomes. If fusion were simply a less probable event than hemifusion, requiring a larger number of identical fusion machines to occur first, then two predictions can be made. First, fusion should have a shorter average delay time than hemifusion, since there are more machines. Second, fusion should have a longer execution time of lipid mixing after it begins than hemifusion, since the full event cannot be faster than the partial event. Using a new automated video microscopy technique, we simultaneously monitored many HA-expressing cells fusing with erythrocytes and identified individual cell pairs with either full or only partial redistribution of fluorescent lipids. The full lipid mixing phenotype also showed contents mixing, i.e., fusion. Kinetic screening of the digitized fluorescence data showed that the execution of lipid mixing after the onset is faster for fusion than hemifusion. We found no correlation between the delay times before the onset of lipid mixing and the final fusion phenotype. We also found that the execution time for fusion was faster than that for hemifusion. Thus, we provide the first experimental evidence for fusion and hemifusion arising from different machines.
INTRODUCTION
Biological membrane fusion can be described as the protein mediated merging of the membrane bilayers and the opening of an expanding fusion pore to join the aqueous volumes. Using many assays, three basic phenotypes have been defined in the literature. The fusion phenotype is defined traditionally by lipid and contents mixing. The unrestricted hemifusion phenotype has been defined as lipid mixing without contents mixing (Kemble et al., 1994; Melikyan et al., 1997; Chernomordik et al., 1998; Qiao et al., 1999; Kozerski et al., 2000). The restricted hemifusion phenotype has been defined when lipid flow between membranes is restricted, but can be transformed to fusion (lipid- and content-mixing) with treatments known to destabilize the hemifusion structures(Chernomordik et al., 1998; Leikina and Chernomordik, 2000).
These distinct phenotypes are caused by multiprotein aggregates (Bentz, 2000b; Leikina and Chernomordik, 2000), often referred to as fusion machines (Malhotra et al., 1988). For the prototype membrane fusion reaction mediated by influenza hemagglutinin (HA) (White, 1996; Skehel and Wiley, 2000; Bentz and Mittal, 2003), the necessity of HA aggregation and estimates for the numbers of HAs in the aggregates has been deduced with increasing precision by kinetic studies (Ellens et al., 1990; Danieli et al., 1996; Blumenthal et al., 1996; Bentz, 2000a; Mittal and Bentz, 2001; Mittal et al., 2002b). It is known that reducing the surface density of activated HAs will shift the phenotypes from fusion toward hemifusion (Chernomordik et al., 1998; Leikina and Chernomordik, 2000; Mittal et al., 2002a). The question is: Are the distinct phenotypes due to different fusion machines or due to different probabilistic outcomes from different numbers of essentially identical fusion machines?
For influenza, different fusion machines mean different numbers of activated HAs in the aggregate and/or different aggregate geometries. For the analysis of calcium-triggered exocytosis, the assumption has been used that the number of fusion machines (comprised of the same number of proteins) and the corresponding probabilities of fusion fully determine fusion efficiency (Blank et al., 2001). If this were the case for HA-mediated fusion, i.e., if fusion simply requires more of the same machines that generate hemifusion, then the delay for the onset of lipid mixing would be shorter for fusion than for hemifusion, simply because there are more machines when fusion occurs. Furthermore, fusion should have a longer execution time of lipid mixing after it begins than hemifusion, since the full event cannot be faster than the partial event.
We have investigated this question using a novel, simple and rigorous computer driven kinetic analysis of video fluorescence microscopy of HA expressing cells and fluorescently labeled erythrocytes, using both lipid and aqueous fluorophores. For a large number of individually fusing pairs we can extract the delay time for initiation of dye spread and the time required for dye spread to be complete. For each cell pair, we can determine whether lipid mixing is complete or partial and correlate the phenotype with contents mixing. This allowed the novel observation that dye spread was complete only when pores occur. Incomplete lipid dye spread meant no pore formation.
Our data unambiguously demonstrate, for the first time, that different and independent machines generate the fusion and hemifusion phenotypes since we found no correlation between the waiting times for the onset of lipid mixing and the final fusion phenotypes. Moreover, the kinetic analysis shows a single type of machine for fusion, but suggests several different hemifusion machines. Interestingly, at least for the full lipid mixing phenotypes, our analysis suggests that the observed fluorescence spreading is due to the flow through a single channel, i.e., the first successful fusion site, rather than partial flow from many sites. Finally, we found that lipidic connections in unrestricted hemifusion phenotype spontaneously dissociate and thus even after onset of lipid mixing hemifusion remains a transient rather than a stable structure.
MATERIALS AND METHODS
RBC labeling, cell preparation, and fusion
Human RBCs, freshly isolated from whole blood, were labeled with fluorescent lipid R18 or PKH26 (Sigma Chemical, St. Louis, MO) and, in some experiments, with an aqueous dye, 6-carboxyfluorescein, as described (Chernomordik et al., 1998). To label RBC membranes with R18 (Hoekstra et al., 1984), we rapidly added 15 μl of R18 solution in ethanol (1 mg/ml) to 10 ml of a RBC suspension (1% hematocrit) in phosphate-buffered saline (PBS). RBCs were labeled with PKH26 as described previously (Chernomordik et al., 1998). Unbound R18 or PKH26 was removed by washing RBC with complete medium (15 min at room temperature) followed by four washings with PBS. Under these conditions, R18 or PKH26 incorporated into RBC membranes to a high enough concentration to cause significant self quenching of fluorescence. HAb2 cells expressing HA of Japan strain (A/Japan/305/57) used for fusion experiments were grown to ∼50% confluent monolayers. Expressed HA0 at cell surfaces was cleaved into its fusion-competent HA1-S-S-HA2 form with 10 μg/ml trypsin for 15 min at room temperature. We terminated the reaction by washing cells twice with the complete medium. Cells were washed twice by phosphate-buffered solution (PBS) and then incubated for 15 min with a 1 ml suspension of RBCs or RBC ghosts (0.01% hematocrit). The unbound RBC were removed by three washings with PBS. HA cells with only one bound RBC were then used for experiments. Under the above conditions, most of the HA cells eventually had only 1–2 RBCs bound.
Fusion of HAb2 cells with human red blood cells (RBC) labeled by membrane dye R18 or PKH26, in some experiments, with an aqueous dye, 6-carboxyfluorescein, was triggered by application of the low pH medium (PBS titrated by citrate to acidic pH supplemented with 1 mM n-propyl gallate), and assayed with fluorescence microscopy as dye redistribution from RBC to HA cells. All experiments were performed at room temperature (23–25°C).
In some experiments, a low pH pulse of 5 or 10 min was followed by 0.5mM chlorpromazine (CPZ) application for 1 min. Fusion was quantified by counting cells that showed any lipid mixing and/or contents mixing 30 min after the low pH pulse and after CPZ application. In some experiments, CPZ was applied for 1 min after 30 min of the low pH pulse and then fusion was quantified.
Image analysis
Image acquisition was done as described previously (Mittal et al., 2002a). Briefly, redistribution of R18 or PKH26 from RBC to HAb2 cells was observed using a CCD camera and recorded on videotape. The videos were digitized using the WinTV-USB (Hauppauge Computer Works) at a frame capture rate of one frame per second, and converted to Microsoft AVI format files using software AMCAP (Hauppauge Computer Works, Inc.). From the digitized video files, images were extracted at the resolution of one image frame per second.
The images were analyzed with Scion Image software (Scion Corp., Frederick, MD). The fluorescence microscopy was dark background, with RBCs appearing as bright particles due to the fluorescent dye incorporated in their membrane, as shown in Fig. 1. The underlying HAb2 cells are not visible. The spread of the dye is clear as a function of time. The key times we need are the delay time before dye spread begins and the kinetics of the dye spread.
FIGURE 1.

Kinetics of lipid mixing event for a single RBC-HA expressing cell pair. Open circles show the area of a bright spot, which is a fluorescent RBC bound to a HA cell (in the dark background), as a function of time. As the RBC fuses with the HA cell, the fluorescent dye starts redistributing due to lipid mixing. The slight noise in the area measurements is due to image acquisition. Since complete kinetics of a single cell pair lipid mixing event can be measured using this technique (shown by the solid line drawn through the data), it is possible to define the “delay time” consistently for each fusing cell pair. The delay time, denoted by td, is defined by the intersection of the tangent to the steepest part of the smooth curve (drawn through the data) with the x axis. It is also possible to consistently define a time of completion of each single cell pair fusion event. This time of completion, denoted by ts, is defined by the intersection of the tangent to the steepest part of the curve with the asymptotic/saturation value of the increase in fluorescence area. ts − td denotes the time taken for the execution of a single cell pair lipid mixing event after the onset of dye redistribution.
The images were color inverted, as analysis in Scion image does not allow dark backgrounds. Note that color inversion does not modify any characteristics of the data and just reverses the pixel values on a scale of 0–256. Due to image-acquisition noise, appropriate thresholds were selected based on histogram analysis of at least 10 random images of the same field. After fixing the threshold values, the area for each RBC in the field being monitored was obtained from Scion Image for every frame. As each RBC fuses with the HA cells, the membrane dye spreads, which is quantified by the increase in the fluorescent area of the particle. Note that the threshold value does not affect the final outcome, since we are interested only in the increase in area of the fluorescence (i.e., the threshold is just a means to filter and minimize the image acquisition noise). The total area of fluorescence spread coupled with bright field observations of the fields indicated that size/shape of the underlying HAb2 cells was quite uniform.
Extracting single cell pair fusion kinetics in MATLAB
The collected data (area measurements for each RBC in the field per second) were then transferred to MATLAB (The MathWorks) for extracting fusion kinetics for each RBC fusing with its corresponding HA cell. Area measurements, normalized by subtracting the initial area of the RBC attached to a single HAb2 cell (not visible in the dark background) from all the subsequent measurements (hence data start from zero), were collected as shown by the open symbols in Fig. 1. The normalization was achieved by subtracting average initial areas, i.e., areas measured in the first few seconds, due to noise in image acquisition. Theoretically, given a perfect microscope and no fluctuations in its lamp, we would need to subtract only the very first area measurement. To uniformly quantify the kinetic characteristics (e.g. delay time) for dye redistribution between the fusing membranes, the change in the fluorescence area curve was fit with an empirical equation:
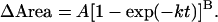 |
(1) |
This equation is an analytical solution for a mass-action based model for HA-mediated fusion and has been exhaustively tested for fitting first fusion pore formation data for HA-mediated fusion (Bentz, 2000a). However, for the purpose of studying lipid mixing, this formalism is just a convenient way of fitting the experimental data. All fitting was done in MATLAB (The MathWorks, Inc.) using the function “fmins” as done previously (Bentz, 2000a; Mittal and Bentz, 2001; Mittal et al., 2002b). The aim of this fitting was to consistently define the kinetic parameters for the dye redistribution curve (represented by the area measurements). Each area data curve was fit to 16 initial conditions (for A, B, and k) and the best fit final values for A, B, and k, i.e., those giving the smallest root mean squared error (rmse), were selected to represent the actual data curve. The solid sigmoidal line in Fig. 1 shows the best fit to the area data for a particular case.
Using the fitted curve, we analytically defined the delay time (td) as the x intercept of the tangent to the steepest part of the curve (Bentz, 1992). By symmetry, we also defined an effective saturation time of dye redistribution (ts) as the intercept of this same tangent with the asymptote to the lipid mixing curve. Finally, we define the difference of these times, ts − td, as the time to execution of the lipid mixing. These two parameters, ts and td, completely define the kinetics of area change. Any other delay or completion time definitions would only be proportional and would not change any conclusions.
To avoid artifacts pertaining to extrapolating the fitted curve, only those curves were selected for which the area data (open symbols) visually crossed the inflection point on the latter part of the curve, which amounted to roughly 70% of the RBC-cell pairs which attempted fusion. This yielded reliable estimates (i.e., reliable sigmoidal extrapolations) for the extent of increase in fluorescence areas. These cells are our population of fusing cells which show lipid mixing.
RESULTS
Monitoring single cell-cell fusion using automated video fluorescence microscopy
As seen in real time fluorescence microscopy, the onset of the lipid mixing, as well as its rate and final extent differ from cell pair to cell pair in the same field of view. To simultaneously follow these multiple fusion reactions we digitized the recorded video with 1s resolution. For each field we identified the background fluorescence (see Methods) and quantified the fluorescent area for each individual RBC/HAb2 pair at each time point. This allowed us to individually measure the fluorescence redistribution for each of the multiple cell pairs in the field. This way, if there were ten cell pairs in the field, we collected ten samples of lipid mixing kinetics.
Open symbols in Fig. 1 show area measurements for a single RBC/HAb2 pair collected from image analysis. Solid line shows the fit to the data to extrapolate those curves that did not reach saturation. Delay times (td) and times of completion for the lipid mixing (ts) were defined consistently for each cell pair as shown (see figure legend and Methods for details). The spread of fluorescent lipids from the RBC to the HAb2 cell (in the dark background) due to lipid mixing is shown by the images A, B, C, and D taken at various time points on the kinetic curve. Note that we monitor only the spread of fluorescence, without regard to the relative intensities of the different cells.
Closed symbols in Fig. 2 show the cumulative distributions of delay times for lipid mixing between HA cells and R18 labeled RBCs at pH 4.8 (circles) and pH 5.2 (squares) respectively, obtained manually (Mittal et al., 2002a). The manual method involves finding the delay in onset of dye redistribution by eye. Open symbols show the cumulative distributions of td for the same data using the automated method. Although the former method was extremely labor intensive and took months of data analysis, our new method took only a few days of operation time. Clearly, the distributions are nearly identical, thus validating the automated method.
FIGURE 2.

Comparison of kinetics of lipid mixing using the automated measurements (of delay time described in Fig. 1) and manual measurements. Closed symbols show the manually obtained cumulative distributions (Mittal et al., 2002a) of delay times for RBCs fusing with HA cells at pH 4.8 (circles) and pH 5.2 (squares). Open symbols show the cumulative distributions obtained for the same video microscopy data using the automated measurements. Although the former took months of data analysis to obtain these cumulants manually, the latter kinetic distributions were obtained only in few days of data analysis.
Existence of kinetically distinct lipid mixing events
If the HA fusion machines are identical, then we would expect a single distribution of delay times and a single distribution of execution times, ts − td, i.e., the time to execution of lipid mixing after the onset. Simply put, a single fusion machine would be expected to take a similar average time to finish its job after starting it for every individually fusing cell pair.
Fig. 3 shows the above distributions for R18-labeled RBCs and for PKH26-labeled RBCs at pH 4.8 (A and B, respectively). Clearly, there are apparent subsets shown by the “staircase” kind of structure of the distributions. These have been collected into kinetic subsets denoted by S1–S5, with demarcations simply being the apparent end of one stair and the beginning of the next. Similar distributions were obtained at pH 5.2 for RBCs labeled with either dye (C and D) indicating that the kinetic subsets are observed for different membrane probes and at different pH. The staircase structure is not as prominent for pH 5.2 with R18 (Fig. 3 C), as compared to other data sets in Fig. 3. Nevertheless, we subdivided the distribution in Fig. 3 C based on the mild visual demarcations, since we were interested primarily in the fastest and slowest subsets in the staircase structure, as addressed in the results below.
FIGURE 3.

Distributions of the start-to-finish (execution) times of lipid mixing. Cumulative ts − td (defined in Fig. 2) distributions for lipid mixing were obtained at pH 4.8 (A and B) and 5.2 (C and D) using R18 (A and C) and PKH26 (B and D) respectively. These cumulative distributions represent the fraction of cells with a completed lipid mixing event, with 100% being all the cells which show any lipid mixing. If the mechanisms for the lipid mixing in all the individually fusing cell pairs is the same, i.e., lipid mixing in all the cell pairs proceeds through similar lipid connections, one would expect the time taken from the onset of lipid dye redistribution to completion of dye redistribution event to be similar for all the cell pairs and follow a smooth curve. However, clear kinetic subsets of cell pairs are seen in terms of the visible staircase structures. The staircase structure is divided into kinetic subsets S1 to S5 based on visual inflection points. Statistically, Kolmogorov-Smirnov tests show that these staircase structures in all the four data sets cannot be explained by single normal distributions having the respective means and standard deviations, i.e., multiple distinct distributions are required to explain these (see text for details).
While the presence of staircases was an indication of kinetically distinct events, we were interested to see whether these kinds of structures can be explained by a single random normal distribution with noise in the data. To test the above, we did Kolmogorov-Smirnov (KS) tests for the ts − td distributions for both lipid dyes at both pH values. KS tests were used here to compare two distributions with the same mean and standard deviation, with equal sample size, for similarity or difference. Here we compared theoretical normal distributions having the same mean and standard deviation with each of the ts − td distributions. The results of these tests are that there is less than 5% chance for these kinetic subsets to result from a single normal distribution for R18 data. For PKH26 data, there is less than 1% chance for the kinetic subsets to result from a single normal distribution. Therefore, a single normal distribution by chance is very unlikely to result in a staircase like structure obtained for any data set here. Although it remains possible that the data could be explained by some other single distribution, the phenotypes comprising the kinetic subsets show different lipid mixing events. The td distributions (Fig. 2) were explained by single normal distributions using the KS tests, as expected. The fact that distributions of execution times could not be a result of a single distribution implies that all the fusing cell pairs do not end up at the same completion point.
Two phenotypes of lipid mixing
From the new kinetic measurement defined by our automation, i.e., ts, we observed that some cells showed complete redistribution of the membrane dye whereas some showed only incomplete redistribution of the membrane dye after ts time i.e., at the end of the lipid mixing process. These different patterns of lipid mixing cannot be explained by the variations in either focal angle for different RBCs or image acquisition or in the size/shape of the HA cells. Whatever the ratio between the sizes of contacting HA cell and RBC, partial lipid mixing phenotype is seen clearly as more fluorescent RBC bound to less fluorescent HA cell.
Fig. 4 shows four different cell-RBC pairs, at pH 4.8 using R18 and PKH26 as membrane dyes, before the defined delay time and after the defined finishing time of the fusion process. Clearly, the observed lipid mixing in the upper panel is phenotypically different from that in the lower panel for both the dyes.
FIGURE 4.

Full and partial lipid mixing. Two phenotypes of lipid mixing are observed based on the quality of lipid mixing. For some RBCs fusing with HA cells, complete lipid probe redistribution is seen after defined time of completion of the lipid mixing event. Complete lipid dye redistribution is defined by lack of any remaining boundary of the RBC, i.e., lack of a distinct circular shape of the RBC at the end of the experiment. This is shown by the upper four panels, in which the first column shows R18 redistribution and the second column shows PKH26 redistribution. First row shows the RBC appearance at the time the pH is lowered, and second row shows the image of the same RBC at the end of the experiment. This lipid mixing phenotype, for which the end point is complete lipid dye redistribution, is defined as full lipid mixing. For some RBCs, incomplete lipid probe redistribution is seen well after the defined time of completion of the lipid mixing event. A clear boundary of the initial RBC membrane is seen in the lowest two panels signifying only partial exchange of the lipid dye between the membranes. This lipid mixing phenotype, for which the end point is incomplete dye redistribution, is defined as partial lipid mixing.
We define partial lipid mixing as the event where one can still see the initial boundary of the fusing RBC (i.e., incomplete redistribution of the membrane dye) and full lipid mixing as the event where no remaining boundary of the fusing RBC is visible (i.e., complete redistribution of the membrane dye). This phenotypic divide was surprising, since the partial lipid mixing phenotype persisted well beyond the defined finishing time.
Fig. 5 shows the percentages of the fusing cells with full lipid mixing phenotype at pH 4.8 and 5.2 for both R18 and PKH26. For both dyes, 25–30% of the cells showed full lipid mixing phenotype at pH 4.8, as compared to ∼5% of the cells showing the same phenotype at pH 5.2. Similar phenotypes and similar fractions for the phenotypes with two different dyes strongly suggested that the observations were real. Since ts essentially represents completion of the lipid mixing process, this could mean either that the cells with partial lipid mixing phenotype were stuck at some fusion intermediate stage with a blocked lipid connection or this phenotype was a result of reversible merger of membranes (Leikina and Chernomordik, 2000). Our results discussed below suggested that the cells with partial lipid mixing phenotype are somehow involved in a fundamentally different mechanism for the lipid mixing event as compared to the full lipid mixing phenotype.
FIGURE 5.

Full lipid mixing phenotype as a function of pH. Black bars show the percentage of cells with complete R18 redistribution and gray bars show the percentage of cells with complete PKH26 redistribution. N denotes the total number of cells that showed any lipid mixing (i.e., either of the two phenotypes in Fig. 4), i.e., N denotes 100%*.
Full lipid mixing phenotype implies contents mixing
Our result that there were ∼5 times more cells with full lipid mixing phenotype at pH 4.8 as compared to pH 5.2 was reminiscent of the earlier observation that the shift from lipid and content mixing to only lipid mixing upon raising pH was about the same (Chernomordik et al., 1998; Leikina and Chernomordik, 2000). Could the full lipid mixing phenotype represent content mixing?
To test whether contents mixing correlated with the lipid mixing phenotypes, we fused HA cells and double-labeled RBCs (with PKH26 as membrane dye and carboxyfluorescein (CF) as aqueous dye) at pH 4.8. Fig. 6 shows the percentages of cell pairs with different lipid mixing phenotypes and the percentage of cells with CF redistribution. Clearly, the percentage of cells with full lipid mixing phenotype was nearly the same as the percentage of cells with CF redistribution. The obvious next question was whether the identical percentages also meant that the same cell pairs were showing full lipid mixing and CF redistribution.
FIGURE 6.

Lipid mixing phenotypes versus contents mixing. Double-labeled RBC ghosts were fused with HA cells at pH 4.8 using PKH26 as the lipid probe and CF as the aqueous probe. Clearly, the percentage of cells with full lipid mixing phenotype, i.e., percentage of cells showing complete PKH26 redistribution, is nearly the same as the percentage of cells with contents mixing, i.e., percentage of cells with CF redistribution. As in Fig. 5, N denotes the total number of cells that showed any lipid mixing, i.e., N denotes 100%*.
Out of 112 cell pairs monitored in this experiment, phenotypes of 108 cell pairs correlated, as predicted above, with fusion (full lipid mixing implies contents mixing) and hemifusion (partial lipid mixing implies no contents mixing). The four exceptions were not quantifiable due to either RBC lysis, photobleaching, or dye leakage. Fig. 7 shows some typical examples of this correlation. The images were taken ∼4 min after lowering the pH to 4.8. Fig. 2 shows that all observed lipid mixing events start well within 3 min at pH 4.8, and our results discussed below (see Figs. 3 and 8) show that full lipid mixing is completed within ∼1 min after the onset of lipid mixing at this pH. Clearly, CF spreading is seen only for the cell pairs with fully redistributed PKH26, i.e., aqueous dye redistribution is seen for full lipid mixing phenotypes only. Regardless of the degree of the incompleteness of lipid dye redistribution, provided that the initial boundary of the RBC was still visible, none of cell pairs showed contents mixing. Similar results were obtained in the experiments with R18 and CF labeled RBCs (not shown). Therefore, full lipid mixing phenotype corresponds essentially exactly to contents mixing and partial lipid mixing phenotype corresponds essentially exactly to unrestricted hemifusion. Strictly speaking, our content mixing assay does not resolve fusion pores too small to allow the passage of CF( Chernomordik et al., 1998). Therefore some of the cell pairs scored as unrestricted hemifusion likely represent the case of small nonexpanding fusion pores.
FIGURE 7.

Full lipid mixing phenotype correlates with contents mixing, and partial lipid mixing phenotype correlates with hemifusion. Representative images of double labeled RBC ghosts fusing with HA cells at pH 4.8 were taken after ∼4 min of lowering the pH. From the waiting time cumulants at pH 4.8, it was known that within ∼3 min, nearly all the possible lipid mixing events have initiated (see Fig. 2) and cells showing full lipid mixing phenotype complete their dye redistribution within 50–75 s (see text for details). Looking at cells after longer times would not change the conclusion of this figure. Each box in the figure has an upper panel of images showing the double-labeled RBC for PKH26 and CF at the time of lowering the pH. The lower panel shows the respective dye redistribution (or lack of) at the end of the experiment. Clearly, cells having full lipid mixing phenotype (i.e., complete PKH26 redistribution) show contents mixing (i.e., CF redistribution) and cells having partial lipid mixing phenotype show no contents mixing. For all qualities of partial lipid mixing, no contents mixing is observed.
FIGURE 8.

Lipid mixing phenotypes in different kinetic subsets defined in Fig. 3. Representative images are shown for RBCs fusing with HAb2 cells at pH 4.8 using R18 and PKH26 as lipid mixing probes. Images of different fluorescent RBCs before onset of lipid mixing and after completion of the all the dye redistribution are shown, as in Fig. 5, for all the kinetic subsets. Clearly, full lipid mixing events belong to the initial kinetic subsets (defined in Fig. 4) and partial lipid mixing events are completely dominant in the later kinetic subsets.
After the onset lipid mixing in fusion is faster than in hemifusion
It is now clear that we have identified both phenotypically and kinetically distinct lipid mixing events. The next question was how are they correlated to each other. Fig. 8 shows some examples of the phenotypes of RBCs from the subsets of cell pairs defined as S1–S5 in Fig. 3 at pH 4.8 for both the membrane dyes used. Each kinetic subset has two panels. The upper panel shows the RBC appearance before onset of dye redistribution, and the lower panel shows that RBC's appearance ∼50 s after ts It is apparent that the kinetic subset with the fastest execution of lipid mixing after onset, S1, shows the full lipid mixing phenotype. The slower subsets show various degrees of partial lipid mixing phenotypes, which we have not attempted to subdivide phenotypically. At pH 5.2, with fewer activated HAs, primarily the hemifusion phenotype was observed. These results are quantified in Tables 1 and 2.
TABLE 1.
Number of cells with different lipid mixing phenotypes at pH 4.8 with R18 and PKH26 as lipid probes
| Probe | S1 | S2 | S3–S5 |
|---|---|---|---|
| Number of cells with full lipid mixing phenotype in kinetic subsets | |||
| R18 (n = 20) | 15 | 5 | — |
| PKH26 (n = 22) | 9 | 10 | 3 |
| Number of cells with partial lipid mixing phenotype in kinetic subsets | |||
| R18 (n = 55) | 1 | 17 | 37 |
| PKH26 (n = 55) | 1 | 4 | 50 |
TABLE 2.
Number of cells with different lipid mixing phenotypes at pH 5.2 with R18 and PKH26 as lipid probes
| Probe | S1 | S2–S5 |
|---|---|---|
| Number of cells with full lipid mixing phenotype in kinetic subsets | ||
| R18 (n = 3) | 3 | — |
| PKH26 (n = 4) | 3 | 1 |
| Number of cells with partial lipid mixing phenotype in kinetic subsets | ||
| R18 (n = 55) | 1 | 54 |
| PKH26 (n = 48) | 1 | 48 |
Clearly, full lipid mixing has a faster time to execution than any of the partial lipid mixing phenotypes. This strongly suggested that both events follow separate kinetic pathways, if only because a partial event cannot be slower than the full event.
Lipid mixing phenotype is independent of delay times
One may expect that fusion machines yielding different patterns of lipid mixing take different time to form. Perhaps those machines yielding full lipid mixing, with the shortest times to execution, would have consistently shorter (or longer) delay times. However, a complete lack of correlation between td and ts − td (Fig. 9) argues against this hypothesis. Cell pairs with an earlier onset of lipid dye redistribution do not necessarily finish earlier. On the other hand, the half time for all the data shown in Fig. 9 correlates quite well with ts (0.95 > regression coefficients > 0.5, not shown). Thus, a decision is made during the delay time for the end point of the fusion event and, once started, the event finishes roughly on time.
FIGURE 9.

The lack of correlation between the delay time and the execution time of lipid mixing. ts − td, the execution time for lipid mixing after the onset of dye redistribution, is independent of td, the delay time in the onset. All the data from all the kinetic subsets in Fig. 3 were pooled together for the plot. The dashed lines show rough demarcations between fusion and hemifusion phenotypes based on execution times. The data points below the dashed lines in each of the panels correspond to the fusion phenotype and above the lines correspond to hemifusion phenotype (see text and Tables 1 and 2 for more details).
Cells in partial lipid mixing phenotype do not stay hemifused
Lipid flow can be blocked at a stage of partial lipid mixing because lipidic junction between the hemifused cells dissociates. To test this possibility we took advantage of the earlier work indicating that hemifusion connections can be transformed into expanding fusion pores by application of an amphiphilic agent chlorpromazine, CPZ (Melikyan et al., 1997; Chernomordik et al., 1998; Grote et al., 2000; Leikina and Chernomordik, 2000; Armstrong et al., 2000). In the experiments presented in Fig. 10, RBCs double labeled with PKH26 and CF were fused with HAb2 cells at pH 5.3 at 24°C, i.e., under conditions that would yield predominantly partial lipid mixing phenotype. Applying CPZ immediately after a low pH pulse promoted the full lipid mixing phenotype and contents mixing (both had one to one phenotypic correspondence in these experiments) as shown by gray bars, compared to the limited extent of full lipid mixing phenotype (and contents mixing) without any CPZ treatment as shown by the black bars. If CPZ was applied after 30 min of the low pH pulse, it had no effect on the percentage of the cell pairs showing the full lipid mixing phenotype, as shown by the blank bars, compared to the black bar. In agreement with (Leikina and Chernomordik, 2000), the CPZ-sensitive lipidic connections that can be affected by CPZ were lost in the post low pH pulse period of 30 min Note that the percentages in Fig. 10 are based only on those cells that showed any lipid mixing. The actual percentages for all the cell pairs (i.e., including those that did not show any dye redistribution) were less than 50%, with full lipid mixing phenotype accounting for <5% without CPZ (see Fig. 5).
FIGURE 10.

Dissociation of hemifusion connections in partial lipid mixing phenotype. Under conditions where predominantly partial lipid mixing phenotype is observed, we tested effect of CPZ application on the percentage of cells with full lipid mixing phenotype. Black bars show the percentage of cells with full lipid mixing phenotype when pH 5.3 was applied for 5 or 10 min after prebinding CF- and PKH26-labeled RBCs with HAb2 cells, then replacing the medium with PBS at neutral pH and counting cell pairs showing any lipid mixing after 30 min, 0.5 mM CPZ in PBS was applied for 1 min immediately after the pH 5.3 pulse (gray bars) or after 30 min at neutral pH (blank bars) and cell pairs with any lipid mixing were counted. Full lipid mixing phenotype was also confirmed by contents mixing observed by CF redistribution.
DISCUSSION
We wanted to determine whether fusion and unrestricted hemifusion arose from different fusion machines or whether a single type of fusion machine could yield both phenotypes as distinct probabilistic outcomes. If there were only one identical machine for hemifusion and fusion, then hemifusion would be an earlier outcome and fusion a later, more advanced or complex, outcome. To investigate the above, we used the newly developed automated video fluorescence microscopy to segregate and analyze the lipid mixing kinetics in two major fusion phenotypes: content and full lipid mixing (fusion) vs. only partial lipid mixing (unrestricted hemifusion). Our main technical strength is comparison of the different phenotypes using the same variable (i.e., lipid mixing), in contrast to the different assays required for individual phenotypes in prior studies. This allowed the novel observation that dye spread was complete only when pores occur. Incomplete lipid dye spread meant no pore formation. Another novel observation was that the delay time before the onset of lipid mixing was uncorrelated with phenotype, Fig. 9. If fusion were due to more identical fusion machines achieving a less likely outcome, then the average delay time for fusion would have to be less than that for hemifusion, which is not the case.
We used the automated analysis to screen lipid mixing in real time for many cell pairs thereby quantitatively characterizing the kinetics for the individual pairs yielding different phenotypes. This automated analysis decreased the data analysis time ∼100-fold, compared with visual analysis (Mittal et al., 2002a). Without this analytical package, assessing delay times would have been laborious and assessing execution times would have been impractical. The implications of these execution times will be discussed below.
The full lipid mixing phenotype, which was proven to include aqueous contents mixing, was found in about a third of the cases at pH 4.8 and was reduced to ∼5% at pH 5.2, Fig. 5. The fact that raising the pH led to fewer full lipid mixing pairs and more partial lipid mixing pairs suggests that the hemifusion machines have fewer activated HAs, as originally suggested (Chernomordik et al., 1998). Further, since the same single normal distribution described the pool of delay times for both phenotypes, assembly of fusion machines likely takes about the same average time as assembly of hemifusion machines. Since HA aggregation is rapid compared with first pore formation (Bentz, 2000a), assembly implies a postaggregation event.
Partial lipid mixing phenotype in unrestricted hemifusion
There is also the question of why membrane probe redistribution stops in the partial lipid mixing phenotype. Since the partial lipid mixing was observed for two different lipid probes, it is unlikely that probe redistribution between merged membranes is limited by a very low solubility of the probes in some large membrane domains surrounding the fusion machine either at RBC or HA cell sides. An alternative suggestion would be that the dye, which stays unredistributed, is the probe in the inner monolayer of RBC membrane. This seems unlikely since R18 is known to rapidly partition between both of the monolayers of the labeled membrane (Melikyan et al., 1996).
Leikina and Chernomordik (2000) found that after the unrestricted hemifusion phenotype was established, proteolysis of low pH-activated HAs dissociated the RBC-HA-cell connections and released RBC. Thus, at least after proteolysis, the membrane connection was broken. Our results present the first evidence that lipidic connections yielding unrestricted hemifusion dissociate spontaneously. This explains why unrestricted hemifusion is a functional dead-end. Assuming that the gradual dissociation of hemifusion connections reflects the inactivation of HAs, our finding is consistent with the recent theoretical studies predicting that the hemifusion state for lipid bilayers is energy-intensive (Kuzmin et al., 2001; Kozlovsky and Kozlov, 2002; Kozlovsky et al., 2002; Markin and Albanesi, 2002). These intermembrane connections remain energetically favorable only while activated HAs support them.
Thus, hemifusion is the outcome when outer monolayers mix transiently, but the inner monolayers of the potential fusion site are not engaged. The free energy released by the conformational changes of HA are needed hold the lipidic contact together and to cause this engagement of the inner monolayer. Peptides and FHA2 are able to perturb only the outer monolayers. Perturbing the outer monolayers appears fairly easy, requiring only peptide binding and disturbing the packing of the lipids. Tieleman and Bentz (2002) presented a molecular simulation that suggested one mechanism by which the inner monolayers could be engaged.
Execution times for fusion and hemifusion
Does mixing of the outer monolayers in hemifusion and fusion proceed through the same path? Our data show that the execution of lipid mixing in fusion is faster than that in unrestricted hemifusion and there is (at least) one path leading to full lipid mixing and several paths leading to unrestricted hemifusion (staircase(s) in Fig. 3). It may be that during fusion, lipid mixing is accelerated by fast expansion of the fusion pore. It is clear that the hemifusion machine only engages the outer monolayers. In contrast, the machine yielding fusion engages the inner monolayers of both target and HA-containing membranes in the process. Molecular dynamic simulations have just begun the task of trying to understand what could cause this engagement (Tieleman and Bentz, 2002). Another intriguing question is whether lipid mixing proceeds through many channels, each contributing a portion of the spreading of fluorescence signal, or whether it proceeds from a portal formed from the first successful site. Our kinetic analysis of the first fusion pore formation and of lipid mixing between erythrocytes and HA-expressing cells always predicted hundreds of potential fusion sites, but the rate constants predicted that only one of these sites would accomplish the bulk of the lipid mixing before the second successful site would open (Bentz, 2000a; Mittal and Bentz, 2001; Mittal et al., 2002b). Direct experiment evidence supporting this kinetic analysis was shown in Fig. 6 of Mittal et al. (2002a). Electron microscopy has shown many contact points between erythrocytes and HA-expressing cells, however statistical analysis suggested that lipid mixing and pore formation take place only through a very small fraction of the total number contact points (Frolov et al., 2000). Given that the full lipid mixing phenotype in this work was found in only about a third of the cell pairs at pH 4.8 and was reduced to ∼5% at pH 5.2, Fig. 5, we also suggest that very few of the many contact sites lead to fusion. The relationship between the microscopically visualized contact points and the fusion sites characterized by the kinetics analyses remains to be clarified.
Our automated video fluorescence microscopy analysis can provide powerful insight into this question. It provides both the delay time distribution(s) and the execution time distribution(s). If lipid mixing were through a single portal, then our finding that the execution time for fusion was faster than that for unrestricted hemifusion would prove that different machines were involved. Since it is clear that the decision for hemifusion or fusion is made during the delay time, the only way that execution of fusion could be faster than hemifusion after that delay time is if they were due to different machines. The execution time would simply be the time for the portal to equalize the fluorophore concentration in the merged membranes.
If lipid mixing is due to partial contributions from many identical machines, then the interpretation of execution time is more complex, but would still require different machines for fusion and hemifusion. If all of the machines opened simultaneously and the lipid flow through each was identical, i.e., each machine moved a small fraction of the whole lipid pool, then the flow would be identical to a single site moving all of the lipid pool. However, even if the machines are identical, there is no mechanism to support the postulate that they would all open at once. Such synchrony would be completely at odds with the current landscape theory of protein folding. The postulate of partial contributions of lipid flow toward lipid mixing would be that the machines which yield fusion, which are the most interesting, are identical and have a single distribution of opening times and of flow rates (i.e., execution times). The same would hold true for hemifusion events also. Thus, in this case also, multiple distributions for execution times of the pooled lipid mixing events mean that different machines cause fusion and hemifusion.
Implications for fusion mechanisms
Our results contradict the simple hypothesis that different numbers of identical fusion machines generate different fusion phenotypes with different probabilities, at least for the cases of fusion and unrestricted hemifusion. Raising the number of activated HAs would increase the number of identical fusion machines allowing realization of less probable and more advanced fusion phenotypes. If this hypothesis were correct, fusion would require a higher number of hemifusion machines and thus, one would expect partial lipid mixing (unrestricted hemifusion) to develop after shorter waiting time than full lipid mixing (fusion). This is clearly not the case as shown by the lack of correlation between the waiting time before the onset of the lipid mixing and the final fusion phenotype.
The staircase distributions clearly reflect multiple kinetic subsets of lipid mixing events. Although phenotypic classification remains qualitative (based on visual identification of the remaining boundary of the RBC in partial lipid mixing phenotype), our results prove the existence of two, or more, distinct kinetic subsets. The kinetic subsets could be explained by the size of the fusion machines, i.e., the number of HAs (Ellens et al., 1990; Blumenthal et al., 1996; Danieli et al., 1996; Markovic et al., 2001; Roche and Gaudin, 2002), coupled with the cooperativity found in HA activation as the number of HAs in the area of contact increases (Markovic et al., 2001; Mittal et al., 2002b). In addition to size, the machines can differ in the distribution of HA conformations within the machine (Bentz, 2000a,b; Bentz and Mittal, 2000; Mittal and Bentz, 2001; Mittal et al., 2002b). Note that no assumption of HA surface density homogeneity over cells has been made or was required to explain the staircases. We have single distributions of delay times, thus this parameter is independent of any underlying HA surface density heterogeneity. The staircase shows that there is a distribution of execution times, and hence different machines. If the staircase was due to a heterogeneous HA surface density distribution amongst the cells, then the simple explanation would be different HA aggregate sizes as a function of HA surface density, as predicted by our previous kinetic analysis (Bentz, 2000a). We believe that further applications of our new technique along with theoretical modeling and characterization of HA conformations for different subsets will be necessary to elucidate the structures of the different sites of HA mediated membrane destabilization, as well as different fusion systems.
Acknowledgments
We thank Drs. Thomas Twardowski for very helpful advice on image analysis and Michael O'Connor for help on statistical analysis. We also acknowledge enjoyable discussions with Drs. Shivanthi Anandan, James Lear, Jeremy Lee, Kamran Melikov, Corinne Ramos, and Joshua Zimmerberg.
References
- Armstrong, R. T., A. S. Kushnir, and J. M. White. 2000. The transmembrane domain of influenza hemagglutinin exhibits a stringent length requirement to support the hemifusion to fusion transition. J. Cell Biol. 151:425–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentz, J. 1992. Intermediates and kinetics of membrane fusion. Biophys. J. 63:448–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentz, J. 2000a. Minimal aggregate size and minimal fusion unit for the first fusion pore of influenza hemagglutinin-mediated membrane fusion. Biophys. J. 78:227–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentz, J. 2000b. Membrane fusion mediated by coiled coils: a hypothesis. Biophys. J. 78:886–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentz, J., and A. Mittal. 2000. Deployment of membrane fusion protein domains during fusion. Cell Biol. Int. 24:819–838. [DOI] [PubMed] [Google Scholar]
- Bentz, J., and A. Mittal. 2003. Architecture of the influenza hemagglutinin membrane fusion site. Biochim. Biophys. Acta. 1614:24–35. [DOI] [PubMed] [Google Scholar]
- Blank, P. S., S. S. Vogel, J. D. Malley, and J. Zimmerberg. 2001. A kinetic analysis of calcium-triggered exocytosis. J. Gen. Physiol. 118:145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenthal, R., D. P. Sarkar, S. Durell, D. E. Howard, and S. J. Morris. 1996. Dilation of the influenza hemagglutinin fusion pore revealed by the kinetics of individual cell-cell fusion events. J. Cell Biol. 135:63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernomordik, L. V., V. A. Frolov, E. Leikina, P. Bronk, and J. Zimmerberg. 1998. The pathway of membrane fusion catalyzed by influenza hemagglutinin: restriction of lipids, hemifusion, and lipidic fusion pore formation. J. Cell Biol. 140:1369–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danieli, T., S. L. Pelletier, Y. I. Henis, and J. M. White. 1996. Membrane fusion mediated by the influenza virus hemagglutinin requires the concerted action of at least three hemagglutinin trimers. J. Cell Biol. 133:559–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellens, H., J. Bentz, D. Mason, F. Zhang, and J. M. White. 1990. Fusion of influenza hemagglutinin-expressing fibroblasts with glycophorin-bearing liposomes: role of hemagglutinin surface density. Biochemistry. 29:9697–9707. [DOI] [PubMed] [Google Scholar]
- Frolov, V. A., M. S. Cho, P. Bronk, T. S. Reese, and J. Zimmerberg. 2000. Multiple local contact sites are induced by GPI-linked influenza hemagglutinin during hemifusion and flickering pore formation. Traffic. 1:622–630. [DOI] [PubMed] [Google Scholar]
- Grote, E., M. Baba, Y. Ohsumi, and P. J. Novick. 2000. Geranylgeranylated SNAREs are dominant inhibitors of membrane fusion. J. Cell Biol. 151:453–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoekstra, D., T. de Boer, K. Klappe, and J. Wilschut. 1984. Fluorescence method for measuring the kinetics of fusion between biological membranes. Biochemistry. 23:5675–5681. [DOI] [PubMed] [Google Scholar]
- Kemble, G. W., T. Danieli, and J. M. White. 1994. Lipid-anchored influenza hemagglutinin promotes hemifusion, not complete fusion. Cell. 76:383–391. [DOI] [PubMed] [Google Scholar]
- Kozerski, C., E. Ponimaskin, B. Schroth-Diez, M. F. Schmidt, and A. Herrmann. 2000. Modification of the cytoplasmic domain of influenza virus hemagglutinin affects enlargement of the fusion pore. J. Virol. 74:7529–7537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlovsky, Y., and M. M. Kozlov. 2002. Stalk model of membrane fusion: Solution of the energy crisis. Biophys. J. 82:882–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlovsky, Y., L. Chernomordik, and M. Kozlov. 2002. Lipid intermediates in membrane fusion: formation, structure, and decay of hemifusion diaphragm. Biophys. J. 83:2634–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmin, P. I., J. Zimmerberg, Y. A. Chizmadzhev, and F. S. Cohen. 2001. A quantitative model for membrane fusion based on low-energy intermediates. Proc. Natl. Acad. Sci. USA. 98:7235–7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leikina, E., and L. V. Chernomordik. 2000. Reversible merger of membranes at the early stage of influenza hemagglutinin-mediated fusion. Mol. Biol. Cell. 11:2359–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra, V., L. Orci, B. S. Glick, M. R. Block, and J. E. Rothman. 1988. Role of an N-ethylmaleimide-sensitive transport component in promoting fusion of transport vesicles with cisternae of the Golgi stack. Cell. 54:221–227. [DOI] [PubMed] [Google Scholar]
- Markin, V. S., and J. P. Albanesi. 2002. Membrane fusion: stalk model revisited. Biophys. J. 82:693–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markovic, I., E. Leikina, M. Zhukovsky, J. Zimmerberg, and L. V. Chernomordik. 2001. Synchronized activation and refolding of influenza hemagglutinin in multimeric fusion machines. J. Cell Biol. 155:833–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikyan, G. B., B. N. Deriy, D. C. Ok, and F. S. Cohen. 1996. Voltage-dependent translocation of R18 and DiI across lipid bilayers leads to fluorescence changes. Biophys. J. 71:2680–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikyan, G. B., S. A. Brener, D. C. Ok, and F. S. Cohen. 1997. Inner but not outer membrane leaflets control the transition from glycosylphosphatidylinositol-anchored influenza hemagglutinin-induced hemifusion to full fusion. J. Cell Biol. 136:995–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal, A., and J. Bentz. 2001. Comprehensive kinetic analysis of influenza hemagglutinin-mediated membrane fusion: role of sialate binding. Biophys. J. 81:1521–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal, A., E. Leikina, J. Bentz, and L. V. Chernomordik. 2002a. Kinetics of influenza hemagglutinin-mediated membrane fusion as a function of technique. Anal. Biochem. 303:145–152. [DOI] [PubMed] [Google Scholar]
- Mittal, A., T. Shangguan, and J. Bentz. 2002b. Measuring pKa of activation and pKi of inactivation for influenza hemagglutinin from kinetics of membrane fusion of virions and of HA expressing cells. Biophys. J. 83:2652–2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao, H., R. T. Armstrong, G. B. Melikyan, F. S. Cohen, and J. M. White. 1999. A specific point mutant at position 1 of the influenza hemagglutinin fusion peptide displays a hemifusion phenotype. Mol. Biol. Cell. 10:2759–2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche, S., and Y. Gaudin. 2002. Characterization of the equilibrium between the native and fusion- inactive conformation of rabies virus glycoprotein indicates that the fusion complex is made of several trimers. Virology. 297:128–135. [DOI] [PubMed] [Google Scholar]
- Skehel, J. J., and D. C. Wiley. 2000. Receptor binding and membrane fusion in virus entry: The Influenza Hemagglutinin. Annu. Rev. Biochem. 69:531–569. [DOI] [PubMed] [Google Scholar]
- Tieleman, D. P., and J. Bentz. 2002. Hydrophobic defects in membrane fusion. Biophys. J. 83:1501–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White, J. 1996. Membrane fusion: the influenza paradigm. Cold Spring Harbour Symposium. 60:581–588. [DOI] [PubMed]