

Abstract
The photophysical properties of synthetic compounds derived from the imidazolidinone chromophore of the green fluorescent protein were determined. Various electron-withdrawing or electron-donating substituents were introduced to mimic the effect of the chromophore surroundings in the protein. The absorption and emission spectra as well as the fluorescence quantum yields in dioxane and glycerol were shown to be highly dependent on the electronic properties of the substituents. We propose a kinetic scheme that takes into account the temperature-dependent twisting of the excited molecule. If the activation energy is low, the molecule most often undergoes an excited-state intramolecular twisting that leads it to the ground state through an avoided crossing between the S1 and S0 energy surfaces. For a high activation energy, the torsional motion within the compounds is limited and the ground-state recovery will occur preferentially by fluorescence emission. The excellent correlation between the fluorescence quantum yields and the calculated activation energies to torsion points to the above-mentioned avoided crossing as the main nonradiative deactivation channel in these compounds. Finally, our results are discussed with regard to the chromophore in green fluorescent protein and some of its mutants.
INTRODUCTION
The green fluorescent protein (GFP) from the jellyfish Aequorea victoria has become a widely used marker in molecular and cell biology due to its strong intrinsic visible fluorescence, which is easily detectable by fluorescence spectroscopy (for reviews, see Cubitt et al., 1995; Tsien, 1998; Palm and Wlodawer, 1999; Prendergast, 1999; Zimmer, 2002). This protein of around 27 kDa can be fused to many other proteins, allowing their visualization in living cells without interfering with their function. Therefore, it becomes possible to follow signaling and trafficking in cells (Chalfie et al., 1994; Prasher, 1995) and to study protein-protein interactions (Mitra et al., 1996; Park and Raines, 1997; Ozawa et al., 2000). Some GFP variants are also used as noninvasive intracellular pH biosensors (Kneen et al., 1998; Llopis et al., 1998; Robey et al., 1998; Hanson et al., 2002) or fluorescent indicators for local Ca2+ concentrations (Miyawaki et al., 1997; Romoser et al., 1997).
The chromophore of this soluble globular protein of 238 amino acid residues is a p-hydroxybenzylidene-imidazolidinone derivative formed by an autocatalytic, posttranslational cyclization and oxidation of the polypeptide backbone, involving Ser-65, Tyr-66, and Gly-67 residues (Shimomura, 1979; Cody et al., 1993; Heim et al., 1994). The GFP crystal structure as a monomer (Brejc et al., 1997), as a dimer (Yang et al., 1996), and of several mutants (Ormö et al., 1996; Palm et al., 1997; Wachter et al., 1997) show that the chromophore is always located in the middle part of a central helix inside an 11-stranded β-barrel. It is thus totally embedded in the protein matrix and isolated from the bulk solvent. The chromophore of the wild type GFP (wt-GFP) possesses peculiar spectroscopic properties: two absorption peaks at 395 and 475 nm, usually attributed respectively to the neutral and anionic forms of the chromophore (Warren and Zimmer, 2001), and one emission peak at 508 nm, with a high fluorescence quantum yield (φ = 0.79) (Ward and Bokman, 1982; Niwa et al., 1996). The substitution of one or more amino acids within the chromophore or in its immediate proximity has allowed the development of GFP mutants with specific absorption and emission properties (Palm et al., 1997; Wachter et al., 1998; Ito et al., 1999).
The photophysical parameters that define the GFP spectroscopic properties are currently under investigation. Vibrational spectroscopy has provided information on the ground-state structure of the chromophore and its spectral properties when the protein environment is modified (van Thor et al., 1998; Bell et al., 2000). Some information was also obtained on the ground-state and first excited-state dynamics by time-resolved fluorescence spectroscopy (Chattoraj et al., 1996; Lossau et al., 1996; Didier et al., 2002; Winkler et al., 2002) and by resonance Raman spectroscopy (Esposito et al., 2001; Schellenberg et al., 2001; Tozzini and Nifosi, 2001). The excited-state decay kinetics is consistent with an excited-state proton transfer (EPST) from the chromophore to the protein (Chattoraj et al., 1996; Lossau et al., 1996; Wachter et al., 1997). Two deprotonated excited-state species of the chromophore, I* and B*, with similar lifetimes (3.3 ns and 2.8 ns, respectively) are thought to be mainly responsible for the GFP fluorescence (Striker et al., 1999).
One question still remains open: why do the denatured GFP and the chromophore isolated by enzymatic digestion of GFP lose totally their emissive properties (Shimomura, 1979; Cody et al., 1993; Niwa et al., 1996)? This could be due to structural properties of the chromophore itself, such as limited intramolecular rotations about the exocyclic double bond (Niwa et al., 1996; Chen et al., 2001) or to an increased conformational freedom of the chromophore that results in a speeding up of the fast internal conversion pathway (Kummer et al., 1998). Quantum mechanical calculations suggest that internal rotation, which distorts the coplanarity of both rings of the molecule, increases the nonradiative decay rate of the excited chromophore (Voityuk et al., 1998; Weber et al., 1999). Certain environmental influences have also been invoked, such as the collisional quenching effect of oxygen and water (Prendergast, 1999), and a modification of the electron flux (Kummer et al., 1998) due to the absence of the hydrogen bonding network (Ormö et al., 1996).
In this paper, we report an exhaustive investigation of the influence of these different parameters on the fluorescence of synthetic compounds derived from the chromophore imidazolidinone. These compounds were substituted at the imidazolidinone level with electron-withdrawing or electron-donating groups to modify the spatial electron flux in the chromophore. Low-temperature experiments were conducted on some compounds to block the intramolecular rotations and largely increased fluorescence quantum yields were obtained. The kinetic scheme was then solved by introducing a temperature-dependent twisting in the excited state. The potential energy surfaces were calculated semiempirically for all the compounds. The curves showed an “avoided crossing” (Bearpark et al., 1996; Voityuk et al., 1998) between the S1 and S0 states when the molecules are twisted, which allows a radiationless decay. It appeared that the theoretical activation energy (Ea) of the twisting plays an important role because a strong correlation between Ea and the fluorescence quantum yields is observed. Some environmental factors known to influence the photophysical kinetics, such as hydrophobicity or viscosity of the solvent were also studied. New criteria could be defined for obtaining a fluorescent product at room temperature. These bright compounds will further be used to study hydrophobic protein pockets, to mark cellular compartments and to follow the destiny of intrinsic cellular proteins.
MATERIALS AND METHODS
Materials
Dimethylsulfoxide (DMSO), 1,4-dioxane, glycerol, absolute ethanol, and H2SO4 were purchased from Merck (Darmstadt, Germany). Ether, fluorescein, quinine sulfate, and chemical reactants were from Aldrich (Sigma-Aldrich, St. Louis, MS) and isopentane from Fluka (Sigma-Aldrich) All these chemicals and the starting material for the imidazolone synthesis had the highest purity available commercially and were used without further purification. A solvent mixture (EPA) of ether: isopentane: absolute ethanol in volume proportions of 5:5:2 was made extemporarily for the low-temperature experiments (Frisch et al., 1998). Ultrapure water (Milli Q Instrument, Millipore-Waters Corp., Billerica, MA) was used for the aqueous solutions.
Methods
Synthetic compounds
Four different approaches have been used to reach the GFP chromophores derivatives. The first synthetic pathway (compounds I-1, I-2, I-4, I-6, I-7, and I-10) started from corresponding oxazolones that were allowed to react with various primary amines following procedures described in the literature (Kumar and Mukerjee, 1981; Tanaka et al., 1988; Kojima et al., 1998; Tripathy and Mukerjee, 1985). These oxazolones were obtained by cyclization of commercially available n-aroylglycines with aromatic aldehydes in acetic anhydride (Kojima et al., 1998). An alternative approach is based on the condensation of aromatic amidine with ethyl chloroacetate and an appropriate aldehyde in the presence of NaI (compound I-16, Shafi, 1985) or in the absence of NaI (compounds I-5, I-9, I-17–I-23 and I-26, Devasia, 1976). The last method (compounds I-3, I-8, I-11, I-15, I-24, I-25, I-27, I-28, and I-29) consists in replacing the amidine by an imidic acid ester that can react with glycine ethyl ester and an aldehyde according to Kidwai and Devasia (1962). The compound I-3 was alkylated using potassium hydroxide in MeOH as described in the literature (Badr et al., 1979) to give I-12, I-13, and I-14. All the new products are specified in Table 1.
TABLE 1.
Photophysical characteristics of the imidazolone derivatives in dioxane
| Derivative | R1 | R2 | R3 | Extinction coefficient (M−1·cm−1) | λmaxabs (nm) | λmaxemis (nm) | φ* |
|---|---|---|---|---|---|---|---|
| I-1 | OH | Me | Me | 25500 | 370 | 440 | 0.0001 |
| I-2† | OH | Me | (CH2)3Me | 26600 | 372 | 436 | 0.0001 |
| I-3 | OH | Ph | H | 32600 | 399 | 467 | 0.0002 |
| I-4† | OH | 3,4-diMeOPh | Me | 28700 | 398 | 476 | 0.0002 |
| I-5 | H | Ph | H | 13100 | 384 | 444 | 0.0012 |
| I-6 | H | Ph | OH | 19700 | 385 | 443 | 0.0002 |
| I-7 | H | Ph | Ph | 19500 | 380 | 453 | 0.001 |
| I-8† | H | 4-MeOPh | H | 26100 | 387 | 452 | 0.032 |
| I-9† | H | 3,4-diMeOPh | H | 23000 | 390 | 457 | 0.034 |
| I-10† | H | 3,4-diMeOPh | Me | 25900 | 386 | 470 | 0.045 |
| I-11 | MeO | Ph | H | 15400 | 398 | 464 | 0.0002 |
| I-12† | MeO | Ph | Me | 16800 | 394 | 471 | 0.0002 |
| I-13† | MeO | Ph | CH2COOEt | 25900 | 389 | 465 | 0.0002 |
| I-14† | MeO | Ph | CH2Ph | 20600 | 393 | 468 | 0.0002 |
| I-15† | MeO | 4-NO2Ph | H | 22200 | 428 | 573 | 0.060 |
| I-16† | OCH2COOEt | Ph | H | 19100 | 397 | 461 | 0.0003 |
| I-17 | H, 2-MeO | Ph | H | 26300 | 400 | 465 | 0.0022 |
| I-18† | N(Me)2 | Me | H | 39900 | 415 | 483 | 0.0009 |
| I-19 | N(Me)2 | Ph | H | 38800 | 454 | 520 | 0.0020 |
| I-20† | N(Me)2 | 4-MeOPh | H | 37000 | 450 | 507 | 0.0023 |
| I-21 | N(Me)2 | 3,4-diMeOPh | H | 50300 | 455 | 515 | 0.0025 |
| I-22† | CF3 | Ph | H | 22000 | 383 | 451 | 0.135 |
| I-23† | CN | Ph | H | 24800 | 393 | 464 | 0.158 |
| I-24† | COOMe | Ph | H | 28800 | 394 | 461 | 0.159 |
| I-25† | CN | 4-MeOPh | H | 29200 | 401 | 482 | 0.159 |
| I-26† | CN | 3,4-diMeOPh | H | 26500 | 405 | 482 | 0.189 |
| I-27† | COOMe | 3,4-diMeOPh | H | 32200 | 403 | 478 | 0.295 |
| I-28† | CN | 4-NO2Ph | H | 20700 | 406 | 508 | 0.220 |
| I-29† | COOMe | 4-NO2Ph | H | 25300 | 405 | 510 | 0.258 |
Fluorescence quantum yields φ determined at 293 K. SEM of 0.001 for φ between 0.010 and 0.099 and of 0.005 for φ between 0.100 and 0.300.
New products.
The imidazolones were characterized by 1H-NMR through their typical styryl proton signal at 7.1 ± 0.2 ppm and their purity was determined by elemental analysis.
Highly concentrated solutions of each analog of ∼3 × 10−3 M were first prepared by dissolution either in dioxane or DMSO, depending on their solubility. Measurements were performed on ∼1000-fold diluted solutions, either in dioxane, H2O, EPA, or glycerol.
Spectroscopic measurements
Steady-state absorption spectra were recorded on a Cary IV spectrophotometer. When necessary, correction for light scattering by excimers was made according to Shih and Fasman (1972). The extinction coefficients of the compounds were determined at the absorption maximum.
Steady-state fluorescence spectra were obtained on an SLM 48000 spectrofluorometer, with 4-nm excitation and emission bandwidths. The fluorescence quantum yields were determined at 20°C from the area under the fluorescence spectra, taking as reference a solution of quinine sulfate in 0.1 M H2SO4 (φQS = 0.51 at this pH) (Velapoldi and Mielenz, 1980) or fluorescein in 0.1 M NaOH (φF = 0.95) (Brannon and Magde, 1978) of the same absorbance at the excitation wavelength. The excitation wavelength was chosen as the best compromise between the absorption bands of the compound and the reference. At this wavelength, the concentrations were adjusted to an absorbance of 0.1, so that the fluorescence intensities were proportional to the absorbances of the solutions. For low-temperature experiments, the fluorescence spectra were measured in a 4- × 10-mm quartz cuvette with the sample temperature controlled by means of an Oxford Instrument Cryostat DN 1704 with liquid nitrogen. Steady-state absorption and fluorescence spectra were taken before and after the measurements to check that the samples were not degenerated during the experiments.
Potential energy surfaces
All model calculations of the electronic ground and excited states were performed at the semiempirical level of theory using the MOPAC 93 program. The semiempirical Hamiltonian AM1 was employed and the configuration interaction included all single and double electron excitations. For triplet states, an active space of seven molecular orbitals and four electrons was used. The dihedral angles β and γ were defined by the bond connectivities N2-CA2-CB2-CG2 and CA2-CB2-CG2-CD1, respectively. Energy mapping was conducted by 10° increment rotations of the imidazolone ring about its torsion angles. Rotations around the C1-CA1 bond were also considered but did not influence the potential energy mapping.
RESULTS
Photophysical properties of the imidazolone derivatives
The two main bands at ∼395 and 475 nm in the absorption spectrum of wt-GFP are due to the neutral and anionic forms of the chromophore, respectively (Bell et al., 2000). Both forms are in equilibrium in the protein and correspond to the A and B forms in the four-state model elaborated by Weber et al. (1999) for the photophysical behavior of wt-GFP. The intense green fluorescence of the imidazolone chromophore at 508 nm seems to arise from the anionic B* form of the chromophore or from an intermediate I*. This last form results from the neutral A* after deprotonation by excited-state proton transfer from the imidazolone to the protein matrix (Lossau et al., 1996; Chattoraj et al., 1996). As the photophysical properties of the native chromophore are pH sensitive, the electronic charge repartition in the molecule must play an important role in the fluorescence emission. Therefore, we synthesized several derivatives of the chromophore imidazolone with various electronic substituents and investigated the relationship between their electron distributions and photophysical properties.
In this study, the 4-hydroxybenzilidene-2,3-dimethyl-imidazolidinone (Fig. 1) was selected as the reference compound because it represents the smallest fluorescent moiety of GFP. The OH of the Tyr residue was conserved at atom CZ and the two aliphatic carbons of the peptidic backbone were replaced in CA1 and CA3 by methyl groups (compound I-1 called HBDI by Esposito et al., 2001). We substituted different radicals R1, R2, and R3 in place of HOy, CA1, and CA3, respectively (Table 1). First, derivatives of this molecule were synthesized with a phenyl as R2 to mimic both the steric hindrance and the hydrophobic environment of part of the protein backbone. Then the hydroxyl in position R1 was replaced by H to get free of the pH effect in this part of the molecule. Finally, various derivatives were obtained by substitutions in the positions R1 and R2 by electron-donating groups, such as MeO or N(Me)2, or by electron-withdrawing groups, such as CF3, NO2, CN, or COOMe.
FIGURE 1.

Model chromophore with atom labels according to the GFP crystal structure (Ormö et al., 1996). β and γ are the dihedral angles N2-CA2-CB2-CG2 and CA2-CB2-CG2-CD1, respectively.
The photophysical properties of these compounds (Table 1) were determined in dioxane, an aprotic solvent with a very small low-frequency dielectric constant (ɛ = 2.2 at 25°C), commonly referred to as a good simulator of the hydrophobic environment of a chromophore buried inside a protein matrix. The absorbance and emission spectra of the most fluorescent compounds (φ > 0.1) of Table 1 (I-22–I-29) are described here. Fig. 2 shows the spectra of I-23, I-27, and I-28, which are representative of the three kinds of spectra obtained and which are used to define three classes of derivatives [A], [B], and [C]. I-22 and I-24 show the same kind of spectra as I-23: all three correspond to compounds substituted by only one electron-withdrawing group in R1 (Fig. 2 a). One notices that these compounds have structured absorption spectra, characterized by a main absorption peak with a shoulder on each side. They form class [A]. I-25 and I-26 show spectra similar to I-27 (Fig. 2 b). These compounds, substituted with an electron-acceptor in R1 and a donor in R2, have much less structured spectra than the ones in Fig. 2 a and belong to class [B]. Fig. 2 c shows the spectra of I-28, which are very similar to the spectra of I-29. Both molecules have two electron acceptors in R1 and R2 and are red shifted in comparison with the previous ones. They are not structured at all and their peaks have greatly increased widths at half maximum. They correspond to class [C]. The broadening of the spectra in class [B] and [C] might be attributed to an increasing admixture of 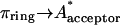 or
or 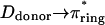 states where πring and πring* represent the bonding and antibonding orbitals of the aromatic rings respectively. In all cases, the S1-S0 fluorescence spectrum is approximately the mirror image of the S0-S1 absorption spectrum, which points to small overall differences between the nuclear configuration of the ground and excited states. However, the low intensity of the 0-0 band indicates considerably different equilibrium nuclear configurations in the ground and first excited states.
states where πring and πring* represent the bonding and antibonding orbitals of the aromatic rings respectively. In all cases, the S1-S0 fluorescence spectrum is approximately the mirror image of the S0-S1 absorption spectrum, which points to small overall differences between the nuclear configuration of the ground and excited states. However, the low intensity of the 0-0 band indicates considerably different equilibrium nuclear configurations in the ground and first excited states.
FIGURE 2.
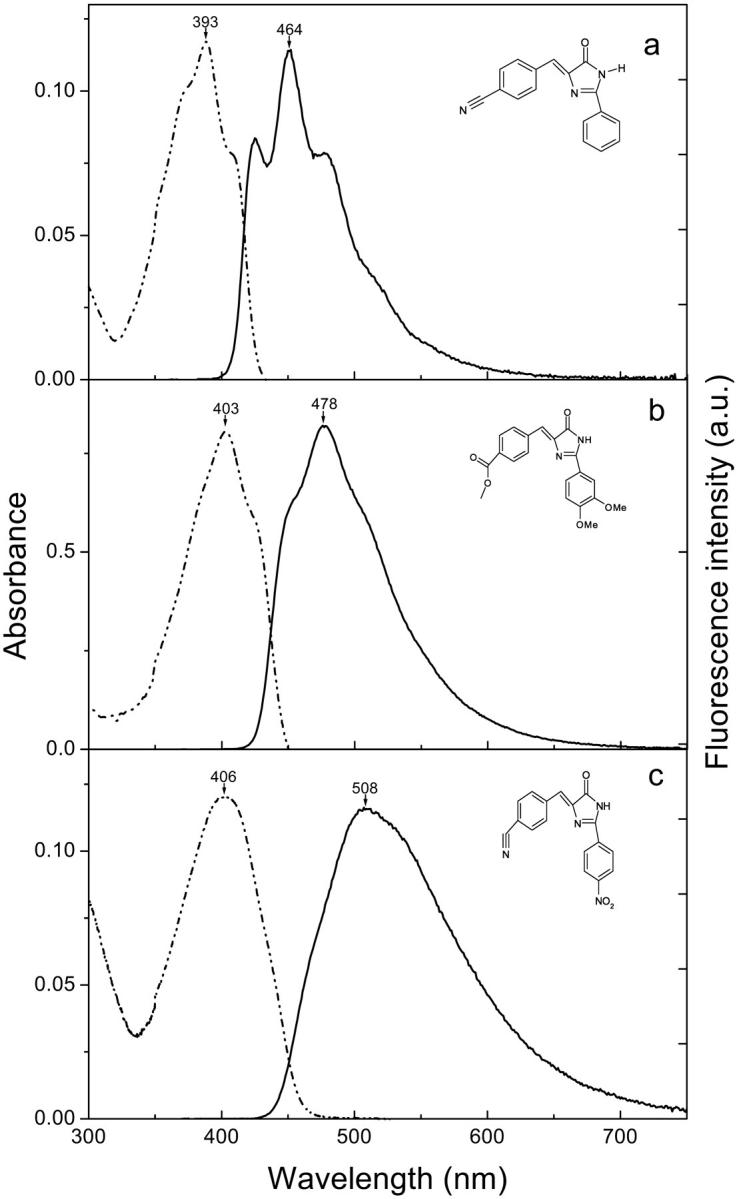
Room-temperature steady-state absorption spectra (dashed) and fluorescence emission spectra (solid) of GFP derivatives in dioxane: I-23 (a); I-27 (b); and I-28 (c). The absorption and fluorescence spectra were normalized to approximately the same maximum amplitude.
The extinction coefficient of the reference molecule I-1 at the absorption maximum is 25,500 M−1 cm−1 (Table 1) and there is a 105-nm blue shift in the absorption and a 70-nm blue shift in the emission compared to the anionic form of the chromophore in GFP. The substitution of R2 by a phenyl ring (I-3) induces an increase in the extinction coefficient and a marked red shift in both the absorption and emission spectra compared to I-1 (∼30 nm) but the fluorescence quantum yield still remains negligible. The replacement of OH by H in compound I-5 induces a fall in the extinction coefficient with a small blue shift. The fluorescence quantum yield becomes nonnegligible when R2 is substituted with the electron-donating groups 4-MeOPh or 3,4-diMeOPh in compounds I-8, I-9, and I-10.
In the literature, it appears that an electron-donating group in R1 could be responsible for some fluorescence properties of the native anionic form of the chromophore (Bell et al., 2000). However, the substitution at R1 by either MeO or N(Me)2, which are two strong electron-donating groups, does not have any influence on the fluorescence quantum yield that remains very low (I-11 to I-21). One can notice that a substitution in R3 with a Me, CH2COOEt or CH2Ph does not significantly modify the spectroscopic characteristics. By contrast, a substitution at R1 with electron-withdrawing groups induces a red shift, compared to the first compounds, both in the absorption maximum and in the emission maximum accompanied by an important increase in φ (from 0.158 to 0.220 for CN in R1 and from 0.159 to 0.295 for COOMe in R1) (I-23, I-25, I-26, I-28, and I-24, I-27, I-29, respectively). Among these fluorescent compounds, I-27 shows the greatest overall brightness, i.e., the product of the extinction coefficient and the quantum yield.
Table 1 clearly shows that the necessary and sufficient conditions for obtaining fluorescent imidazolones in dioxane with a quantum yield φ > 0.1 at 20°C are: i), an electron-withdrawing group in R1 and ii), a phenyl in R2, substituted with either an electron-donating group or an electron-withdrawing group. A large selection of fluorescent compounds is thus obtained with maximum absorption and emission wavelengths ranging from 383 to 406 nm and from 451 to 510 nm, respectively.
Fluorescence quantum yields and activation energies
After the excitation by an adequate radiation, and transition from the ground state S0 to the first singlet excited state S1, the synthetic compounds may follow several theoretical deexcitation pathways. These can be described as one radiative process, the fluorescence, and several nonradiative processes. The nonradiative pathways include either a fast internal conversion or an intersystem crossing from the first electronic excited state S1 to the T1 triplet state. A third nonradiative pathway includes the excited-state intramolecular twisting about the two dihedral angles β (N2-CA2-CB2-CG2) and γ (CA2-CB2-CG2-CD1) followed by fast internal conversion after conical intersection. All these processes compete with each other.
In the following simplified kinetic scheme, two geometric forms of the molecules are considered, a planar P (β = 0°, γ = 0°) and a twisted Tw (β ≠ 0° and/or γ ≠ 0°).
The fluorescence quantum yield φ of a fluorescent compound is thus given by:
Table 6.
| P + hν → P* | S0 → S1 | Excitation | |
| P* → P + hν | S1 → S0 | Fluorescence | (kf) |
| P* → P | S1 → S0 | Radiationless deactivation via internal conversion and/or intersystem crossing | (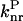 ) ) |
P* → 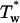 → P → P
|
S1 → S0 | Radiationless deactivation via conical intersection | (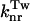 ) ) |
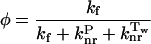 |
(1) |
Low-temperature behavior
Some low-temperature experiments were undertaken with the protic solvent EPA used for glass-forming solutions (see Materials and Methods). Temperature was slowly decreased from 293 K to 83 K in a thermostated cuvette in the fluorescence spectrophotometer, and steady-state fluorescence spectra were recorded every 15 min. The variation of the fluorescence quantum yield φ as a function of temperature is shown in Fig. 3 for three derivatives having different fluorescent properties at 293 K: I-1, which does not present any fluorescence at this temperature in water, EPA or dioxane, and I-24 and I-27, which are fluorescent in EPA and dioxane.
FIGURE 3.

Low-temperature effect on the fluorescence quantum yields of I-1 (▪), I-24 (•), and I-27 (▿). The maximal SEM on the φ values are of 0.005. The inset shows the corresponding Arrhenius plots for the 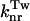 rate constant. Measurements were performed in EPA. For details, see Materials and Methods.
rate constant. Measurements were performed in EPA. For details, see Materials and Methods.
The fluorescence of I-1 is undetectable until 133 K, a temperature near the glass-forming temperature of the solvent. The fluorescence intensity strongly increases with lower temperatures without reaching a maximum. This can be explained by the progressive rigidification of I-1 in the cooled, solution, which induces an important decrease in the nonradiative deexcitation processes. For I-27 and I-24, the fluorescence intensity increases progressively with decreasing temperatures and reaches a constant level corresponding to maximal fluorescence quantum yields of 0.42 and 0.78, respectively. It is noticeable that at 80 K the fluorescence quantum yield of I-24 reaches the high value of 0.79 observed at room temperature for the wt-GFP chromophore in the protein.
At low temperature, the probability of the molecules to be in a twisted conformation is very low in comparison to the probability of the planar conformation. Consequently, 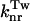 in Eq. 1 becomes negligible. Because the maximal quantum yield φ0 obtained at 80 K corresponds to:
in Eq. 1 becomes negligible. Because the maximal quantum yield φ0 obtained at 80 K corresponds to:
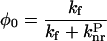 |
(2) |
the variation of the rate constant 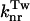 with temperature can be calculated by means of the general formula described by Saltiel and Sun (1989) in the case of transstilbene twisting.
with temperature can be calculated by means of the general formula described by Saltiel and Sun (1989) in the case of transstilbene twisting.
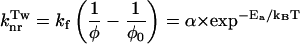 |
(3) |
where α and Ea are the frequency factor and the activation energy of the first excited-state S1. T represents the absolute temperature and kB the Boltzmann constant.
The radiative decay rate constant, kf, equal to the φ/τ ratio, was assumed to be equal to 1.4 × 108 s−1, taking the fluorescence quantum yield of I-27 (Table 1) and its fluorescence lifetime determined for other purposes as 2.1 × 10−9 s. This value of kf was assumed to be temperature and solvent independent, and used for all the compounds based on their structural similarity.
The curves displayed in the inset of Fig. 3 are the corresponding Arrhenius plots for the 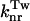 rate constant. However, because the viscosity of the medium increases with decreasing temperatures and, consequently, the activation energy barrier is medium enhanced (Saltiel and Sun, 1989), it is interesting to note the non-Arrhenius behavior and the “characteristic knee” already mentioned by Litvinenko et al. (2003) and attributed to the onset of strong caging at the critical crossover temperature Tc of the glass-forming solvent. In our case, because the solvent is a mixture of three components, the Tc value is not known but the “knee” appears well above the glass transition temperatures Tg of ethanol (95 K) and isopentane (65 K) (Carpentier et al., 1967; Sugisaki et al., 1968) and therefore might well correspond to the Tc value of EPA.
rate constant. However, because the viscosity of the medium increases with decreasing temperatures and, consequently, the activation energy barrier is medium enhanced (Saltiel and Sun, 1989), it is interesting to note the non-Arrhenius behavior and the “characteristic knee” already mentioned by Litvinenko et al. (2003) and attributed to the onset of strong caging at the critical crossover temperature Tc of the glass-forming solvent. In our case, because the solvent is a mixture of three components, the Tc value is not known but the “knee” appears well above the glass transition temperatures Tg of ethanol (95 K) and isopentane (65 K) (Carpentier et al., 1967; Sugisaki et al., 1968) and therefore might well correspond to the Tc value of EPA.
Relationship between activation energies Ea and fluorescence quantum yields
The main nonradiative deexcitation processes from the singlet excited state evoked so far in the literature are a cis-trans photoisomerization of the exo-methylene double bond (Niwa et al., 1996; Weber et al., 1999) or more limited rotations about the two dihedral angles β and γ (Phillips, 1997; Kummer et al., 1998; Voityuk et al., 1998; Chen et al., 2001; Webber et al., 2001). Therefore, the theoretical activation energies of the different conformations obtained after internal rotation about these angles were determined for some 20 compounds by semiempirical calculations with the MOPAC 93 program. The energies of I-1 and I-23 were also calculated with the ab initio Gaussian 98 program (CIS method, STO-3G basis set) (Frisch et al., 1998) and a good correlation was observed (data not shown).
The energy mapping was conducted for the ground and first excited states by simultaneous 10° increments of both dihedral angles. The potential energy surfaces (PES) were identical for all rotations of the R2 substituent around the C1-CA1 σ bond, which could be explained by the axial symmetry of nearly all the substituents. This rotation angle was further kept at 0° to give a planar conformation to the imidazolidinone ring coupled to R2. As an example of the results, the PES of I-23 are represented in Fig. 4 a. For each compound, we checked that the 0-0 transition energies estimated from the intersection of the excitation and emission spectra, are reasonably well predicted by the MOPAC calculations: the differences amount at most to 3%. For β = 90° an “avoided crossing” between the S0 and the S1 PES is observed for all the derivatives (Voityuk et al., 1998).
FIGURE 4.
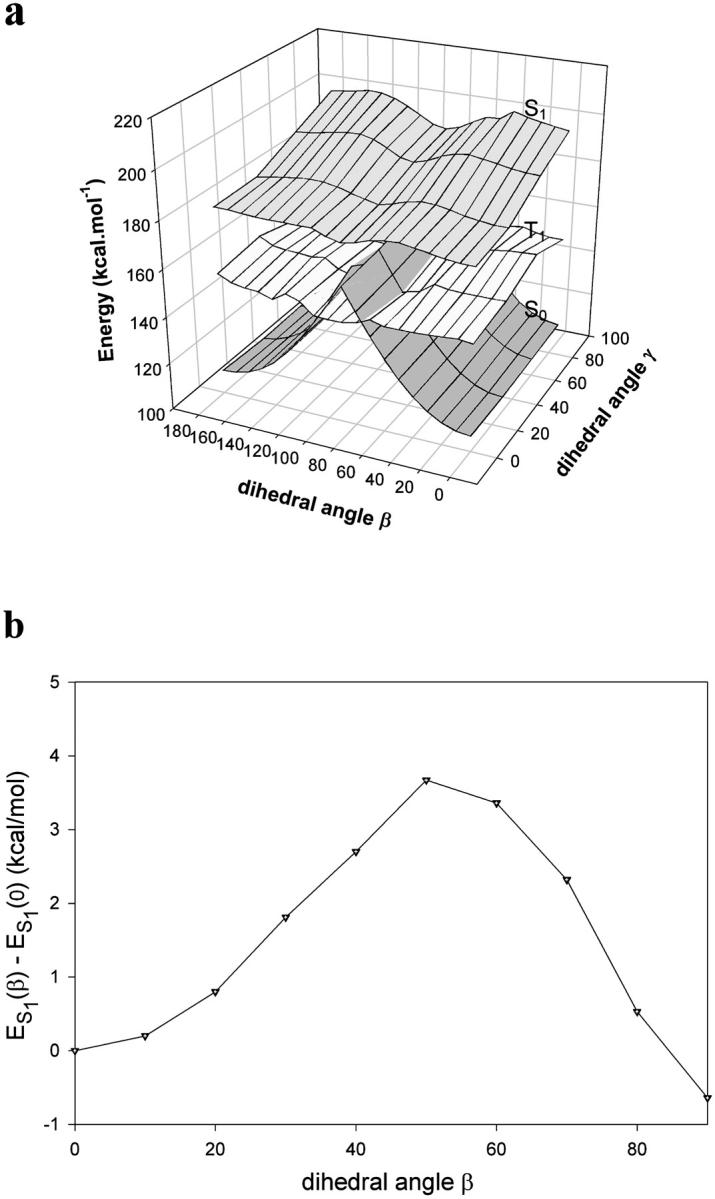
Energy diagrams of I-23: (a) Calculated two-dimensional potential energy surfaces for the S0, S1, and T1 states as a function of the dihedral angles β and γ; (b) Energy profile of the S1 state as a function of the dihedral angle β (from 0° to 90°), with γ held fixed at 0°. The zero value corresponds to the energy of the molecule with (β, γ) equal to (0, 0). Calculations were made with the MOPAC program.
It is possible to restrict the two-dimensional energy diagram of I-23 to an energy profile because i), the energy surfaces present a symmetry around β = 90° and ii), the lowest energies of the excited state are obtained with γ = 0°. The profile plotted in Fig. 4 b represents the relative variation of the first excited-state energy ES1(β) during the rotational motion of the imidazolidinone ring around β, compared to the energy ES1(0) of the planar conformation of the molecule with β = γ = 0°. The same type of profile was obtained for all the derivatives. The maximum of ES1 was always observed for β between 40° and 50°. These maxima, given in Table 2, represent the activation energy, Ea, of the barriers that must be overcome to reach the avoided crossing region, i.e., the nonradiative decay channel. They seem to play a key role in the photophysical properties of the molecules because their height is linked to the fluorescence quantum yield.
TABLE 2.
Theoretical maximum activation energies of various compounds
| Derivative* | R1 | R2 | Energy (kcal mol−1) |
|---|---|---|---|
| I-1 | OH | Me | 1.96 |
| I-3 | OH | Ph | 2.21 |
| I-5 | H | Ph | 2.53 |
| I-8 | H | 4-MeOPh | 1.87 |
| I-22 | CF3 | Ph | 3.27 |
| I-23 | CN | Ph | 3.67 |
| I-25 | CN | 4-MeOPh | 4.26 |
| I-24 | COOMe | Ph | 3.42 |
| I-26 | CN | 3,4-diMeOPh | 4.30 |
| I-27 | COOMe | 3,4-diMeOPh | 7.14 |
| I-28 | CN | 4-NO2Ph | 3.68 |
| I-29 | COOMe | 4-NO2Ph | 5.81 |
In all cases, R3 is H
The relationship between the activation energies Ea of 12 compounds and their fluorescence quantum yields at room temperature in dioxane (Table 1) was investigated.
From Eqs. 1 and 3, one obtains:
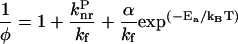 |
(4) |
Assuming that the 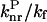 ratio in Eq. 1 does not significantly differ at room temperature from one compound to another because all the compounds show the same basic chemical structure, the semilogarithmic variation of (1/φ − 1 −
ratio in Eq. 1 does not significantly differ at room temperature from one compound to another because all the compounds show the same basic chemical structure, the semilogarithmic variation of (1/φ − 1 − 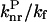 ) was plotted as a function of Ea/kBT (Fig. 5). The constant ratios were solved to fit the experimental data linearly with a slope of −1. The values obtained for
) was plotted as a function of Ea/kBT (Fig. 5). The constant ratios were solved to fit the experimental data linearly with a slope of −1. The values obtained for 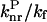 (2.39) and
(2.39) and 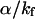 (1470) are consistent with the proposal that the avoided crossing between the ground and first excited state is the main nonradiative channel for these molecules because α ≫
(1470) are consistent with the proposal that the avoided crossing between the ground and first excited state is the main nonradiative channel for these molecules because α ≫ 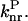 The points corresponding to I-1, I-3, and I-5 that are clearly outside the fitting curve should be fitted by another curve parallel to the first one with a higher Y-intercept, reflecting a higher
The points corresponding to I-1, I-3, and I-5 that are clearly outside the fitting curve should be fitted by another curve parallel to the first one with a higher Y-intercept, reflecting a higher 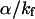 ratio. This variation can be explained by comparing the chemical structures of the different compounds. Actually, I-1, I-3, and I-5 are the simplest derivatives that do not possess any electron-donating or electron-withdrawing substituents in R1 and R2 and thus can freely twist around the exo-methylene bond.
ratio. This variation can be explained by comparing the chemical structures of the different compounds. Actually, I-1, I-3, and I-5 are the simplest derivatives that do not possess any electron-donating or electron-withdrawing substituents in R1 and R2 and thus can freely twist around the exo-methylene bond.
FIGURE 5.

Semilogarithmic variation of 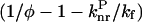 as a function of Ea/kBT. Derivatives with φ ⩾ 0.1 (▿) and φ < 0.1 (•). The fluorescence quantum yields were determined in dioxane at 293 K.
as a function of Ea/kBT. Derivatives with φ ⩾ 0.1 (▿) and φ < 0.1 (•). The fluorescence quantum yields were determined in dioxane at 293 K.
Effect of extrinsic factors on the fluorescence quantum yields
A total rigidification of I-24 leads to the fluorescence yield of 0.79 seen in the wt-GFP chromophore. However, when the chromophore is isolated from the proteic cage, the fluorescence quantum yield falls down. It was thus interesting to study the influence of the environment on the fluorescence quantum yields of the synthetized derivatives by using three solvents of various hydrophobicity, viscosity, or polarity.
As dioxane is miscible in all proportions with highly polar solvents like water, the fluorescence emission of the various derivatives was measured in 1/1000 (v/v) dioxane/water solutions. The spectra obtained (data not shown) are much less structured in the aqueous medium than in pure dioxane and the fluorescence emission maximum shifts to the red by 50–100 nm, possibly as a result of the increase in solvent polarity. However, the fluorescence quantum yields of the compounds never exceed 0.003 (Table 3). One deduces that in water the radiationless relaxation via internal conversion is largely preponderant over the radiative processes. Because all the molecules investigated here possess a large number of lone pair orbitals and hence are good hydrogen-bond acceptors, a quenching process through hydrogen bonding with solvent molecules must occur (see Biczok et al., 1997 and references given therein). To strengthen this conclusion we studied the effect of adding traces of water to a solution of I-27 in dioxan whose quantum yield is 0.295. Adding water, even at a level as low as 1.4% (v/v) to the solution immediately results in a drop of ∼30% in quantum yield.
TABLE 3.
Fluorescence quantum yields in water, glycerol, and dioxane
| Class | Derivative* | R1 | R2 | φwater (ɛ† = 80) | φglycerol (ɛ = 40) | φdioxane (ɛ = 2) | φdiox/φgly‡ |
|---|---|---|---|---|---|---|---|
| [A] | I-22 | CF3 | Ph | 0.003 | 0.118 | 0.135 | 1.14 |
| I-23 | CN | Ph | 0.003 | 0.139 | 0.158 | 1.14 | |
| I-24 | COOMe | Ph | 0.0016 | 0.156 | 0.159 | 1.02 | |
| [B] | I-26 | CN | 3,4-diMeOPh | 0.0006 | 0.069 | 0.189 | 2.74 |
| I-27 | COOMe | 3,4-diMeOPh | 0.0015 | 0.100 | 0.295 | 2.95 | |
| [C] | I-28 | CN | 4-NO2Ph | 0.0015 | 0.023 | 0.220 | 9.56 |
| I-29 | COOMe | 4-NO2Ph | 0.0006 | 0.014 | 0.258 | 18.4 |
In all cases, R3 is H.
ɛ is the low-frequency dielectric constant of the solvent.
Ratio of the fluorescence quantum yields obtained in dioxane and glycerol.
The fluorescence quantum yields were also determined in 100% glycerol solutions for all the derivatives with a fluorescence quantum yield in dioxane φdiox > 0.10 (Table 3). In all cases, we observe a 15- to 100-fold increase in the φgly values in this highly viscous medium relative to the results obtained in water. However, the values are always lower than those obtained in the hydrophobic low polarity solvent dioxane. The ratio φdiox/φgly leads again to the same three classes of derivatives distinguished by their photophysical characteristics in dioxane and that depend mainly on the R2 substituent. The number of hydrogen bonds between the H-bond acceptor atoms of the substituents in R1 and R2 and the solvent as well as the solvent-solute dipolar interactions might explain the differences between the three classes. It appears that hydrogen bonding would have an opposite effect to that induced by the viscosity increase and favor the nonradiative process. In class [A], as both the φdiox and φgly are similar, one can conclude that through a combined viscosity increase and H-bond number decrease from water to glycerol, it is nearly possible to reach the same fluorescence quantum yield as in dioxane. In classes [B] and [C], the φgly are lower than the values obtained in class [A], although I-26 to I-29 are the most fluorescent compounds in dioxane. The more numerous H-bonds between their bulky substituents in R2 (particularly on the 3,4-diMeOPh) and the solvent would limit the fluorescence quantum yield increase. The differences observed between classes [B] and [C] could only be attributed to the opposite electronic properties of the substituents, 4-NO2Ph and 3,4-diMeOPh.
In view of the high fluorescence quantum yield of wt-GFP, free dissolved O2 does not seem to enter into the GFP protein matrix once the β-barrel is formed (Ormö et al., 1996). The chromophore is only in contact with the oxygen atom of structural water molecules through hydrogen bonds. But in the present work, the derivatives are surrounded by the solvent, which may dissolve molecular O2. To check a possible quenching effect of O2 on their fluorescence, some experiments were performed on samples deaerated by bubbling argon. The fluorescence quantum yields of I-27 obtained with and without dissolved O2 are compared (Table 4). This derivative was chosen because it is the most fluorescent compound in both water and dioxane, so that even small quantum yield variations are detectable. Table 4 shows that dissolved O2 has no significant inhibitory effect in EPA or dioxane. In water, the quantum yield slightly increases in the absence of O2 but not significantly, so that dissolved O2 cannot explain the loss of the fluorescence properties of the isolated chromophore.
TABLE 4.
Influence of molecular oxygen on the fluorescence quantum yield of I-27 at 20°C
| Solvent | Water | Glycerol | EPA | Dioxane |
|---|---|---|---|---|
| Dielectric constant | 80 | 40 | 24 | 2 |
| φ (oxygenated solution) | 0.0020 | 0.100 | 0.159 | 0.295 |
| φ (without O2) | 0.0027 | nd* | 0.159 | 0.285 |
Not determined in 100% glycerol because of argon bubbles.
DISCUSSION
In wt-GFP, the fluorescence spectrum is characterized by a strong emission peaking at 508 nm, which seems to originate from two deprotonated states, an environmentally unrelaxed intermediate I* state or a relaxed anionic state B* (Creemers et al., 1999; Chattoraj et al., 1996). Its high quantum yield (φ = 0.79) results from a conjunction of several factors. In the crystal structure of wt-GFP (Protein data bank access code 1ema), it appears that the chromophore is highly protected from the bulk solvent. Indeed, it was shown that the viscosity of the surrounding medium affected the fluorescence decay of the isolated chromophore (Kummer et al., 2002) but not that of the GFP (Suhling et al., 2002a), which is however sensitive to the refractive index (Suhling et al., 2002b). In the protein, the chromophore is surrounded by both apolar and polar residues, including a number of charged residues (Ormö et al., 1996). On one side of the chromophore, there is a large cavity containing only water molecules that contribute to a hydrogen bonding network linking the buried side chains of Glu-222 and Gln-69 (Chen et al., 2001). On the opposite side, there are polar interactions through hydrogen bonds between the phenolic hydroxyl of the chromophore and His-148, Thr-203, and Ser-205. There are also hydrogen bonds bridging the carbonyl of the imidazole ring to Glu-94 and Arg-96; this last residue is in a protonated form, which contributes to the partial negative charge residing on this carbonyl oxygen. All the surrounding residues and this dense hydrogen bonding network contribute to stabilize the chromophore in a nearly planar conformation.
The absence of fluorescence of the isolated chromophore has already given rise to studies on some models of the chromophore (Kojima et al., 1998; Webber et al., 2001; Kummer et al., 2002). Here we studied a large selection of synthetic derivatives of the imidazolidinone with various substituents, introduced both to increase the rigidity of the molecule and to modify the electron repartition in the molecule. Theoretical results with MOPAC 93 show that a crucial parameter as regards fluorescence ability is the activation barrier for a twist around the β angle. In non- or little-substituted compounds, the molecule is free to twist around the β and γ angles. It therefore emits very little fluorescence but undergoes a fast radiationless decay by internal conversion after reaching an avoided crossing. If the molecule is substituted with certain well-defined bulky substituents, the equilibrium between both radiative and nonradiative deexcitation pathways is modified and the compounds are fluorescent. The fluorescence quantum yields of our compounds were linked to the activation energies by introducing a new term corresponding to their twisted form in the classical scheme used for rigid aromatic hydrocarbons (Birks, 1973). Reducing or blocking the rotation about the exo-methylene double bond by either increasing the viscosity of the solvent or by lowering the temperature leads to increased fluorescence quantum yields in all the derivatives tested. Moreover a correlation is found between the inductive effect of the substituents and the quantum yield. It appears that all compounds with a high quantum yield (I-22 to I-29) possess an electron-withdrawing substituent in R1 and a phenyl group in R2 either not substituted or substituted by an electron-withdrawing or an electron-attracting substituent.
Additional information is obtained from the GFP mutants. Mutations of the residues of the β-barrel have various consequences, depending on whether they are within the chromophore or just in its vicinity in the folded protein. When mutations are located within the chromophore (mutations of residues Ser-65, Tyr-66, or Gly-67), the structural formula of the chromophore is completely modified (Cubitt et al., 1995; Wachter et al., 1997; Tsien, 1998). These mutants will not be discussed, because it is obvious that the spectral characteristics will be affected. More interesting are the mutations in the vicinity of the chromophore that indirectly induce i), a modification in the hydrogen bonding network and ii), steric and electrostatic changes in its close environment. These changes induce modifications in the absorption and emission spectra. Table 5 reports the photophysical characteristics of some of these mutants that are fluorescent. The 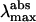 indicate that some mutants exist in the neutral and anionic forms, like wt-GFP. Others are mainly in the anionic form (E222G) or the neutral form (cycle 3, T203I). They are all emitting with a
indicate that some mutants exist in the neutral and anionic forms, like wt-GFP. Others are mainly in the anionic form (E222G) or the neutral form (cycle 3, T203I). They are all emitting with a 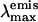 between 502 and 511 nm, which indicates that it is always the anionic form that is fluorescent, probably after a fast excited-state proton transfer from the neutral form to the surrounding protein matrix, as in wt-GFP (Chattoraj et al., 1996; Lossau et al., 1996; Wachter et al., 1997). A comparison with our results in Table 1 leads to the conclusion that compounds I-28 and I-29 (class [C] compounds) show similar spectroscopic characteristics. One could put forward the hypothesis that the rigidity of I-28 and I-29 and their electron distribution are similar to that found in the chromophore of these mutants, i.e., an electron attractive effect of the protein surroundings near the R1 and R2 positions.
between 502 and 511 nm, which indicates that it is always the anionic form that is fluorescent, probably after a fast excited-state proton transfer from the neutral form to the surrounding protein matrix, as in wt-GFP (Chattoraj et al., 1996; Lossau et al., 1996; Wachter et al., 1997). A comparison with our results in Table 1 leads to the conclusion that compounds I-28 and I-29 (class [C] compounds) show similar spectroscopic characteristics. One could put forward the hypothesis that the rigidity of I-28 and I-29 and their electron distribution are similar to that found in the chromophore of these mutants, i.e., an electron attractive effect of the protein surroundings near the R1 and R2 positions.
TABLE 5.
Photophysical characteristics of some mutants of GFP
| Mutations | Extinction coefficient (M−1 cm−1) | λmaxabs (nm) | λmaxemis (nm) | φ |
|---|---|---|---|---|
| None* | 25500 | 396, 475 | 508 | 0.79 |
| I167T† | – | 396, 471 | 502 | – |
| N146I, M153T, V163A* | – | 452 | 505 | – |
| F99S, M153T, V163A (cycle3)‡ | 30000 | 397 | 506 | 0.79 |
| F64L, I167T, K238N§ | 11500 | 395, 468 | 506 | 0.84 |
| E222Q¶ | – | 476 | 506 | 0.5 |
| F64L§ | 18300 | 396, 473 | 508 | 0.85 |
| E222G‖ | – | 475 | 510 | – |
| T203I‖ | – | 399 | 511 | – |
| S202F, T203I* | 20000 | 399 | 511 | 0.60 |
| T203I, S72A, Y145F* | 29000 | 399 | 511 | 0.64 |
Because the chromophore in GFP is linked to its environmental amino acids through hydrogen bonds and is under the electrostatic influence of its neighboring residues, our strategy to obtain further insight in GFP photophysics will be to bury our fluorescent products in hydrophobic protein pockets. Moreover thanks to the dependence of their emission on the presence of hydrogen-bond donors, on viscosity, and on rotational mobility, these synthetic derivatives may also find use as fluoroprobes of the microenvironment of proteins and other biological macromolecules.
Acknowledgments
This work was supported by grants from the Centre National de la Recherche Scientifique and the Université Louis Pasteur.
References
- Badr, M. Z. A., H. A. H. El-Sherief, and M. E. Tadros. 1979. Synthesis of 1,2-disubstituted 4-benzylidene-2-imidazolin-5-ones and their thiones derivatives. Indian J. Chem. 18B:240–242. [Google Scholar]
- Bearpark, M. J., F. Bernardi, M. Olivucci, M. A. Robb, and B. R. Smith. 1996. Can fulvene S1 decay be controlled? A CASSCF study with MMVB dynamics. J. Am. Chem. Soc. 118:5254–5260. [Google Scholar]
- Bell, A. F., X. He, R. M. Wachter, and P. J. Tonge. 2000. Probing the ground state structure of the green fluorescent protein chromophore using Raman spectroscopy. Biochemistry. 39:4423–4431. [DOI] [PubMed] [Google Scholar]
- Biczok, L., T. Berces, and H. Linschitz. 1997. Quenching processes in hydrogen-bonded pairs: interaction of excited fluorenone with alcohols and phenols. J. Am. Chem. Soc. 119:11071–11077. [Google Scholar]
- Birks, J. B. 1973. Organic Molecular Photophysics. Wiley, New York.
- Brannon, J. H., and D. Magde. 1978. Absolute quantum yield determination by thermal blooming fluorescein. J. Phys. Chem. 82:705–709. [Google Scholar]
- Brejc, K., T. K. Sixma, P. A. Kitts, S. R. Kain, R. Y. Tsien, M. Ormö, and S. J. Remington. 1997. Structural basis for dual excitation and photoisomerization of the Aequorea victoria green fluorescent protein. Proc. Natl. Acad. Sci. USA. 94:2306–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpentier, M. R., D. B. Davies, and A. J. Matheson. 1967. Measurements of the glass-transition temperature of simple liquids. J. Chem. Phys. 46:2451–2456. [Google Scholar]
- Chalfie, M., Y. Tu, G. Euskirchen, W. W. Ward, and D. C. Prasher. 1994. Green fluorescent protein as a marker for gene expression. Science. 263:802–805. [DOI] [PubMed] [Google Scholar]
- Chattoraj, M., B. A. King, G. U. Bublitz, and S. G. Boxer. 1996. Ultra-fast excited state dynamics in green fluorescent protein: multiple states and proton transfer. Proc. Natl. Acad. Sci. USA. 93:8362–8367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, M. C., C. R. Lambert, J. D. Urgitis, and M. Zimmer. 2001. Photoisomerization of green fluorescent protein and the dimensions of the chromophore cavity. Chem. Phys. 270:157–164. [Google Scholar]
- Cody, C. W., D. C. Prasher, W. M. Westler, F. G. Prendergast, and W. W. Ward. 1993. Chemical structure of the hexapeptide chromophore of the Aequorea green-fluorescent protein. Biochemistry. 32:1212–1218. [DOI] [PubMed] [Google Scholar]
- Creemers, T. M., A. J. Lock, V. V. Subramaniam, T. M. Jovin, and S. Volker. 1999. Three photoconvertible forms of green fluorescent protein identified by spectral hole-burning. Nat. Struct. Biol. 6:557–560. [DOI] [PubMed] [Google Scholar]
- Cubitt, A. B., R. Heim, S. R. Adams, A. E. Boyd, L. A. Gross, and R. Y. Tsien. 1995. Understanding, improving and using green fluorescent proteins. Trends Biochem. Sci. 20:448–455. [DOI] [PubMed] [Google Scholar]
- Devasia, G. M. 1976. A new method for the synthesis of unsaturated 2,4-disubstituted 2-imidazolin-5-ones. Tetrahedron Lett. 7:571–572. [Google Scholar]
- Didier, P., L. Guidoni, G. Schwalbach, M. Bourotte, A. Follenius-Wund, C. Pigault, and J.-Y. Bigot. 2002. Ultrafast gain dynamics of the green fluorescent protein. Chem. Phys. Lett. 364:503–510. [Google Scholar]
- Ehrig, T., D. J. O'Kane, and F. G. Prendergast. 1995. Green-fluorescent protein mutants with altered fluorescence excitation spectra. FEBS Lett. 367:163–166. [DOI] [PubMed] [Google Scholar]
- Esposito, A. P., P. Schellenberg, W. W. Parson, and P. J. Reid. 2001. Vibrational spectroscopy and mode assignments for an analog of the green fluorescent protein chromophore. J. Mol. Struct. 569:25–41. [Google Scholar]
- Frisch, M. J., G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzeweski, J. A. Montgomery, R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Ciolowski, J. V. Ortiz, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, J. L. Andres, M. Head-Gordon, E. S. Reploge, and J. A. Pople. 1998. Gaussian 98 (Revision A.1). Pittsburgh, PA.
- Hanson, G. T., T. B. McAnaney, E. S. Park, M. E. Rendell, D. K. Yarbrough, S. Chu, L. Xi, S. G. Boxer, M. H. Montrose, and S. J. Remington. 2002. Green fluorescent protein variants as ratiometric dual emission pH sensors. 1. Structural characterization and preliminary application. Biochemistry. 41:15477–15488. [DOI] [PubMed] [Google Scholar]
- Heim, R., D. C. Prasher, and R. Y. Tsien. 1994. Wavelength mutations and posttranslational autoxidation of green fluorescent protein. Proc. Natl. Acad. Sci. USA. 91:12501–12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito, Y., M. Suzuki, and Y. Husimi. 1999. A novel mutant of green fluorescent protein with enhanced sensitivity for microanalysis at 488 nm excitation. Biochem. Biophys. Res. Commun. 264:556–560. [DOI] [PubMed] [Google Scholar]
- Jung, G., S. Mais, A. Zumbusch, and C. Bräuchle. 2000. The role of dark states in the photodynamics of the green fluorescent protein examined with two-color fluorescence excitation spectroscopy. J. Phys. Chem. 104:873–877. [Google Scholar]
- Kidwai, A. R., and G. M. Devasia. 1962. A new method for the synthesis of amino acids. Synthesis of amino acids and their derivatives through 2,4-disubstituted-2-imidazolin-5-ones. J. Org. Chem. 27:4527–4531. [Google Scholar]
- Kneen, M., J. Farinas, Y. Li, and A. S. Verkman. 1998. Green fluorescent protein as a noninvasive intracellular pH indicator. Biophys. J. 74:1591–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima, S., H. Ohkawa, T. Hirano, S. Maki, H. Niwa, M. Ohashi, S. Inouye, and F. I. Tsuji. 1998. Fluorescent properties of model chromophores of tyrosine-66 substituted mutants of Aequorea green fluorescent protein (GFP). Tetrahedron Lett. 39:5239–5242. [Google Scholar]
- Kumar, P., and A. K. Mukerjee. 1981. Synthesis of 4-arylidene-1,2-disubstituted-Δ2-imidazolin-5-ones. Indian J. Chem. B. 20:416–418. [Google Scholar]
- Kummer, A., C. Kompa, H. Lossau, F. Pollinger-Dammer, M. E. Michel-Beyerle, C. M. Silva, E. Bylina, W. Coleman, M. Yang, and D. Youvan. 1998. Dramatic reduction in fluorescence quantum yield in mutants of green fluorescent protein due to fast internal conversion. Chem. Phys. 237:183–193. [Google Scholar]
- Kummer, D. A., C. Kompa, H. Niwa, T. Hirano, S. Kojima, and E. Michel-Beyerle. 2002. Viscosity-dependent fluorescence decay of the GFP chromophore in solution due to fast internal conversion. J. Phys. Chem. 106:7557–7559. [Google Scholar]
- Litvinenko, K. L., N. M. Webber, and R. Meech. 2003. Internal conversion in the chromophore of the green fluorescent protein: temperature dependence and isoviscosity analysis. J. Phys. Chem. 107:2616–2623. [Google Scholar]
- Llopis, J., J. M. McCaffery, A. Miyawaki, M. G. Farquhar, and R. Y. Tsien. 1998. Measurement of cytosolic, mitochondrial, and Golgi pH in single living cells with green fluorescent proteins. Proc. Natl. Acad. Sci. USA. 95:6803–6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lossau, H., A. Kummer, R. Heineke, F. Polliner-Dammer, C. Kompa, G. Bieser, T. Jonsson, C. M. Silva, M. M. Yang, D. C. Youvan, and M. E. Michel-Beyerle. 1996. Time-resolved spectroscopy of wild-type and mutant green fluorescent proteins reveals excited state deprotonation consistent with fluorophore-protein interactions. Chem. Phys. 231:1–16. [Google Scholar]
- Mitra, R., C. Silva, and D. Youvan. 1996. Fluorescence resonance energy transfer between blue-emitting and red-shifted excitation derivatives of the green fluorescent protein. Gene. 173:13–17. [DOI] [PubMed] [Google Scholar]
- Miyawaki, A., J. Llopis, R. Heim, J. M. McCaffery, J. A. Adams, M. Ikura, and R. Y. Tsien. 1997. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature. 388:882–887. [DOI] [PubMed] [Google Scholar]
- Niwa, H., S. Inouye, T. Hirano, T. Matsuno, S. Kojima, M. Kubota, M. Ohashi, and F. I. Tsuji. 1996. Chemical nature of the light emitter of the Aequorea green fluorescent protein. Proc. Natl. Acad. Sci. USA. 93:13617–13622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormö, M., A. B. Cubitt, K. Kallio, L. A. Gross, R. Y. Tsien, and S. J. Remington. 1996. Crystal structure of the Aequorea victoria green fluorescent protein. Science. 273:1392–1395. [DOI] [PubMed] [Google Scholar]
- Ozawa, T., S. Nogami, M. Sato, Y. Ohya, and Y. Umezawa. 2000. A fluorescent indicator for detecting protein-protein interactions in vivo based on protein splicing. Anal. Chem. 21:5151–5157. [DOI] [PubMed] [Google Scholar]
- Palm, G. J., A. Zdanov, G. A. Gaitanaris, R. Stauber, G. N. Pavlakis, and A. Wlodawer. 1997. The structural basis for spectral variations in green fluorescent protein. Nat. Struct. Biol. 4:361–365. [DOI] [PubMed] [Google Scholar]
- Palm, G. T., and A. Wlodawer. 1999. Spectral variants of green fluorescent protein. Methods Enzymol. 302:378–394. [DOI] [PubMed] [Google Scholar]
- Park, S. H., and R. T. Raines. 1997. Green fluorescent protein as a signal for protein-protein interactions. Protein Sci. 6:2344–2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson, G. H., S. M. Knobe, W. D. Sharif, S. R. Kain, and D. W. Piston. 1997. Use of the green fluorescent protein and its mutants in quantitative fluorescence microscopy. Biophys. J. 73:2782–2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips, G. N., Jr. 1997. Structure and dynamics of green fluorescent protein. Curr. Opin. Struct. Biol. 7:821–827. [DOI] [PubMed] [Google Scholar]
- Prasher, D. C. 1995. Using GFP to see the light. Trends Genet. 11:320–323. [DOI] [PubMed] [Google Scholar]
- Prendergast, F. G. 1999. Biophysics of the green fluorescent protein. Methods Cell Biol. 58:1–18. [DOI] [PubMed] [Google Scholar]
- Robey, R. B., O. Ruiz, A. V. Santos, J. Ma, F. Kear, L. J. Wang, C. J. Li, A. A. Bernardo, and J. A. Arruda. 1998. pH-dependent fluorescence of a heterologously expressed Aequorea green fluorescent protein mutant: in situ spectral characteristics and applicability to intracellular pH estimation. Biochemistry. 37:9894–9901. [DOI] [PubMed] [Google Scholar]
- Romoser, V. A., P. M. Hinkle, and A. Persechini. 1997. Detection in living cells of Ca2+-dependent changes in the fluorescence emission of an indicator composed of two green fluorescent protein variants linked by a calmodulin-binding sequence. A new class of fluorescent indicators. J. Biol. Chem. 272:13270–13274. [DOI] [PubMed] [Google Scholar]
- Saltiel, J., and Y. P. Sun. 1989. Intrinsic potential energy barrier for twisting in the trans-stilbene S1 state in hydrocarbon solvents. J. Phys. Chem. 93:6246–6250. [Google Scholar]
- Schellenberg, P., E. Johnson, A. P. Esposito, P. J. Reid, and W. W. Parson. 2001. Resonance raman scattering by the green fluorescent protein and an analogue of its chromophore. J. Phys. Chem. 105:5316–5322. [Google Scholar]
- Shafi, P. M. 1985. Quantitative synthesis of 2-aryl-4-arylidene-2-imidazolin-5-ones. Curr. Sci. 54:1231–1232. [Google Scholar]
- Shih, T. Y., and G. D. Fasman. 1972. Circular dichroism studies of histone-deoxyribonucleic acid complexes. A comparison of complexes with histone I (f-1), histone IV (f2al), and their mixtures. Biochemistry. 11:398–404. [DOI] [PubMed] [Google Scholar]
- Shimomura, O. 1979. Structure of the chromophore of Aequorea green fluorescent protein. FEBS Lett. 104:220–222. [Google Scholar]
- Striker, G., V. Subramaniam, C. A. M. Seidel, and A. Volkmer. 1999. Photochromicity and fluorescence lifetimes of green fluorescent protein. J. Phys. Chem. 103:8612–8617. [Google Scholar]
- Sugisaki, M., K. Adachi, H. Suga, and S. Seki. 1968. Bull. Chem. Soc. Jpn. 41:593–594. [Google Scholar]
- Suhling, K., D. M. Davis, and D. Phillips. 2002a. The influence of solvent viscosity on the fluorescence decay and time-resolved anisotropy of green fluorescent protein. J. Fluoresc. 12:91–95. [Google Scholar]
- Suhling, K., J. Siegel, D. Phillips, P. M. W. French, S. Lévêque-Fort, S. E. D. Webb, and D. M. Davis. 2002b. Imaging the environment of green fluorescent protein. Biophys. J. 83:3589–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka, K., K. Matsuo, A. Nakanishi, Y. Kataoka, K. Takase, and S. Otsuki. 1988. Syntheses of cyclic hydroxamic acid derivatives, and their chelating abilities and biological activities. Chem. Pharm. Bull. (Tokyo). 36:2323–2330. [DOI] [PubMed] [Google Scholar]
- Tozzini, V., and R. Nifosi. 2001. Ab initio molecular dynamics of the green fluorescent protein (GFP) chromophore: an insight into the photoinduced dynamics of green fluorescent proteins. J. Phys. Chem. B. 105:5797–5803. [Google Scholar]
- Tripathy, K. P., and A. K. Mukerjee. 1985. A facile synthesis of N-substituted 2-acylamino-2-alkenamides. Synthesis. 285–288.
- Tsien, R. Y. 1998. The green fluorescent protein. Annu. Rev. Biochem. 67:509–544. [DOI] [PubMed] [Google Scholar]
- van Thor, J. J., A. J. Pierik, I. Nugteren-Roodzant, A. Xie, and K. J. Hellingwerf. 1998. Characterization of the photoconversion of green fluorescent protein with FTIR spectroscopy. Biochemistry. 37:16915–16921. [DOI] [PubMed] [Google Scholar]
- Velapoldi, R. A., and K. D. Mielenz. 1980. A fluorescence standard reference material: quinine sulfate dihydrate. In NBS Special Publication: Standard Reference Materials. Washington, DC. 260–264.
- Voityuk, A. A., M. E. Michel-Beyerle, and N. Rosch. 1998. Structure and rotation barriers for ground and excited states of the isolated chromophore of the green fluorescent protein. Chem. Phys. Lett. 296:269–276. [Google Scholar]
- Wachter, R. M., B. A. King, R. Heim, K. Kallio, R. Y. Tsien, S. G. Boxer, and S. J. Remington. 1997. Crystal structure and photodynamic behavior of the blue emission variant Y66H/Y145F of green fluorescent protein. Biochemistry. 36:9759–9765. [DOI] [PubMed] [Google Scholar]
- Wachter, R. M., M. A. Elsliger, K. Kallio, G. T. Hanson, and S. J. Remington. 1998. Structural basis of spectral shifts in the yellow-emission variants of green fluorescent protein. Structure. 6:1267–1277. [DOI] [PubMed] [Google Scholar]
- Ward, W. W., and S. H. Bokman. 1982. Reversible denaturation of Aequorea green-fluorescent protein: physical separation and characterization of the renatured protein. Biochemistry. 21:4535–4540. [DOI] [PubMed] [Google Scholar]
- Warren, A., and M. Zimmer. 2001. Computational analysis of Thr203 isomerization in green fluorescent protein. J. Mol. Graphics Modell. 19:297–303. [DOI] [PubMed] [Google Scholar]
- Webber, N. M., K. L. Litvinenko, and S. R. Meech. 2001. Radiationless relaxation in a synthetic analogue of the green fluorescent protein chromophore. J. Phys. Chem. 105:8036–8039. [Google Scholar]
- Weber, W., V. Helms, J. A. McCammon, and P. W. Langhoff. 1999. Shedding light on the dark and weakly fluorescent states of green fluorescent proteins. Proc. Natl. Acad. Sci. USA. 96:6177–6182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler, K., J. Lindner, V. Subramaniam, T. M. Jovin, and P. Vöhringer. 2002. Ultrafast dynamics in the excited state of green fluorescent protein (wt) studied by frequence-resolved femtosecond pump-probe spectroscopy. Phys. Chem. Chem. Phys. 4:1072–1081. [Google Scholar]
- Yang, F., L. G. Moss, and G. M. Phillips. 1996. The molecular structure of green fluorescent protein. Nat. Biotechnol. 14:1246–1252. [DOI] [PubMed] [Google Scholar]
- Zimmer, M. 2002. Green fluorescent protein (GFP): applications, structure, and related photophysical behavior. Chem. Rev. 102:759–781. [DOI] [PubMed] [Google Scholar]