

Abstract
The main potential of intrinsically fluorescent proteins (IFPs), as noninvasive and site-specific markers, lies in biological applications such as intracellular visualization and molecular genetics. However, photophysical studies of IFPs have been carried out mainly in aqueous solution. Here, we provide a comprehensive analysis of the intracellular environmental effects on the steady-state spectroscopy and excited-state dynamics of green (EGFP) and red (DsRed) fluorescent proteins, using both one- and two-photon excitation. EGFP and DsRed are expressed either in the cytoplasm of rat basophilic leukemia (RBL-2H3) mucosal mast cells or anchored (via LynB protein) to the inner leaflet of the plasma membrane. The fluorescence lifetimes (within ∼10%) and spectra in live cells are basically the same as in aqueous solution, which indicate the absence of both IFP aggregation and cellular environmental effects on the protein folding under our experimental conditions. However, comparative time-resolved anisotropy measurements of EGFP reveal a cytoplasmic viscosity 2.5 ± 0.3 times larger than that of aqueous solution at room temperature, and also provide some insights into the LynB-EGFP structure and the heterogeneity of the cytoplasmic viscosity. Further, the oligomer configuration and internal depolarization of DsRed, previously observed in solution, persists upon expression in these cells. DsRed also undergoes an instantaneous three-photon induced color change under 740-nm excitation, with efficiently nonradiative green species. These results confirm the implicit assumption that in vitro fluorescence properties of IFPs are essentially valid for in vivo applications, presumably due to the β-barrel protection of the embodied chromophore. We also discuss the relevance of LynB-EGFP anisotropy for specialized domains studies in plasma membranes.
INTRODUCTION
Wild-type (wt) green fluorescent protein (GFP) and red fluorescent protein (DsRed), isolated from the jellyfish Aequorea victoria and Discosoma corallimorpharian, respectively, have become invaluable fluorescent markers for biological studies (Chalfie and Kain, 1998; Tsien, 1998; Baird et al., 2000; Gross et al., 2000; Fradkov et al., 2000; Belmont, 2001; Chiesa et al., 2001; Wahlfors et al., 2001; Ayoob et al., 2001; Day et al., 1999, 2001; Zimmer, 2002). The wide range of absorption/emission wavelengths of these intrinsically fluorescent proteins (IFPs) and their mutants allows for multicolor labeling, negligible cellular intrinsic fluorescence, and the creation of fluorescence donor-acceptor pairs for Förster resonance energy transfer (FRET), a popular technique in cell biology for studying protein-protein interactions (Day et al., 1999, 2001; Jones et al., 2000; Mizuno et al., 2001; Schuttrigkeit et al., 2001). Genetic encoding of IFPs allows for site-specific labeling within cells for visualization of gene expression and cellular functions and opens many exciting opportunities in biological and biomedical research. So far, however, photophysical studies of IFPs have been carried out mainly in aqueous solution. A comprehensive analysis of IFP fluorescence in the intracellular environment is crucial for reliable quantitative in vivo applications; this is the objective of the research reported here.
IFPs exhibit interesting fluorescence spectroscopy and complex excited-state molecular dynamics that have been the focus of numerous studies in solution (Tsien, 1998; Chalfie and Kain, 1998; Chattoraj et al., 1996; Lossau et al., 1996; Heikal et al., 2000, 2001; Cotlet et al., 2001c). The IFP fluorescence flickers on μs to ms timescales as revealed by fluorescence correlation spectroscopy (FCS). One mechanism of pH-dependent fluorescence flickering, which is observed in most GFPs, is external proton exchange with the bulk buffer (Haupts et al., 1998; Widengren et al., 1999; Schwille et al., 2000; Heikal et al., 2000, 2001, 2002). An additional pH-independent flicker, with a rate that depends on illumination intensity, was also observed and attributed to an internal proton displacement associated with conformational rearrangements of the protein (Haupts et al., 1998; Heikal et al., 2001, 2002) and photoisomerization (Widengren et al., 1999; Schwille et al., 2000; Malvezzi-Campeggi et al., 2001). Such complex photophysics of GFPs makes their use as intracellular pH indicators challenging (Kneen et al., 1998), particularly on the single molecule level (Iino et al., 2001; Kubitscheck et al., 2000; Garcia-Parajo et al., 2000). Our objective here is to consider some of these challenges to evaluate the risk of systematic errors in quantitative analysis of in vivo studies using IFP markers.
The most stable configuration of wt-DsRed in solution is an oligomer (a tetramer) according to analytical ultracentrifugation (Gross et al., 2000), FCS and time-resolved fluorescence anisotropy (Heikal et al., 2000), x-ray crystallography (Yarbrough et al., 2001; Wall et al., 2000), and single-molecule studies (Lounis et al., 2001; Cotlet et al., 2001b,c; Garcia-Parajo et al., 2001; Harms et al., 2001). Such tetramerization of DsRed has greatly hindered its use as a genetically encoded fusion tag. In addition to the slow maturation of DsRed (Baird et al., 2000; Terskikh et al., 2000), a multiphoton color change of DsRed has also been reported in live mammalian cells under intense illumination of ≤760 nm (Marchant et al., 2001). Based on the intensity dependence of DsRed photobleaching, the color change was attributed to a three-photon process, and the green emission persists for ∼30 h without affecting the cell viability. However, the red fluorescence was reported to exhibit less than a quadratic dependence on the excitation intensity (slope = 1.5 on a log-log scale), which was attributed to photobleaching (Marchant et al., 2001). From this earlier work, it was not clear whether the mechanism for the light-induced color change of DsRed is parallel (i.e., green and red emission are generated instantaneously) or sequential (i.e., red emitting species was converted to green emitting species). If it is a sequential mechanism, what is the time delay before the generation of the green species? Finally, how does the fluorescence quantum yield of the green DsRed emission compare with that of the red species? Our answers to these questions in this report establish the degree to which quantitative interpretation of in vivo results may be made using DsRed.
IFP photophysical measurements have been carried out primarily in aqueous solution; however, their main potential lies in quantitative in vivo applications. There are several complications that could invalidate the solution results for quantitative in vivo applications if not recognized and taken into account. These include possible IFP aggregate formation inside cells, binding to cellular constituents, and/or the integrity of protein folding in cellular environments. Additionally, we can compare the rotational and translational diffusion, which allows us to assess the role of viscosity (versus collision or binding with intracellular constituents) in hindering the mobility of IFP markers in living cells. In addition to pH, the viscosity (e.g., in subcellular environments) is also expected to affect the kinetic rate of external proton exchange of GFP with the surrounding buffer, according to FCS studies (Haupts et al., 1998). These previous observations motivated the research presented here on EGFP in living cells. However, cellular effects on GFP folding and aggregation must first be excluded to use them quantitatively with confidence as pH-indicators.
In this report, we present a comprehensive characterization of one (1P) and two (2P) photon fluorescence spectroscopy and excited anionic state dynamics of EGFP and DsRed both in aqueous solution and in living rat basophilic leukemia (RBL) cells, a mucosal mast cell line. These IFPs were expressed in the RBL cytoplasm or anchored to the inner leaflet of the plasma membrane via LynB protein, a member of the Src family kinase that plays an important role in immunoreceptor signaling (Field et al., 1995; Kovarova et al., 2001), which one of us has studied extensively in this cell line (Sheets et al., 1999). A new DsRed variant (DsRed2) also was studied to investigate the effects of mutations (that reduced its maturation time, tendency for aggregation and toxicity) (Yanushevich et al., 2002) on its fluorescence properties as compared with the original DsRed (Matz et al., 1999; Baird et al., 2000; Gross et al., 2000; Fradkov et al., 2000). The present research investigates the living-cell environmental effects on the emission spectra and excited-state dynamics of IFPs, including their rotational mobility. The results provide interesting insights on the rotational mobility restrictions of the protein in either the cytoplasm or when anchored to the inner leaflet of the plasma membrane. Since the light-induced color change can lead to misinterpretation of fluorescence-based biological studies, we also examined the mechanism of laser-induced color change of DsRed under 740-nm excitation. The quantitative results on IFP behavior in live cells are essential first steps toward quantitative in vivo applications.
MATERIALS AND METHODS
Cell and sample preparation
RBL-2H3 mast cells were maintained as described previously (Pierini et al., 1996). The day before transfection, cells that had been resuspended (∼2 × 105 cells/ml) in phenol-red free medium (minimal essential medium, 20% fetal bovine serum, 4 mM l-glutamine, and 50 μg/ml gentamicin) were plated in glass-bottomed 35-mm petri dishes (MatTek Corp., Ashland, MA). One to two days before measurements, RBL cells were transfected using Lipofectamine 2000 (GibcoBRL, Gaithersburg, MD) following the protocol provided by the manufacturer. The only exception was that 100 nM phorbol 12,13-dibutyrate (Sigma Chemical Co., St. Louis, MO) in Opti-Mem I (GibcoBRL) was added to the cells immediately before DNA/Lipofectamine incubation to increase transfection yield (David Holowka, Cornell University; unpublished observations). After incubation with the DNA/Lipofectamine complexes (for 5–9 h), the cells were washed with phenol-red free medium and incubated with 1 μg/ml IgE. Immediately before an experiment, the transfected cells were washed twice and maintained in BSA-containing buffered saline solution (BSA/BSS; 20 mM HEPES, pH 7.4, 135 nM NaCl, 5 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 5.6 mM glucose, and 1 mg/ml BSA).
To express EGFP, DsRed, and DsRed2 proteins in the RBL cytoplasm, the cells were transfected with the vectors EGFP-N1, DsRed-N1, and DsRed2-N1 (Clontech Laboratories, Palo Alto, CA). The new variant DsRed2, with mutations of basic residues (R2T, K5E, and K9T) in the original DsRed, was designed to shorten maturation time, lower the tendency for aggregation, and decrease toxicity (Yanushevich et al., 2002). The LynB-IFP fusion proteins were generated by polymerase chain reaction using the cDNA encoding wild-type rat LynB as the template (Vonakis et al., 1997). Labeling the C-terminal of the Src family kinase with IFPs was carried out by cloning the Lyn protein into the EGFP-N1 and DsRed-N1 vectors. The LynB-IFP constructs were anchored to the inner leaflet of the plasma membrane via N-terminal myristoylation and palmitoylation at positions Gly-2 and Cys-3 of LynB, respectively (Kovarova et al., 2001).
Recombinant EGFP protein was purchased from Clontech and used as received for the solution studies. This EGFP has no additional tag and should be identical to the EGFP expressed in RBL cells, after their transfection with the EGFP-N1 vector. DsRed was a generous gift from Professor Roger Tsien (University of California, San Diego). EGFP or DsRed were diluted into 100 mM potassium phosphate at pH 7.2 (final concentration of ∼5 μM) with negligible pH effects on the fluorescence of EGFP (pKa = 5.9 ± 0.1; Haupts et al., 1998; Heikal et al., 2001) and DsRed (Baird et al., 2000; Heikal et al., 2000; Malvezzi-Campeggi et al., 2001).
Confocal laser scanning microscopy
Confocal fluorescence images were acquired with a BioRad 1024MP laser scanner (Bio-Rad Laboratories, Hercules, CA) coupled to a Zeiss Axiovert 135TV microscope (Zeiss, Jena, Germany), and an argon/krypton laser. A Zeiss Plan Neo-Fluor (40×, 1.3 NA) objective was used for acquiring images in direct slow scanning mode. Cells transfected with EGFP constructs were imaged with the 488-nm laser line and a 515LP emission filter, whereas DsRed-expressing cells were imaged with the 568-nm laser line and a 585LP emission filter. The average laser power used for imaging cytoplasmic EGFP and DsRed was ∼5 μW at the sample, whereas higher power was needed to image the lower expression levels of LynB-EGFP (∼50 μW) and LynB-DsRed (∼100 μW).
Time- and wavelength-resolved fluorescence
The excited-state fluorescence decays of the IFP chimera were measured using time-correlated single photon counting, which is described in detail elsewhere (Heikal et al., 2001) and modified to fit the IM-35 microscope. Briefly, the infrared or the second harmonic of Ti:sapphire (Spectra Physics, Mountain View, CA) femtosecond laser pulses (4 MHz) were directed through the light-inlet port of an inverted IM-35 microscope (Zeiss, Jena, Germany), after pulse picking with an electro-optic modulator (Conoptics, Danbury, CT). The epifluorescence polarization was selected using a Glan-Thompson prism (Melles Giriot, Rochester, NY) mounted at the top exit port of the microscope before being detected using a microchannel plate photomultiplier. The excited-state lifetime was measured using magic angle (54.7°) polarization and then analyzed using a deconvolution with instrumental response function (full width half maximum, FWHM ∼60 ps) and an iterative nonlinear least-squares algorithm (O'Connor and Phillips, 1984). The 2P-instrumental response function was measured using the second harmonic generated from monobasic potassium phosphate (powder crystals), whereas scattering medium (dairy milk) was used for measuring 1P-response with weak satellites near the zero time due to weak reflections. Under these conditions, the estimated temporal resolution for our measurements is ∼15 ps (∼25% of the FWHM of the system response) considering the signal-to-noise ratio and decay time constants. This temporal resolution will decrease for multiexponential decays as well as for time constants that are smaller than the FWHM of the response function. For multiexponential fluorescence F(t) decays with time constants  and preexponential factor
and preexponential factor 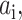 the following fitting functional form was used (Heikal et al., 2001):
the following fitting functional form was used (Heikal et al., 2001):
 |
(1) |
The reported relative amplitudes  and the average lifetime 〈τfl〉 are defined as:
and the average lifetime 〈τfl〉 are defined as:
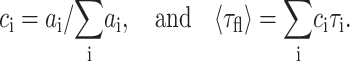 |
(2) |
For time-resolved fluorescence anisotropy, parallel 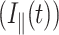 and perpendicular
and perpendicular 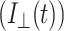 1P-fluorescence decays were measured for equal acquisition time during which there was no significant change in the signal level. The anisotropy decay of a free rotating molecule (e.g., EGFP in solution) in an isotropic environment is described by a single exponential (O'Connor and Phillips, 1984; Lakowicz, 1999):
1P-fluorescence decays were measured for equal acquisition time during which there was no significant change in the signal level. The anisotropy decay of a free rotating molecule (e.g., EGFP in solution) in an isotropic environment is described by a single exponential (O'Connor and Phillips, 1984; Lakowicz, 1999):
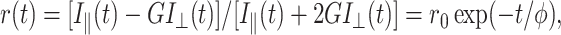 |
(3) |
where φ is the rotational correlation time of the protein and G describes the sensitivity of detection optics and micro-channel plate to polarization. The G-factor was determined experimentally using tail-matching for fluorescein decays whose rotational time (∼150 ps) is much smaller than the corresponding excited-state lifetime (∼3.9 ns). The initial anisotropy  which yields the angle (δ) between the absorption and emission dipoles, is given by (Volkmer et al., 2000):
which yields the angle (δ) between the absorption and emission dipoles, is given by (Volkmer et al., 2000):
 |
(4) |
Ultrafast processes, such as internal conversion or energy transfer, would complicate the interpretation of the  -value, especially if the timescale involved is beyond the temporal resolution of the instrument. Based on the number of photons absorbed (α), the maximal theoretical
-value, especially if the timescale involved is beyond the temporal resolution of the instrument. Based on the number of photons absorbed (α), the maximal theoretical  -values are 0.4 and 0.57 (for collinear dipoles without depolarizing processes) using 1P and 2P measurements, respectively. Under our experimental conditions, the optical depolarization due to the microscope objective (Axelrod, 1979; 1989; Gautier et al., 2001) was negligible using underfilled objective (40×, 1.15 NA, Olympus, Melville, NY). Further, the average baseline signals (before time 0) of both parallel and perpendicular fluorescence decays were subtracted before calculating the anisotropy decay curve to rule out a polarization-biased scattered light contribution.
-values are 0.4 and 0.57 (for collinear dipoles without depolarizing processes) using 1P and 2P measurements, respectively. Under our experimental conditions, the optical depolarization due to the microscope objective (Axelrod, 1979; 1989; Gautier et al., 2001) was negligible using underfilled objective (40×, 1.15 NA, Olympus, Melville, NY). Further, the average baseline signals (before time 0) of both parallel and perpendicular fluorescence decays were subtracted before calculating the anisotropy decay curve to rule out a polarization-biased scattered light contribution.
The hydrodynamic volume (V = υ + h) of a spherical protein also can be calculated using the Stokes-Einstein equation (Lakowicz, 1999):
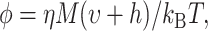 |
(5) |
where M, υ, and h are the molecular mass, specific volume of the protein, and hydration volume, respectively. The environmental absolute temperature is T, and 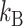 is the Boltzmann constant. In contrast to the free rotator, the angular range of rotational mobility in (or on) organized structures (e.g., membranes) is limited due to local steric hindrance, which usually imposes certain restrictions on the probe orientations. Restricted rotational diffusion (e.g., LynB-EGFP anchored to the inner leaflet to the plasma membrane) can be described as a biexponential decay:
is the Boltzmann constant. In contrast to the free rotator, the angular range of rotational mobility in (or on) organized structures (e.g., membranes) is limited due to local steric hindrance, which usually imposes certain restrictions on the probe orientations. Restricted rotational diffusion (e.g., LynB-EGFP anchored to the inner leaflet to the plasma membrane) can be described as a biexponential decay:
 |
(6) |
In this case, the initial anisotropy r0 = A1 + A2. The same equation also describes the anisotropy decay of DsRed, except when fused to the LynB protein where an additional residual anisotropy 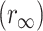 was needed for a better fit.
was needed for a better fit.
For spectral measurements, the epifluorescence was imaged on an optical fiber (500 μm diameter) at the side exit port of the microscope and then recorded on a cooled CCD camera mounted at the exit window of a PC-controlled spectrometer (Spex270M, Olympus, Edison, NJ). The average power of a parked beam (i.e., nonscanning mode) at the sample is ≤5 μW for 485 nm and ≤500 μW for 970 nm. Dichroic mirrors 500LP and 720SP (Chroma Technology Corp., Brattleboro, VT) were used for 1P- and 2P-excitation, respectively. The cellular morphology was examined before and after each measurement to rule out cellular photodamage. Furthermore, cells of varying brightness were selected to compare their photophysical characteristics. The background signal from cellular autofluorescence and the surrounding buffer was negligible. Finally, an overfilled objective (PlanApo60/1.2 NA, water immersion) was used for studying the laser-induced color changes of DsRed under 740-nm illumination.
RESULTS
Cellular localization of IFP constructs
To investigate the effects of intracellular environments on IFP photophysics, we transfected RBL cells with EGFP and DsRed to either target the cytoplasm or anchor to the inner leaflet of the plasma membrane via the LynB protein (Fig. 1). As expected, EGFP and DsRed are located throughout the cytoplasm (Fig. 1, B and C) due to the absence of a specific targeting sequence. For some experiments, we also transfected RBL cells with DsRed2, which is also expressed throughout the cytoplasm similar to the wt-DsRed as observed with confocal microscopy (data not shown). The LynB-IFP constructs (Fig. 1, E and F) are anchored to the inner leaflet of the plasma membrane and thus sample the immediately adjacent cytoplasmic environment. Expression levels of the Lyn constructs are highly variable from cell to cell, and thus are difficult to compare with the endogenous Lyn protein expression in the plasma membrane. However, Kovarova et al. (2001) have estimated an expression level of ∼31% for LynA-GFP in lipid raft fractions, compared with ∼55% of the endogenous Lyn protein, using quantitative immunoblotting analysis. Furthermore, the LynB-GFP construct mediates stimulated phosphorylation of the IgE receptor when expressed in bone marrow derived mast cells from Lyn-1-mice (Kovarova et al., 2001) or in Chinese hamster ovary (CHO) cells (Julie Gosse and David Holowka, Cornell University, unpublished results) indicating that the GFP label does not interfere with the principal function of this tyrosine kinase.
FIGURE 1.

Genetic encoding of IFPs allows for site-specific visualization of living cells. (A and D) Transmission images reveal typical RBL mast cell morphologies that were used in the fluorescence measurements. The corresponding confocal fluorescence images are also shown for EGFP (B) and DsRed (C) proteins expressed in RBL cytoplasm. The LynB-EGFP (E) and LynB-DsRed (F) constructs are anchored to the RBL plasma membrane. The average excitation laser power used for imaging the cytoplasmic EGFP (488 nm) and DsRed (568 nm) was ∼5 μW at the sample, whereas higher power was needed for imaging LynB-EGFP (∼50 μW) and LynB-DsRed (∼100 μW) to compensate for the low concentrations. Bar, 10 μm.
For the fluorescence measurements, transfected cells were selected based on morphology (Fig. 1, A and D represent typical RBL cells) and IFP expression levels, which allowed us to probe possible concentration effects (e.g., aggregation) on the photophysics. The cells were also examined visually before and after an experiment to ensure negligible photodamage under the reported illumination intensities. The measurements were carried out on cells of various expression levels, one to two days posttransfection, and were repeated using multiple transfections for reproducibility. Furthermore, our control experiments on nontransfected cells confirm negligible contributions from the cellular autofluorescence.
Steady-state fluorescence spectra
The 2P-fluorescence spectra of EGFP in solution (Fig. 2, curve 1, lines) and in the cytoplasm of RBL cells (Fig. 2, curve 1, circles) are similar, independent of the expression level, due to the β-barrel protection of the chromophore. DsRed also exhibits a similar 2P-emission spectrum in either solution (Fig. 2, curve 2, lines) or RBL cytoplasm (Fig. 2, curve 2, circles). The lack of significant emission shifts (Chalfie and Kain, 1998) rules out the formation of IFP aggregates in living RBL cells, under our experimental conditions. These measurements were also repeated at λx = 485 nm to examine the effects of excitation pathway on the fluorescence spectrum. The emission profile is the same (within ∼4 nm) under 1P-illumination of IFPs in both solution and cells (cytoplasm and plasma-membrane–anchored) (data not shown). (Note that 1P-excitation without a confocal aperture provides a larger observation volume, as compared to 2P-excitation, enabling us to collect more fluorescence photons from LynB-IFP on the thin cell lamella.) Interestingly, the mutations that cause much faster maturation of DsRed2 than the original DsRed do not affect its advantageous red emission (at ∼583 nm) or its fluorescence quantum yield (see below). Importantly, these results also demonstrate that the in vitro steady-state fluorescence spectroscopy for IFPs can be directly applied to in vivo applications for detection optimization in both microscopy and FRET studies.
FIGURE 2.

The cellular environment does not significantly affect the 2P-emission spectra of EGFP and DsRed (either in the cytoplasm or anchored to the inner leaflet of the plasma membrane). Representative fluorescence emission spectra of EGFP (curve 1) and DsRed (curve 2) in the RBL cytoplasm (circles) and in solution (pH 7.2, lines) using λx = 970 nm. The 1P-fluorescence emission of LynB-DsRed is slightly red-shifted (∼4 nm) in subcellular environments compared with that in solution. These spectra were not corrected for the wavelength sensitivity of the fiber, spectrometer grating, or the CCD camera.
To examine the mechanism for the multiphoton-induced color change of DsRed under 740-nm excitation, we measured the 2P-fluorescence spectra (using a 720-nm short pass filter) as a function of the average excitation power (Fig. 3, A and B). Below saturation, the red emission (585 ± 20 nm) intensity increases quadratically (slope = 2.02 ± 0.06 on a log-log scale) with excitation intensity while the spectral profile remains unchanged (Fig. 3 B, curve 1). This is in contrast to the less-than-quadratic dependence of the fluorescence (at 605 ± 45 nm) of DsRed in CHO cells (slope = 1.50 ± 0.04) reported earlier by Marchant et al. (2001). The intensity of the laser-induced green emission tail (480 ± 45 nm) increases as the excitation intensity increases, with a slope of 2.95 ± 0.06 on a log-log scale (Fig. 3 B, curve 3) above the red-emission saturation. A quadratic intensity dependence of EGFP fluorescence was observed (Fig. 3 B, curve 2) with a negligible blue tail of the emission band at 510 nm (data not shown).
FIGURE 3.

Illumination of DsRed (pH 7.2) with intense 740 nm introduces a green (∼475 nm) emission in addition to the usual red (∼583 nm) fluorescence. (A) A semilogarithmic plot of the DsRed (pH 7.2) emission spectra as a function of 740-nm intensity with 0.1 OD steps. The arrows indicate an increase of the fluorescence emission with the illumination intensity. These spectra were not corrected for the wavelength sensitivity of the fiber, spectrometer grating, or the CCD camera. (B) Intensity dependence of DsRed (red emission: solid squares, curve 1; green emission: solid triangles, curve 3) and EGFP (solid circles, curve 2) fluorescence on log-log scales. The intensities are estimated in units of W/cm2. The red (585 ± 20 nm) and green (480 ± 45 nm) emission of DsRed exhibit a slope of 2.02 ± 0.06 and 2.95 ± 0.06, respectively, compared with 1.96 ± 0.05 for EGFP emission (515 ± 15 nm). Notice the relationship between the red fluorescence saturation of DsRed and the threshold of green emission generation.
Excited anionic state fluorescence decay
The first excited-state fluorescence decay is an excellent probe of the local environment as compared to the steady-state fluorescence spectrum, and allows for quantitative assessment of the cellular environmental effects on the chromophore. We investigated the 1P- and 2P-excited anionic state fluorescence decays of cytoplasmic and plasma-membrane–anchored IFPs. The results are summarized in Table 1, and representative decays are shown in Fig. 4.
TABLE 1.
The excited-state fluorescence decays of selected IFPs in solution (pH 7.2) and in live RBL cells (either in the cytoplasm or anchored to the inner leaflet of the plasma membrane via LynB protein)
| Protein | Environment | λx (nm) | λfl (nm) | τ1 (ns) | c1 | τ2 (ns) | c2 | 〈τfl〉 (ns) | n* |
|---|---|---|---|---|---|---|---|---|---|
| EGFP | Solution (pH 7.2) | 970 | 700SP | 2.90(5) | 0.80(5) | 1.85(5) | 0.24(3) | 2.68(1) | 3 |
| 485 | 585 ± 75 | 2.88(5) | 0.82(6) | 1.84(6) | 0.21(1) | 2.68(3) | 7 | ||
| 740 | 515 ± 15 | 2.88(1) | 0.76(2) | 0.34(2) | 0.24(2) | 2.27(5) | 2 | ||
| 740† | 450 ± 40 | 0.11(1) | 0.64(1) | 0.43(1) | 0.32(1) | 0.31(1) | 2 | ||
| Cytoplasm | 970 | 700SP | 2.71(4) | 0.70(5) | 1.85(6) | 0.30(5) | 2.46(1) | 7 | |
| 485 | 580 ± 75 | 2.72(2) | 0.76(3) | 1.86(9) | 0.24(3) | 2.53(2) | 13 | ||
| 2.55(2) | — | — | — | 2.55(2) | 5 | ||||
| LynB-EGFP | Plasma membrane | 485 | 585 ± 75 | 2.76(5) | 0.75(3) | 1.8(1) | 0.25(3) | 2.53(3) | 15 |
| DsRed | Solution (pH 7.2) | 970 | 700SP | 3.41(4) | 0.80(2) | 1.32(4) | 0.22(3) | 3.00(3) | 5 |
| 485 | 585 ± 75 | 3.49(2) | 0.84(1) | 1.45(9) | 0.16(1) | 3.15(2) | 6 | ||
| 740 | 585 ± 20 | 3.50(2) | 0.78(2) | 1.39(6) | 0.22(4) | 3.04(5) | 4 | ||
| 740‡ | 480 ± 45 | 0.03(1) | 0.62(5) | 0.16(1) | 0.30(4) | 0.15(4) | 4 | ||
| Cytoplasm | 970 | 700SP | 3.29(6) | 0.85(2) | 0.8(5) | 0.15(2) | 2.9(2) | 12 | |
| 485 | 585 ± 75 | 3.38(2) | — | — | — | 3.38(2) | 10 | ||
| 585 ± 75§ | 3.0(1) | 0.74(6) | 1.2(1) | 0.26(7) | 2.6(2) | 5 | |||
| LynB-DsRed | Plasma membrane | 485 | 585 ± 75 | 3.5(1) | 0.81(6) | 1.6(3) | 0.19(6) | 3.11(7) | 6 |
| DsRed2 | Cytoplasm | 480 | 580 ± 75 | 3.09(3) | 0.80(3) | 0.64(3) | 0.20(2) | 2.84(9) | 4 |
The fitting parameters are defined in Eqs. 1 and 2. The standard deviation in the last digit of the fitting parameters is written in parentheses.
The number of measurements (n), in either solution or live RBL cells, includes different cells and transfections.
Three exponential decays were measured with τ3 = 2.61 ± 0.03 ns and c3 ∼ 0.04.
Three exponential decays were measured with τ3 = 718 ± 49 ps and c3 ∼ 0.08.
These measurements were carried out two days posttransfection and, in some cases (n = 3 cells), three exponential decays were measured with τ3 = 120 ± 40 ps, c3 ∼ 0.10 ± 0.07 and an average lifetime of 2.4 ± 0.4 ns.
FIGURE 4.

The living-cell environment reveals minimal effects on the excited-state fluorescence decays (i.e., fluorescence quantum yield) of IFPs under 2P-excitation (λx = 970 nm). (A, curve 1) The 2P-fluorescence of cytoplasmic EGFP decays as a biexponential (τ1 = 2.70 ns (c1 = 0.70), τ2 = 1.85 ns (c2 = 0.30) and χ2 = 1.12) with an average lifetime of ∼2.45 ns. (A, curve 2) Under the same conditions, the 2P-fluorescence of EGFP in solution (pH 7.2) decays also as a biexponential (τ1 = 2.91 ns (c1 = 0.78), τ2 = 1.81 ns (c2 = 0.22)) with a slightly different average lifetime of ∼2.67 ns, that is ±8% larger than the cytoplasmic fluorescence decay. (B, curve 1) In the RBL cytoplasm, the DsRed 2P-fluorescence decay parameters (τ1 = 3.23 ns (c1 = 0.78), and τ2 = 370 ps (c2 = 0.22)) are slightly different from that in buffered solution (pH 7.2) where τ1 = 3.4 ns (c1 = 0.78) and τ2 = 1.38 ns (c2 = 0.22) (B, curve 2).
EGFP
In the RBL cytoplasm, the total 2P-fluorescence (λfl ≤ 700 nm) of EGFP (λx = 970 nm) decays biexponentially (Fig. 4 A, curve 1; Table 1), which is similar to solution measurements (Fig. 4 A, curve 2). However, the cytoplasmic average decay time constant (〈τfl〉 = 2.46 ± 0.01 ns) is slightly faster than the solution value (〈τfl〉 = 2.68 ± 0.01 ns). Assuming that the radiative rate constant is the same for in vitro and in vivo EGFP, the fluorescence quantum yield  of EGFP is ∼8% lower in living cells as compared to solution (Φfl = 0.6; Tsien, 1998). By comparison, smaller differences (∼6%) in the averaged fluorescence lifetimes were observed for EGFP under 1P-excitation (λx = 485 nm, λfl = 585 ± 75 nm) of EGFP (Table 1). In a few cases (for n = 4 cells), the cytoplasmic EGFP fluorescence decays as a single exponential with τfl = 2.54 ± 0.01 ns, good χ2, and acceptable residuals. Finally, the 1P-fluorescence of LynB-EGFP decays biexponentially with 〈τfl〉 = 2.53 ± 0.02 ns (Table 1), which is similar to the cytoplasmic EGFP results. These results suggest that the cellular environment and fusion to the LynB protein have negligible effects on the excited-state fluorescence lifetime (i.e., quantum yield) of EGFP.
of EGFP is ∼8% lower in living cells as compared to solution (Φfl = 0.6; Tsien, 1998). By comparison, smaller differences (∼6%) in the averaged fluorescence lifetimes were observed for EGFP under 1P-excitation (λx = 485 nm, λfl = 585 ± 75 nm) of EGFP (Table 1). In a few cases (for n = 4 cells), the cytoplasmic EGFP fluorescence decays as a single exponential with τfl = 2.54 ± 0.01 ns, good χ2, and acceptable residuals. Finally, the 1P-fluorescence of LynB-EGFP decays biexponentially with 〈τfl〉 = 2.53 ± 0.02 ns (Table 1), which is similar to the cytoplasmic EGFP results. These results suggest that the cellular environment and fusion to the LynB protein have negligible effects on the excited-state fluorescence lifetime (i.e., quantum yield) of EGFP.
DsRed
The cytoplasmic DsRed fluorescence (λx = 970 nm) decays as a biexponential (Fig. 4 B, curve 1; Table 1) with an average fluorescence lifetime of 2.9 ± 0.2 ns, which is ∼14% faster than that in solution of 3.38 ± 0.02 ns (Fig. 4 B, curve 2; Table 1). Assuming a similar radiative decay rate in both environments, one would conclude that the fluorescence quantum yield of the cytoplasmic DsRed is ∼14% lower than in solution (Φfl = 0.75; Baird et al., 2000). By comparison, the 1P-fluorescence of DsRed decays mainly as a single exponential with an excited-state lifetime of 3.15 ± 0.02 ns (n = 11 cells; Table 1). However, two days posttransfection, we observed faster decays with average lifetimes of 2.6 ± 0.2 ns (n = 5 cells, biexponential decays) and 2.4 ± 0.4 ns (n = 5 cells, three exponential decays) as summarized in Table 1. These results indicate that the excited-state dynamics of DsRed is sensitive to posttransfection time as observed previously (Jakobs et al., 2000). Interestingly, the S1-state fluorescence of the cytoplasmic DsRed2 variant (Table 1) also decays as a biexponential with τ1 = 3.09 ns (c1 = 0.80) and τ2 = 640 ps (c2 = 0.20) with an average fluorescence lifetime of 2.84 ± 0.09 ns. Accordingly, the estimated fluorescence quantum yield of DsRed2 is ∼0.68 in RBL cytoplasm, as compared to the parent DsRed in solution (Φfl = 0.75; Baird et al., 2000). DsRed2 is a good substitute for the original DsRed due to its fast maturation and lower tendency for aggregation. The excited-state 1P-fluorescence of LynB-DsRed chimera decays as a biexponential with 〈τfl〉 = 3.11 ± 0.07 ns (Table 1).
Our solution results of DsRed, using the microscope set-up described here, are somewhat different from those in our previous report in which we used right-angle fluorescence detection and observed a single exponential decay with 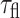 ∼ 3.6 ns (Heikal et al., 2000). To reconcile these results, we carried out several control experiments. First we repeated the same measurements on a newly prepared and mature DsRed to eliminate the possibility of sample degradation. Our control measurements on fluorescein and wt-GFP fluorescence decays, which are single exponentials, ruled out the possibility of experimental artifacts. The background signals of the in vivo and in vitro experiments were also negligible. As a result of these various controls, we infer that the second component in DsRed fluorescence decays can be attributed to a minor contribution (∼16%) from a sparsely populated, short-lived DsRed configuration or state.
∼ 3.6 ns (Heikal et al., 2000). To reconcile these results, we carried out several control experiments. First we repeated the same measurements on a newly prepared and mature DsRed to eliminate the possibility of sample degradation. Our control measurements on fluorescein and wt-GFP fluorescence decays, which are single exponentials, ruled out the possibility of experimental artifacts. The background signals of the in vivo and in vitro experiments were also negligible. As a result of these various controls, we infer that the second component in DsRed fluorescence decays can be attributed to a minor contribution (∼16%) from a sparsely populated, short-lived DsRed configuration or state.
To investigate the mechanism involved in the in vitro DsRed photoconversion under 740-nm excitation, we measured the excited-state fluorescence decays of the red and green species. The red emission of DsRed (585 ± 20 nm) decays as a biexponential with 〈τfl〉 = 3.04 ± 0.05 ns (Fig. 5 A, curve 1; Table 1), which is fairly similar to fluorescence decay measured under 970 nm excitation (〈τfl〉 = 3.00 ± 0.03 ns). However, the three-photon–induced green emission (480 ± 45 nm) decays on an ultrafast timescale with 〈τfl〉 = 150 ± 40 ps (Fig. 5 A, curve 2), which indicates efficient nonradiative relaxation channel(s) competing with the green fluorescence pathway. This fluorescence decay resembles that of the neutral excited-state fluorescence of EGFP excited under 405 nm (Heikal et al., 2001) and 740 nm (Table 1), especially with narrow band detection (450 ± 30 nm). Furthermore, even with the smallest time per channel available (3.26 ps), both the red and green fluorescence build up instantaneously (within our temporal resolution) suggesting a parallel mechanism for the photoactivation of both the red and green species.
FIGURE 5.

The multiphoton-induced green emission of DsRed (pH 7.2) suggests an efficiently nonradiative species/state as compared to the red emitting state. (A, curve 1) The red 2P-fluorescence (585 ± 20 nm) of DsRed in aqueous solution (pH 7.2) decays as a biexponential with τ1 = 3.51 ns (c1 = 0.76), τ2 = 1.31 ns (c2 = 0.24), and χ2 = 1.04 under 740-nm illumination. (A, curve 2) By comparison, the green 2P-fluorescence (480 ± 45 nm) of DsRed (pH 7.2) decays as a triple exponential with τ1 = 25 ps (c1 = 0.62), τ2 = 165 ps (c2 = 0.30), τ3 = 777 ps (c2 = 0.08), and χ2 = 2.1. The average fluorescence lifetime of the green emission is ∼5% of the red emission. (B, curve 1) The fluorescence anisotropy, associated with the green emission (485 ± 40 nm) of DsRed (pH 7.2) under 740-nm excitation, decays as a biexponential with φ1 ∼ 153 ps (A1 = 0.09) and φ2 ∼ 11.9 ns (A2 = 0.35). (B, curve 2) Under the same excitation conditions, the fluorescence (450 ± 45 nm) anisotropy of EGFP (pH 7.2) decays as a biexponential with φ1 ∼ 649 ps (A1 = 0.04) and φ2 ∼ 12.4 ns (A2 = 0.42). The anisotropy associated with the long wavelength emission of both DsRed and EGFP (pH 7.2) are fairly similar to that measured at 485-nm excitation (Fig. 6, A and B; curves 2).
Time-resolved fluorescence anisotropy
The rotational correlation times (φ) of EGFP (Fig. 6 A) and DsRed (Fig. 6 B), in the RBL cytoplasm (curves 1) and in solution (curves 2), were measured and the results are summarized in Table 2. The anisotropy decay of cytoplasmic EGFP is mostly a single exponential (Fig. 6 A, curve 1) with an estimated overall rotational time φ2 = 42 ± 4 ns and an amplitude A2 = 0.36 ± 0.02 (Table 2). This overall rotational time for cytoplasmic EGFP is 2.5 ± 0.3 times slower than EGFP in solution (φ = 17 ± 1 ns; Fig. 6 A, curve 2; Table 2), suggesting a 2.5-fold larger cytoplasmic viscosity as compared to aqueous solution at room temperature. The fitting residual of cytoplasmic EGFP anisotropy was enhanced significantly with an additional minor (A3 = 0.018) decay component (1.6 ± 0.5 ns). The single exponential anisotropy decay and the rotational time of EGFP, in solution, are in agreement with previous studies on the rotational diffusion of wt-GFP and S65T (Volkmer et al., 2000), and Citrine (Heikal et al., 2000), indicating a rigidly constrained chromophore within the protein polypeptide (Ormö et al., 1996; Brejc et al., 1997).
FIGURE 6.

The cytoplasmic environment of RBL retards the rotational mobility of EGFP, by comparison with buffered solution at room temperature, whereas it does not affect the oligomer configuration and intraoligomer depolarization of DsRed. (A) Time-resolved fluorescence anisotropy of EGFP (λx = 485 nm) in RBL cytoplasm (42 ± 4 ns and an amplitude A2 ∼ 0.36, curve 1) and in solution (17 ± 1 ns, curve 2) decays mainly as a single exponential. A minor (A2 ∼ 0.018) second decay component with 1.6 ± 0.5 ns time constant enhanced the cytoplasmic fit significantly. The initial anisotropy 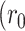 ∼ 0.38) is comparable with the theoretical limit (0.4), suggesting a negligible optical depolarization effects due to the microscope objective. (B, curve 1) The anisotropy of cytoplasmic DsRed decays as a biexponential with slower overall rotational time (φ2 = 134 ± 31 ns with A2 ∼ 0.26) than the excited-state lifetime (∼3.4 ns). The second decay component (φ1 = 290 ± 70 ps with A1 ∼ 0.05) is significantly faster than the overall rotation of the oligomer. (B, curve 2) By comparison, the anisotropy of DsRed in solution (pH 7.2) decays also as a biexponential with a slightly faster overall rotational time (φ2 = 76 ± 5 ns and A2 ∼ 0.25) than that in RBL cytoplasm, with a second decay component of φ1 = 278 ± 53 ps and A1 ∼ 0.06.
∼ 0.38) is comparable with the theoretical limit (0.4), suggesting a negligible optical depolarization effects due to the microscope objective. (B, curve 1) The anisotropy of cytoplasmic DsRed decays as a biexponential with slower overall rotational time (φ2 = 134 ± 31 ns with A2 ∼ 0.26) than the excited-state lifetime (∼3.4 ns). The second decay component (φ1 = 290 ± 70 ps with A1 ∼ 0.05) is significantly faster than the overall rotation of the oligomer. (B, curve 2) By comparison, the anisotropy of DsRed in solution (pH 7.2) decays also as a biexponential with a slightly faster overall rotational time (φ2 = 76 ± 5 ns and A2 ∼ 0.25) than that in RBL cytoplasm, with a second decay component of φ1 = 278 ± 53 ps and A1 ∼ 0.06.
TABLE 2.
Time-resolved fluorescence anisotropy of EGFP and DsRed in both solution (pH 7.2) and in live RBL cells as a function of excitation and detection wavelengths
| Protein | Environment | λx/nm | λfl/nm | φ1/ns | A1 | φ2/ns | A2 | r0 | n* |
|---|---|---|---|---|---|---|---|---|---|
| EGFP | Solution | 485 | 585 ± 75 | — | — | 17(1) | — | 0.383(8) | 6 |
| 740 | 515 ± 15 | — | — | 17.9(2) | 0.45(1) | 0.45(1)† | 2 | ||
| 450 ± 40 | 0.58(4) | 0.03 | 11.6(9) | 0.40(1) | 0.43(1)† | 2 | |||
| EGFP | Cytoplasm | 485 | 580 ± 75 | 1.6(5) | 0.018(6) | 42(4) | 0.36(2) | 0.38(1) | 7 |
| LynB-EGFP | Plasma membrane | 485 | 585 ± 75 | 1.2(8) | 0.017(5) | 80(14) | 0.35(2) | 0.37(1) | 8 |
| DsRed | Solution | 485 | 580 ± 75 | 0.29(3) | 0.06(1) | 76(5) | 0.26(2) | 0.32(1) | 5 |
| 740 | 585 ± 20 | 0.9(1) | 0.05(1) | 70(5) | 0.34(1) | 0.39(1)† | 3 | ||
| 480 ± 45 | 0.21(3) | 0.06(1) | 11.6(5) | 0.34(1) | 0.40(1)† | 2 | |||
| DsRed | Cytoplasm | 485 | 585 ± 20 | 0.29(7) | 0.05(1) | 134(31) | 0.26(1) | 0.31(1) | 4 |
| LynB-DsRed | Plasma membrane‡ | 485 | 585 ± 75 | 0.21(2) | 0.04(1) | 3.5(9) | 0.04(1) | 0.29(2) | 3 |
| DsRed2 | Cytoplasm | 485 | 580 ± 75 | 0.39(7) | 0.04(1) | 111(11) | 0.21(1) | 0.25(1) | 3 |
The fitting parameters are defined in Eq. 6.
The number of measurements (n), in either solution or live RBL cells, includes different cells and transfections.
The maximal theoretical value for the initial 2P-anisotropy is 0.57 as compared with 0.4 for 1P-excitation.
The anisotropy decays as a biexponential plus a residual anisotropy r∞ = 0.21 ± 0.02.
The anisotropy of cytoplasmic DsRed (Fig. 6 B, curve 1) decays as a biexponential (Table 2) with a much slower overall rotational time (φ2 = 134 ± 31 ns with A2 = 0.26 ± 0.01) than the excited-state lifetime (∼3.38 ns, Table 1), which increases the uncertainty in the rotational time estimation. The second decay component (φ1 = 290 ± 70 ps with A1 = 0.05 ± 0.01) is significantly faster than the overall rotation of the oligomer. Although the anisotropy of DsRed in solution (pH 7.2, Fig. 6 B, curve 2, Table 2) decays also as a biexponential (Heikal et al., 2000), the overall rotational time is slightly faster (φ2 = 76 ± 5 ns, A2 = 0.26 ± 0.02) than that in RBL cytoplasm. The second decay component of DsRed anisotropy in solution (φ1 = 290 ± 30 ps with A1 = 0.06 ± 0.01) and the reduced initial anisotropy (0.32 ± 0.01) were previously attributed to intraoligomer energy transfer (Heikal et al., 2000; Lounis et al., 2001; Cotlet et al., 2001b,c; Garcia-Parajo et al., 2001; Harms et al., 2001). These results readily demonstrate the persistence of the oligomer configuration and internal depolarization of DsRed inside live cells. Furthermore, the long rotational time and the internal depolarization preclude the use of DsRed as an intracellular viscosity probe. The anisotropy decays of the cytoplasmic DsRed2 mutant and the parent DsRed were also comparable (Table 2).
The anisotropy measurements of in vivo LynB-IFP provide an opportunity for investigating structural aspects of the constructs as well as probing the microenvironment adjacent to the inner leaflet of the plasma membrane. The anisotropy of LynB-EGFP decays as a biexponential with an overall rotational time φ2 = 80 ± 14 ns and an amplitude A2 = 0.35 ± 0.02 (Fig. 7 A, curve 1; Table 2). The second minor anisotropy decay component (amplitude A1 = 0.017 ± 0.005) has a 1.2 ± 0.8 ns time constant suggesting a segmental motion between the EGFP and Lyn proteins (see Discussion). The overall rotational time of LynB-EGFP is slightly faster than the theoretical prediction based on both its molecular mass (∼84 kDa, see Discussion) and the cytoplasmic viscosity (∼2.5 times larger than that of buffered solution). Similar measurements were carried out on LynB-DsRed with some difficulties due to the low transfection level and the low excitation cross section at 485 nm. The anisotropy decays of plasma membrane-anchored LynB-DsRed (Table 2) are satisfactorily described as biexponential with φ1 = 210 ± 20 ps (A1 = 0.04 ± 0.01), φ2 = 3.5 ± 0.9 ns (A2 = 0.04 ± 0.01), and residual anisotropy 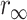 ∼ 0.21 (Table 2).
∼ 0.21 (Table 2).
FIGURE 7.

The fluorescence anisotropy of LynB-EGFP construct, anchored to the inner leaflet plasma membrane, reveals intramolecular flexibility in addition to the overall restricted rotational mobility. (A, curve 1) The anisotropy of LynB-EGFP decays as a biexponential with an overall rotational time φ2 = 80 ± 14 ns and A2 ∼ 0.36. The second minor (an amplitude A1 ∼ 0.021) anisotropy decay component is on the order of 1.5 ± 0.8 ns. (A, curve 2) For visual comparison, the single exponential anisotropy decay of EGFP in solution (pH 7.2) is also shown with 17 ± 1 ns rotational time. (B) A schematic representation of a wobbling-in-cone motion of the LynB-EGFP around its anchor to the inner leaflet of the plasma membrane, with an estimated cone semiangle of ∼13°. This measured overall rotational time of LynB-EGFP construct is consistent with the theoretical prediction (see text) based on both its molecular mass and the cytoplasmic viscosity (∼2.5 times larger than that in solution) adjacent to the plasma membrane.
The structural identity of DsRed species, associated with both the green and red emissions, can also be elucidated by measuring the rotational anisotropy under 740-nm excitation at which color change was observed. The DsRed anisotropy associated with the red emission (585 ± 20 nm) is fairly similar to that under 485-nm illumination (Fig. 6 B, curve 2, Table 2). However, the anisotropy of the green DsRed emission (480 ± 45 nm) decays as a biexponential (Fig. 5 B, curve 1) with fast depolarization of 210 ± 30 ps (A1 = 0.06 ± 0.01) and an overall slow rotational time of 11.6 ± 0.5 ns (A2 = 0.34 ± 0.01), which is much shorter than the overall rotational time of the DsRed oligomer (Fig. 6 B, Table 2). For comparison, similar measurements were carried out on EGFP (pH 7.2) under the same excitation conditions (Fig. 5 B, curve 2). The fluorescence (515 ± 15 nm) anisotropy of EGFP decays as a single exponential with an estimated rotational time of 17.9 ± 0.2 ns, similar to that shown in Fig. 6 A, curve 2. However, the anisotropy associated with the blue emission shoulder (450 ± 40 nm) of EGFP decays as a biexponential (Fig. 5 B, curve 1) with a fast component of ∼580 ps (A1 ∼ 0.04) and an overall slow rotational time of ∼12 ns (A2 ∼ 0.40). These results suggest that the green emission might be assigned to a subunit (i.e., a monomer) within the DsRed oligomer.
DISCUSSION
Genetic encoding of IFPs, one of their major advantages as fluorescent markers for biological research, allows for site-specific monitoring of cellular functions with minimal interference. IFPs also exhibit advantageous fluorescence and thermodynamic properties that can be exploited for quantitative biological applications such as FRET and cellular pH measurements. Until now, however, the photophysics of these molecules has been studied mainly in aqueous solution. There are a number of reasons that might invalidate or complicate the direct application of these results for in vivo use. First, IFPs might aggregate in living cells, potentially causing a shift of the emission spectrum (Chalfie and Kain, 1998). Second, binding of IFPs to cellular constituents might also modify the chromophore's immediate surroundings and, therefore, its fluorescence properties. Finally, the folding of the IFP construct might also be compromised in cellular environments, especially upon fusion with membrane proteins such as Lyn. Knowledge of the cellular effects on the folding and aggregation of GFPs is critical for the effective use of IFPs as pH-indicators for quantitative in vivo applications. In this report, we compare the fluorescence spectroscopy and excited-state dynamics of EGFP and DsRed in aqueous solution and living RBL mast cells. For in vivo studies, these IFPs were located in either the cytoplasm (Fig. 1, B and C) or anchored to the plasma membrane inner leaflet via an associated LynB protein (Fig. 1, E and F).
Assessment of the cellular background signal
Both EGFP and DsRed are excellent 2P-fluorescent markers due to their large 2P-excitation cross sections,  (Heikal et al., 2000, 2001, 2002; Blab et al., 2001; Tsai et al., 2002) and emission peaks (Fig. 2), which help to minimize the intrinsic cellular fluorescence. Much of the autofluorescence in tissues or cultured cells is due to reduced pyridine nucleotides (e.g., NADH), flavin adenine dinucleotide (FAD) and mononucleotides (FMN), and polymeric extracellular proteins including collagen and elastin. The observed maxima (at 730 nm) of
(Heikal et al., 2000, 2001, 2002; Blab et al., 2001; Tsai et al., 2002) and emission peaks (Fig. 2), which help to minimize the intrinsic cellular fluorescence. Much of the autofluorescence in tissues or cultured cells is due to reduced pyridine nucleotides (e.g., NADH), flavin adenine dinucleotide (FAD) and mononucleotides (FMN), and polymeric extracellular proteins including collagen and elastin. The observed maxima (at 730 nm) of  -values 0.035 (NADH), 0.085 (FAD), 0.9 (FMN) GM, (Xu et al., 1996; Huang et al., 2002) are orders of magnitude smaller than EGFP and DsRed (∼100 GM at 970 nm) used in the present work (Heikal et al., 2000, 2001; Blab et al., 2001; Tsai et al., 2002). Therefore, the autofluorescence contribution to our observed signal is expected to be negligible, which is confirmed with our experimental controls on nontransfected cells.
-values 0.035 (NADH), 0.085 (FAD), 0.9 (FMN) GM, (Xu et al., 1996; Huang et al., 2002) are orders of magnitude smaller than EGFP and DsRed (∼100 GM at 970 nm) used in the present work (Heikal et al., 2000, 2001; Blab et al., 2001; Tsai et al., 2002). Therefore, the autofluorescence contribution to our observed signal is expected to be negligible, which is confirmed with our experimental controls on nontransfected cells.
Steady-state spectroscopy
The 2P-fluorescence spectra indicate that EGFP and DsRed retain their emission profiles in the two cellular locations as compared to aqueous solution (Fig. 2), which can be attributed to the chromophore protection by the β-barrel. This observation is particularly important for several applications that require accurate knowledge of the absorption and emission spectra, such as FRET (Day et al., 1999, 2001; Jones et al., 2000; Mizuno et al., 2001; Schuttrigkeit et al., 2001; Gautier et al., 2001), dual-color cross-correlation FCS (Koltermann et al., 1998), and dual-channel imaging using 1P- and 2P-photon fluorescence microscopy, where fluorescence detection can be optimized without ambiguity. Such fluorescence detection optimization allows for minimal illumination intensities to be used to avoid photodamage of biological samples. Furthermore, the lack of spectral shift in cellular environments indicates the absence of IFP aggregation.
Excited-state fluorescence dynamics
Although the 2P-fluorescence spectra of EGFP in live RBL cells and buffered solution are almost indistinguishable (Fig. 2), the average fluorescence lifetime in the living cells was slightly faster (∼8%) than in solution (Fig. 4 A; Table 1). The fluorescence quantum yield is dependent on the fluorescence decay rate,  where
where  and
and  are the radiative and nonradiative rate constants, respectively. Assuming the same
are the radiative and nonradiative rate constants, respectively. Assuming the same 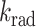 in both environments, we therefore conclude that the 2P-fluorescence quantum yield of cellular EGFP is ∼9% lower than that in solution (
in both environments, we therefore conclude that the 2P-fluorescence quantum yield of cellular EGFP is ∼9% lower than that in solution (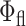 = 0.60; Tsien, 1998). Even smaller differences (∼6%) between the excited-state fluorescence decays of EGFP in live cells and in solution were observed under 1P-excitation, independent of the local RBL cellular environments. Our findings are in agreement with recent studies on the in vivo molecular brightness of EGFP using 2P-FCS (at λx = 895 nm), which was the same in the cytoplasm and nucleus of HeLa cells as in aqueous solution (Chen et al., 2001). These results provide yet another indication that the chromophore is well protected by the β-barrel and that EGFP itself is not aggregated in the cellular environment under these experimental conditions.
= 0.60; Tsien, 1998). Even smaller differences (∼6%) between the excited-state fluorescence decays of EGFP in live cells and in solution were observed under 1P-excitation, independent of the local RBL cellular environments. Our findings are in agreement with recent studies on the in vivo molecular brightness of EGFP using 2P-FCS (at λx = 895 nm), which was the same in the cytoplasm and nucleus of HeLa cells as in aqueous solution (Chen et al., 2001). These results provide yet another indication that the chromophore is well protected by the β-barrel and that EGFP itself is not aggregated in the cellular environment under these experimental conditions.
Similarly, the 2P-excited-state fluorescence decay (Fig. 4 B; Table 1) and emission spectrum (Fig. 2) of DsRed in live cells and buffered solution are comparable, which indicate that the oligomeric configuration of DsRed persists in both environments. Furthermore, the fluorescence quantum yields of DsRed in solution (Baird et al., 2000) and inside cells are basically the same (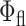 = 0.75). However, Jakobs et al. (2000) reported a slightly different excited-state lifetime of EGFP (2.4 ns) and DsRed (2.7 to 3.1 ns) in Escherichia coli cells, as compared to solution studies (Cotlet et al., 2001a; Heikal et al., 2000, 2001, 2002), using fluorescence lifetime imaging microscopy (FLIM). The high temporal resolution reported here and the fitting routine, which includes deconvolution with the system response function, enable us to resolve minor lifetime differences and provide a quantitative comparison between cellular and solution environments under the same experimental conditions. Fluorescence decays observed under 1P-excitation of DsRed in RBL cytoplasm (Table 1) were more divergent and exhibited sensitivity to posttransfection time. Two days posttransfection, we observed no indications of an immature DsRed species in our steady-state fluorescence and excited-state dynamics measurement in RBL cells, in contrast to the DsRed mutants used as “molecular timers” (Terskikh et al., 2000; Verkhusha et al., 2001). Finally, the 1P-excited-state fluorescence decays (i.e., quantum yield) of DsRed did not change when fused to the LynB protein in the inner leaflet of the plasma membrane of RBL or free in solution (Table 1).
= 0.75). However, Jakobs et al. (2000) reported a slightly different excited-state lifetime of EGFP (2.4 ns) and DsRed (2.7 to 3.1 ns) in Escherichia coli cells, as compared to solution studies (Cotlet et al., 2001a; Heikal et al., 2000, 2001, 2002), using fluorescence lifetime imaging microscopy (FLIM). The high temporal resolution reported here and the fitting routine, which includes deconvolution with the system response function, enable us to resolve minor lifetime differences and provide a quantitative comparison between cellular and solution environments under the same experimental conditions. Fluorescence decays observed under 1P-excitation of DsRed in RBL cytoplasm (Table 1) were more divergent and exhibited sensitivity to posttransfection time. Two days posttransfection, we observed no indications of an immature DsRed species in our steady-state fluorescence and excited-state dynamics measurement in RBL cells, in contrast to the DsRed mutants used as “molecular timers” (Terskikh et al., 2000; Verkhusha et al., 2001). Finally, the 1P-excited-state fluorescence decays (i.e., quantum yield) of DsRed did not change when fused to the LynB protein in the inner leaflet of the plasma membrane of RBL or free in solution (Table 1).
Environmental effects on rotational diffusion of IFPs
The fluorescence anisotropy of EGFP decays as a single exponential in aqueous solution (φ = 17 ± 1 ns) with large (∼0.38) initial anisotropy (Fig. 6 A, Table 2), which indicates that the chromophore is held rigidly within the β-barrel (Ormö et al., 1996; Brejc et al., 1997; Hink et al., 2000). Based on the measured rotational time, the estimated average diameter for a spherical EGFP (in aqueous solution at room temperature) agrees well with the theoretical prediction of its hydrodynamic volume using M ∼ 28 kDa and a hydrated volume h ∼ 1.0 cm3 g−1 (Cantor and Schimmel, 1980).
Comparison between the rotational and translational diffusion of IFPs allows for probing the nature of mobility restrictions inside living cells such as the increased cytoplasmic viscosity, collisions with subcellular particles, or intracellular binding interactions (Dayel et al., 1999; Kao et al., 1993). The overall rotational correlation time for EGFP in the RBL cytoplasm (42 ± 4 ns) is 2.5 ± 0.3 times slower than that in aqueous solution (17 ± 1 ns) (Table 2; Fig. 6 A). Because the fluorescence spectrum and lifetime measurements eliminate the possibility of EGFP aggregation in live cells, we conclude that the cytoplasmic viscosity is 2.5 ± 0.3 times larger than buffered solution (Eq. 5) at room temperature. These results are in general agreement with previous Fourier transform fluorometry measurements of the rotational diffusion of EGFP (Dayel et al., 1999) and GFP-S65T (Swaminathan et al., 1997) in CHO cells, where the rotational time is slower by a factor of 3.2 and 1.8, respectively. The translational diffusion coefficient 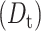 of a molecule is inversely proportional to the viscosity (η) and the molecular diameter (a) according to the relationship,
of a molecule is inversely proportional to the viscosity (η) and the molecular diameter (a) according to the relationship, 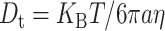 with
with 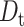 = 6.6 ± 0.2 × 10−7 cm2·s−1 for EGFP in aqueous solution as measured using FCS (data not shown). Using the viscosity implied by the rotational anisotropy measurements, we estimate a translational diffusion coefficient of 2.8 ± 0.1 × 10−7 cm2·s−1 for EGFP in the RBL cytoplasm. This predicted
= 6.6 ± 0.2 × 10−7 cm2·s−1 for EGFP in aqueous solution as measured using FCS (data not shown). Using the viscosity implied by the rotational anisotropy measurements, we estimate a translational diffusion coefficient of 2.8 ± 0.1 × 10−7 cm2·s−1 for EGFP in the RBL cytoplasm. This predicted  -value is in good agreement with fluorescence recovery after photobleaching (FRAP) studies (Dayel et al., 1999) where
-value is in good agreement with fluorescence recovery after photobleaching (FRAP) studies (Dayel et al., 1999) where 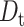 ∼ 3 × 10−7 cm2·s−1 in the CHO cytoplasm, which is ∼3.2 times slower than that in water. Recent FCS studies reveal that the translational diffusion of EGFP is threefold slower in HeLa cytoplasm than that in solution (Chen et al., 2001). Similarly, 1P-FCS studies concluded that the diffusion coefficients of EGFP, S65T, and DsRed in buffer solution are three to four times larger than that in the cytoplasm of HEK 293 cells (Dittrich et al., 2001). From these reported lateral diffusion coefficients using FCS and FRAP techniques, the cytoplasm from many different cell types is roughly (3.4 ± 0.4)-fold more viscous than aqueous solution. The rotational mobility of IFPs in living cells is therefore primarily hindered by viscosity with minor collisional and dynamic-binding effects, particularly on longer observation timescales (e.g., microsecond to millisecond).
∼ 3 × 10−7 cm2·s−1 in the CHO cytoplasm, which is ∼3.2 times slower than that in water. Recent FCS studies reveal that the translational diffusion of EGFP is threefold slower in HeLa cytoplasm than that in solution (Chen et al., 2001). Similarly, 1P-FCS studies concluded that the diffusion coefficients of EGFP, S65T, and DsRed in buffer solution are three to four times larger than that in the cytoplasm of HEK 293 cells (Dittrich et al., 2001). From these reported lateral diffusion coefficients using FCS and FRAP techniques, the cytoplasm from many different cell types is roughly (3.4 ± 0.4)-fold more viscous than aqueous solution. The rotational mobility of IFPs in living cells is therefore primarily hindered by viscosity with minor collisional and dynamic-binding effects, particularly on longer observation timescales (e.g., microsecond to millisecond).
Lyn protein, a member of the Src family of tyrosine kinases, plays a critical role in initiating the allergic response in mast cells and basophils by phosphorylating the IgE receptor. Time-resolved anisotropy measurements provide structural insights on in vivo LynB-IFP constructs as well as the microenvironment adjacent to the inner leaflet of plasma membrane. The same measurements enable us to assess the flexibility of anchors that connect either the LynB-IFP construct to the plasma membrane or IFP to the LynB protein. The LynB protein is anchored to the plasma membrane via N-terminal myristoylation and palmitoylation and is conjugated to either EGFP (Fig. 1 E for LynB-EGFP) or DsRed (Fig. 1 F for LynB-DsRed) at its C-terminus. The combined molecular mass of LynB-EGFP is ∼84 kDa (with ∼56 kDa for LynB (Vonakis et al., 1997) and 28 kDa for EGFP); however, the global structure of the chimera is not known. Taking into account the LynB-EGFP molecular mass and our estimated cytoplasmic viscosity (∼2.5 cp), one would predict an overall rotational time of 128 ± 18 ns, which is slightly slower than the measured slow rotational time φ2 = 80 ± 14 ns. The source of such discrepancy between the two rotational times can be attributed to the limited angular range for rotational mobility due to the tether connecting the LynB-EGFP construct to the plasma membrane. Furthermore, there is a potential for spatial variation of the cytoplasmic viscosity adjacent to or far from the inner leaflet of the plasma membrane. As we demonstrated in this report, such environmental variation can be probed via the relative rotational diffusion of the cytoplasmic EGFP and LynB-EGFP that is anchored to the inner leaflet of the plasma membrane. The observed biexponential anisotropy can be modeled as a wobbling-in-cone around a flexible tether connecting LynB-EGFP to the plasma membrane, in which the amplitude ratio of the two decay components is given by Kinosita et al. (1982), Ameloot et al. (1984), Lakowicz (1999), and Guest et al. (1991):
 |
(7) |
The estimated cone semiangle 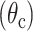 is = 13 ± 3 degrees around a line that is perpendicular to the plasma membrane (Fig. 7 B). This limited angular range would explain the minor differences between the calculated and measured rotational time of LynB-EGFP anchored to the plasma membrane. In addition, the slight flexibility between the two subunits of LynB-EGFP is demonstrated by the minor (A1 = 0.021 ± 0.015) internal depolarization (φ1 = 1.5 ± 0.8 ns) component in the anisotropy decay.
is = 13 ± 3 degrees around a line that is perpendicular to the plasma membrane (Fig. 7 B). This limited angular range would explain the minor differences between the calculated and measured rotational time of LynB-EGFP anchored to the plasma membrane. In addition, the slight flexibility between the two subunits of LynB-EGFP is demonstrated by the minor (A1 = 0.021 ± 0.015) internal depolarization (φ1 = 1.5 ± 0.8 ns) component in the anisotropy decay.
These results undermine the use of LynB-EGFP for probing specialized lipid domains or “rafts” with which Lyn may associate to facilitate IgE receptor signaling. This conclusion is based on the fact that the measured rotational time of LynB-EGFP can be accounted for using both the molecular mass and the immediate cytoplasmic viscosity. Thus, the LynB-EGFP construct undergoes a wobbling motion around its contact points with the plasma membrane and, therefore, is insensitive to the lipid order/disorder. GFP mutants that are attached directly to a membrane-targeting sequence are a viable option for probing the membrane environment and the specialized domains or “rafts” using time-resolved anisotropy. During the final preparation of this manuscript, steady-state fluorescence anisotropy measurements have been reported for GFP expressed and rigidly held with the amino acid sequence of a major histocompatibility complex (MHC) class I molecule (Rocheleau et al., 2003). This labeling scheme would provide a rigid label-membrane contact and, therefore, better sensitivity of the rotational mobility of the label to the surrounding microdomains. Another valuable probe for specialized microdomains is the rotational diffusion of fluorescent markers that are intercalated within the membrane matrix. Thus, our studies provide a guideline for quantitative analysis of specialized domains in plasma membranes using time-resolved anisotropy.
A monomer DsRed would have a molecular weight similar to that of EGFP (Baird et al., 2000) and, therefore, one would expect comparable rotational times. However, the observed overall rotational time is φ2 = 76 ± 5 ns for DsRed in solution with an amplitude A2 = 0.26 ± 0.02 (Table 2, Fig. 6 B, curve 2). In agreement with previous studies (Heikal et al., 2000; Lounis et al., 2001; Cotlet et al., 2001b,c; Garcia-Parajo et al., 2001; Harms et al., 2001), this observation suggests an oligomer configuration with an effective molecular volume 4.4 ± 0.4 times larger than predicted for a monomer. The relatively large uncertainty is due to the difference (more than an order of magnitude) between the excited-state lifetime and the rotational time constant. The fast internal depolarization (φ1 = 290 ± 30 ps with A1 = 0.06 ± 0.01) of DsRed is attributed to energy transfer between nonparallel dipoles of neighboring chromophores that form the oligomer (Heikal et al., 2000). Interestingly, the oligomer configuration and internal depolarization of DsRed persist inside living cells either in the cytoplasm or anchored to the inner leaflet of the plasma membrane as part of LynB-DsRed (Table 2). The increase in the overall rotational time of LynB-DsRed is consistent with higher viscosity in the cellular cytoplasm. Neither DsRed (including its variant DsRed2 in the RBL cytoplasm) expression in living cells nor its being anchored to the plasma membrane via LynB protein fusion seems to prevent its overall rotational freedom (within a limited cone angle), oligomer configuration, or internal depolarization. Recently, a true monomer mutant of DsRed (namely mRFP1) was discovered with a faster (>10 times) maturation with an emission peak around 607 nm (Campbell et al., 2002). However, mRFP1 exhibits a much lower fluorescence quantum yield, extinction coefficient, and photostability than the original DsRed. Thus far, oligomerization of DsRed seems to be critical for its advantageous fluorescence properties (Sacchetti et al., 2002).
Multiphoton-induced color changes of DsRed
Color change of DsRed emission, recently observed in live mammalian CHO cells under intense 740-nm illumination, was attributed to a three-photon process based on the illumination intensity dependence of the photobleaching rate (Marchant et al., 2001). The green DsRed emission persists for more than 30 h without affecting the cell viability. However, less than quadratic dependence (a slope of 1.50 ± 0.04 on a log-log scale) of red fluorescence (at 605 ± 45 nm) on the laser intensity was reported and attributed to photobleaching (Marchant et al., 2001). Below saturation, we observed a quadratic dependence of DsRed fluorescence (585 ± 20 nm) on the excitation intensity (slope = 2.06 ± 0.08 on a log-log scale) (Fig. 3 B, curve 1). Above saturation, less than quadratic dependence of the red emission was also observed, which is consistent with Marchant et al. (2001). Furthermore, the laser-induced green emission (480 ± 45 nm) increases with the excitation intensity (above the red saturation threshold), yielding a slope of 2.95 ± 0.06 (Fig. 3 B, curve 3) supporting the previous photobleaching studies in CHO cells (Marchant et al. 2001).
In time-domain, the ultrafast green fluorescence decay (〈τfl〉 = 150 ± 40 ps) of DsRed (λx = 740 nm) also indicates a distinct transition/species with an efficient nonradiative relaxation pathway that competes with fluorescence (Fig. 5 A). As a result, we conclude that the green emission exhibits a low fluorescence quantum yield as compared with the DsRed red emission (585 ± 20 nm). Furthermore, the red and green fluorescence both build up instantaneously (i.e., within our temporal resolution) after 2P and 3P excitation, respectively, which indicates a parallel photoconversion mechanism. Interestingly, the time-resolved green fluorescence anisotropy of DsRed (in pH 7.2 solution using λx = 740 nm; Fig. 5 B, curve 1) also suggests that the green emission might be assigned to a subunit (i.e., a monomer) of the DsRed oligomer (Fig. 5 B, curve 1). This conclusion is based on the fact that the overall rotational time of this DsRed subunit is much shorter than that of the oligomer configuration (Fig. 6 B, curve 2), yet is fairly similar to that of the monomeric EGFP (Fig. 5 B, curve 2). However, one must keep in mind that the excited-state lifetime is much faster than the overall rotational time associated with the green emission.
Implications on GFP as cellular pH-indicators
Several cellular functions depend on pH gradients across membranes. In addition to the selective targeting of GFPs to specific subcellular locations, their steady-state spectra display pH sensitivity with a wide range of pKa values in aqueous solution (Tsien 1998; Haupts et al., 1998; Widengren et al., 1999; Schwille et al., 2000; Heikal et al., 2000, 2001). For example, the cytosolic, nuclear, and mitochondrial pH values were monitored in intact HeLa cells transfected with enhanced yellow fluorescent protein (Llopis et al., 1998). Other GFP mutants (namely pHluorins) have been used for pH measurements in synaptic vesicles that undergo exocytosis during action potential firing (Miesenböck et al., 1998; Sankaranarayanan et al., 2000). Such pH sensitivity in GFPs is due to external proton exchange (on a timescale of ∼350 μs and a standard free energy of ∼33 kJ/mol) between the chromophore and the surrounding acidic environment (Haupts et al., 1998; Widengren et al., 1999; Schwille et al., 2000; Heikal et al., 2000, 2001). (In contrast to GFPs, DsRed does not exhibit pH sensitivity over a wide range (4.5 < pH < 12), although protein denaturation occurs beyond that pH range (Vrzheshch et al., 2000; Baird et al., 2000; Heikal et al., 2000).) Recently, the excited-state fluorescence average lifetime of EGFP in aqueous solution was measured as a function of pH (Heikal et al., 2001), and the estimated pKa was the same as that measured using steady-state spectroscopy and FCS (Haupts et al., 1998). It is worth mentioning that the pH-dependent flickering fraction of GFP (at nanomolar concentration) is a robust indicator for pH when studied using FCS. The fraction and pKa are determined by statistical thermodynamics of the GFP protonation process and are independent of viscosity and buffer concentration that affects the protonation kinetics (Haupts et al., 1998; Widengren et al., 1999). Our comparative in vitro and in vivo EGFP studies reported here reveal invariant fluorescence decays (within 10%) and spectra, which indicate negligible cellular environmental effects on the protein folding and aggregation. As a result, the observed pH sensitivity of GFPs in solution is still relevant for quantitative in vivo applications.
CONCLUSIONS
We present a comprehensive and quantitative fluorescence analysis of IFP in living cells and in buffered solution under both 1P- and 2P-excitation. The similarity between emission spectra and excited-state dynamics, in both environments, rules out the formation of large IFP aggregates in RBL cells under our experimental conditions. For the same reasons, the cellular environment causes negligible effects on the IFP folding integrity and, therefore, the use of IFPs as intracellular pH indicators. Importantly, the considerable literature on IFPs in solution, characterizing pH sensitivity, fluorescence flicker, excited-state dynamics, and 1P- and 2P- fluorescence spectroscopy are directly relevant for in vivo applications. Extensive studies of all IFPs in both solution and different cell lines would enable us to generalize our findings regarding the similar in vitro and in vivo steady-state spectroscopy and excited-state relaxation of EGFP, DsRed, and DsRed2. Indeed, there are a few steady-state fluorescence studies in the literature (De Angelis et al., 1998) that show similar emission spectra of different GFP mutants in solution and in various cell lines. Accordingly, our generalization of such a trend under similar conditions seems reasonable. These robust fluorescence properties of IFPs are due to the β-barrel protection of the chromophore.
The comparative anisotropy measurements of IFPs in living cells and in solution indicate that the RBL cytoplasmic viscosity is ∼2.5 times larger than that of aqueous solution, which is in agreement with translational diffusion measurements using FRAP and FCS. Whereas the viscosity primarily hinders the rotational mobility (on a faster timescale) of IFPs in living cells, collisions and dynamic adhesion of fluorescent probes with cellular constituents can also retard the translational diffusion on the longer timescales that are specific to the techniques employed. Further, the expression of DsRed constructs in living cells does not prevent the oligomerization of DsRed, which will preclude its use in some cellular applications. Our anisotropy results on LynB-EGFP, anchored to the inner leaflet of the plasma membrane, are an initial step toward elucidating specialized domains in the plasma membrane using time-resolved anisotropy.
The light-induced color change of DsRed is instantaneous after a three-photon excitation mechanism. After 740-nm excitation, the green fluorescence decays on ultrafast timescales, suggesting effective nonradiative relaxation pathways and a greatly reduced quantum yield of the green species compared to the red one. According to our time-resolved anisotropy of DsRed under 740-nm excitation, the green emission can be assigned to a subunit (i.e., monomer) within the oligomer. Finally, we also show that the DsRed2 mutant has fluorescence properties in RBL cytoplasm that are similar to those of DsRed, in addition to its faster maturation time, lower tendency for aggregation, and reduced toxicity.
Acknowledgments
We would like to thank the referees for their valuable comments on this manuscript. The interesting discussion with Prof. Watt Webb and Prof. Barbara Baird is deeply appreciated. The purified DsRed sample was a generous gift from Prof. Roger Tsien and Geoffrey Baird (University of California, San Diego). We are particularly grateful to Julie Gosse for helping with LynB-DsRed preparation and sharing her results with us before publication. We also acknowledge the helpful comments of David Holowka, Raymond Molloy, Mark Williams and Harshad Vishwasrao on this manuscript.
This project was carried out in the Developmental Resource for Biophysical Imaging Opto-Electronics, and the publication is made possible with funding from the National Institutes of Health (NIH) (9-P41-EB0011976-16) and the National Science Foundation (NSF) (BIR 8800278). S.T.H. benefited from an NSF Graduate Research Fellowship and an NIH Molecular Biophysics Training Grant (GM08267). The financial support for E.D.S. was provided by an NIH grant (AI18306). The STC Program of the NSF provided additional support under agreement number ECS-9876771 (Nanobiotechnology Center).
Samuel T. Hess and Erin D. Sheets contributed equally to this work.
Ahmed A. Heikal's present address is 205 Hallowell Building, Department of Bioengineering, Pennsylvania State University, University Park, PA 16802.
Erin D. Sheets' present address is 152 Davey Laboratory, Department of Chemistry, The Pennsylvania State University, University Park, PA 16802.
References
- Ameloot, M., H. Hendrick, W. Herreman, H. Pottel, F. van Cauwelaert, and W. van der Meer. 1984. Effect of orientational order on the decay of the fluorescence anisotropy in membrane suspensions. Experimental verification on unilamellar vesicles and lipid/alpha-lactalbumin complexes. Biophys. J. 46:525–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelrod, D. 1979. Carbocyanine dye orientation in red cell membrane studied by microscopic fluorescence polarization. Biophys. J. 26:557–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelrod, D. 1989. Fluorescence polarization microscopy. Methods Cell Biol. 30:333–352. [PubMed] [Google Scholar]
- Ayoob, J. C., N. C. Shaner, J. W. Sanger, and J. M. Sanger. 2001. Expression of green or red fluorescent protein (GFP or DsRed) linked proteins in nonmuscle and muscle cells. Mol. Biotechnol. 17:65–71. [DOI] [PubMed] [Google Scholar]
- Baird, G. S., D. A. Zacharias, L. A. Gross, R. C. Hoffman, K. K. Baldridge, and R. Y. Tsien. 2000. Biochemistry, mutagenesis, and oligomerization of DsRed, a red fluorescent protein from coral. Proc. Natl. Acad. Sci. USA. 97:11984–11989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmont, A. S. 2001. Visualizing chromosome dynamics with GFP. Trends Cell Biol. 11:250–257. [DOI] [PubMed] [Google Scholar]
- Blab, G. A., P. H. M. Lommerse, L. Cognet, G. S. Harms, and T. Schmidt. 2001. Two-photon excitation action cross-sections of the autofluorescent proteins. Chem. Phys. Lett. 350:71–77. [Google Scholar]
- Brejc, K., T. K. Sixma, P. A. Kitts, S. R. Kain, R. Y. Tsien, M. Ormo, and S. J. Remington. 1997. Structural basis for dual excitation and photoisomerization of the Aequorea victoria green fluorescent protein. Proc. Natl. Acad. Sci. USA. 94:2306–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, R. E., O. Tour, A. E. Palmer, P. A. Steinbach, G. S. Baird, D. A. Zacharias, and R. Y. Tsien. 2002. A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. USA. 99:7877–7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantor, C. R., and P. R. Schimmel. 1980. Biophysical Chemistry II. W. H. Freeman and Company, San Francisco.
- Chalfie, M., and S. Kain. 1998. Green Fluorescent Proteins: Properties, Applications, and Protocols. Wiley-Liss, New York.
- Chattoraj, M., B. A. King, G. U. Bublitz, and S. G. Boxer. 1996. Ultra-fast excited state dynamics in green fluorescent protein: multiple states and proton transfer. Proc. Natl. Acad. Sci. USA. 93:8362–8367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y., J. D. Muller, Q. Q. Ruan, and E. Gratton. 2001. Molecular brightness characterization of EGFP in vivo by fluorescence fluctuation spectroscopy. Biophys. J. 82:133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiesa, A., E. Rapizzi, V. Tosello, P. Pinton, M. de Virgilio, K. E. Fogarty, and R. Rizzuto. 2001. Recombinant aequorin and green fluorescent protein as valuable tools in the study of cell signaling. Biochem. J. 355:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotlet, M., J. Hofkens, S. Habuchi, G. Dirix, M. Van Guyse, J. Michiels, J. Vanderleyden, and F. C. De Schryver. 2001a. Identification of different emitting species in the red fluorescent protein DsRed by means of ensemble and single-molecule spectroscopy. Proc. Natl. Acad. Sci. USA. 98:14398–14403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotlet, M., J. Hofkens, F. Kohn, J. Michiels, G. Dirix, M. Van Guyse, J. Vanderleyden, and F. C. De Schryver. 2001b. Collective effects in individual oligomers of the red fluorescent coral protein DsRed. Chem. Phys. Lett. 336:415–423. [Google Scholar]
- Cotlet, M., J. Hofkens, M. Maus, T. Gensch, M. van der Auweraer, J. Michiels, G. Dirix, M. Van Guyse, J. Vanderleyden, A. J. W. G. Visser, and F. C. De Schryver. 2001c. Excited-state dynamics in the enhanced green fluorescent protein mutant probed by picosecond time-resolved single photon counting spectroscopy. J. Phys. Chem. B. 105:4999–5006. [Google Scholar]
- Day, R. N., S. K. Nordeen, and Y. H. Wan. 1999. Visualizing protein-protein interactions in the nucleus of the living cell. Mol. Endocrinol. 13:517–526. [DOI] [PubMed] [Google Scholar]
- Day, R. N., A. Periasamy, and F. Schaufele. 2001. Fluorescence resonance energy transfer microscopy of localized protein interactions in the living cell nucleus. Methods. 25:4–18. [DOI] [PubMed] [Google Scholar]
- Dayel, M. J., E. F. Y. Hom, and A. S. Verkman. 1999. Diffusion of green fluorescent protein in the aqueous-phase lumen of endoplasmic reticulum. Biophys. J. 76:2843–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Angelis, D. A., G. Miesenböck, B. V. Zemelman, and J. E. Rothman. 1998. PRIM: proximity imaging of green fluorescent protein-tagged polypeptides. Proc. Natl. Acad. Sci. USA. 95:12312–12316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittrich, P., F. Malvezzi-Campeggi, M. Jahnz, and P. Schwille. 2001. Accessing molecular dynamics in cells by fluorescence correlation spectroscopy. Biol. Chem. 382:491–494. [DOI] [PubMed] [Google Scholar]
- Field, K. A., D. Holowka, and B. Baird. 1995. FcɛRI-mediated recruitment of p53/56lyn to detergent-resistant membrane domains accompanies cellular signaling. Proc. Natl. Acad. Sci. USA. 92:9201–9205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fradkov, A. F., Y. Chen, L. Ding, E. V. Barsova, M. V. Matz, and S. A. Lukyanov. 2000. Novel fluorescent protein from Discosoma coral and its mutants possesses a unique far-red fluorescence. FEBS Lett. 479:127–130. [DOI] [PubMed] [Google Scholar]
- Garcia-Parajo, M. F., M. Koopman, E. M. H. P. van Dijk, V. Subramaniam, and N. F. van Hulst. 2001. The nature of fluorescence emission in the red fluorescent protein DsRed, revealed by single-molecule detection. Proc. Natl. Acad. Sci. USA. 98:14392–14397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Parajo, M. F., G. M. J. Segers-Nolten, J.-A. Veerman, J. Greve, and N. F. van Hulst. 2000. Real-time light-driven dynamics of the fluorescence emission in single green fluorescent protein molecules. Proc. Natl. Acad. Sci. USA. 97:7237–7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier, I., M. Tramier, C. Durieux, J. Coppey, R. B. Pansu, J.-C. Nicolas, K. Kemnitz, and M. Coppey-Moisan. 2001. Homo-FRET microscopy in living cells to measure monomer-dimer transition of GFP-tagged proteins. Biophys. J. 80:3000–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross, L. A., G. S. Baird, R. C. Hoffman, K. K. Baldridge, and R. Y. Tsien. 2000. The structure of the chromophore within DsRed, a red fluorescent protein from coral. Proc. Natl. Acad. Sci. USA. 97:11990–11995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guest, C. R., R. A. Hochstrasser, L. C. Sowers, and D. P. Millar. 1991. Dynamics of mismatched base pairs in DNA. Biochemistry. 30:3271–3279. [DOI] [PubMed] [Google Scholar]
- Harms, G. S., L. Cognet, P. H. M. Lommerse, G. A. Blab, and T. Schmidt. 2001. Autofluorescent proteins in single-molecule research: applications to live cell imaging microscopy. Biophys. J. 80:2396–2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupts, U., S. Maiti, P. Schwille, and W. W. Webb. 1998. Dynamics of fluorescence fluctuations in green fluorescent protein observed by fluorescence correlation spectroscopy. Proc. Natl. Acad. Sci. USA. 95:13573–13578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heikal, A. A., S. T. Hess, G. S. Baird, R. Y. Tsien, and W. W. Webb. 2000. Molecular spectroscopy and dynamics of intrinsically fluorescent proteins: coral red (dsRed) and yellow (Citrine). Proc. Natl. Acad. Sci. USA. 97:11996–12001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heikal, A. A., S. T. Hess, E. D. Sheets, and W. W. Webb. 2002. Mutation-photophysics relationships in intrinsically fluorescent proteins. In Femtochemistry and Femtobiology: Ultrafast Dynamics in Molecular Science. A. Douhal and J. Santamaria, editors. World Scientific, Singapore. 774–781.
- Heikal, A. A., S. T. Hess, and W. W. Webb. 2001. Multiphoton molecular spectroscopy and excited state dynamics of enhanced green fluorescent protein (EGFP): acid-base specificity. Chem. Phys. 274:37–55. [Google Scholar]
- Hink, M. A., R. A. Griep, J. W. Borst, A. van Hoek, M. H. M. Eppink, A. Schots, and A. J. W. G. Visser. 2000. Structural dynamics of green fluorescent protein alone and fused with a single chain Fv protein. J. Biol. Chem. 275:17556–17560. [DOI] [PubMed] [Google Scholar]
- Huang, S. H., A. A. Heikal, and W. W. Webb. 2002. Two-photon fluorescence spectroscopy and microscopy on NAD(P)H and flavoprotein. Biophys. J. 82:2811–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iino, R., I. Koyama, and A. Kusumi. 2001. Single molecule imaging of green fluorescent proteins in living cells: E-cadherin forms oligomers on the free cell surface. Biophys. J. 80:2667–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobs, S., V. Subramaniam, A. Schonle, T. M. Jovin, and S. W. Hell. 2000. EGFP and DsRed expressing cultures of Escherichia coli imaged by confocal, two-photon and fluorescence lifetime microscopy. FEBS Lett. 479:131–135. [DOI] [PubMed] [Google Scholar]
- Jones, J., R. Heim, E. Hare, J. Stack, and B. A. Pollok. 2000. Development and application of a GFP-FRET intracellular caspase assay for drug screening. J. Biomol. Screen. 5:307–317. [DOI] [PubMed] [Google Scholar]
- Kao, H. P., J. R. Abney, and A. S. Verkman. 1993. Determinants of the translational mobility of a small solute in cell cytoplasm. J. Cell Biol. 120:175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinosita, K., A. Ikegami, and S. Kawato. 1982. On the wobbling-in-cone analysis of fluorescence anisotropy decay. Biophys. J. 37:461–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneen, M., J. Farinas, Y. X. Li, and A. S. Verkman. 1998. Green fluorescent protein as a noninvasive intracellular pH indicator. Biophys. J. 74:1591–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koltermann, A., U. Kettling, J. Bieschke, T. Winkler, and M. Eigen. 1998. Rapid assay processing by integration of dual-color fluorescence cross-correlation spectroscopy: high throughput screening for enzyme activity. Proc. Natl. Acad. Sci. USA. 95:1421–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovarova, M., P. Tolar, R. Arudchandran, L. Draberova, J. Rivera, and P. Draber. 2001. Structure-function analysis of Lyn kinase association with lipid rafts and initiation of early signaling events after Fcɛ receptor I aggregation. Mol. Cell. Biol. 21:8318–8328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubitscheck, U., O. Kuckmann, T. Kues, and R. Peters. 2000. Imaging and tracking of single GFP molecules in solution. Biophys. J. 78:2170–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowicz, J. R. 1999. Principles of Fluorescence Spectroscopy. Kluwer Academic/Plenum Publishers, New York.
- Llopis, J., J. M. McCaffery, A. Miyawaki, M. G. Farquhar, and R. Y. Tsien. 1998. Measurement of cytosolic, mitochondrial, and Golgi pH in single living cells with green fluorescent proteins. Proc. Natl. Acad. Sci. USA. 95:6803–6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lossau, H., A. Kummer, R. Heinecke, F. Pöllinger-Dammer, C. Kompa, G. Bieser, T. Jonsson, C. M. Silva, M. M. Yang, D. C. Youvan, and M. E. Michel-Beyerle. 1996. Time-resolved spectroscopy of wild-type and mutant green fluorescent proteins reveals excited state deprotonation consistent with fluorophore-protein interactions. Chem. Phys. 213:1–16. [Google Scholar]
- Lounis, B., J. Deich, F. I. Rosell, S. G. Boxer, and W. E. Moerner. 2001. Photophysics of DsRed, a red fluorescent protein, from the ensemble to the single-molecule level. J. Phys. Chem. B. 105:5048–5054. [Google Scholar]
- Malvezzi-Campeggi, F., M. Jahnz, K. G. Heinze, P. Dittrich, and P. Schwille. 2001. Light-induced flickering of DsRed provides evidence for distinct and interconvertible fluorescent states. Biophys. J. 81:1776–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchant, J. S., G. G. Stutzmann, M. A. Leissring, F. M. LaFerla, and I. Parker. 2001. Multiphoton-evoked color change of DsRed as an optical highlighter for cellular and subcelluar labeling. Nat. Biotechnol. 19:645–649. [DOI] [PubMed] [Google Scholar]
- Matz, M. V., A. F. Fradkov, Y. A. Labas, A. P. Savitsky, A. G. Zaraisky, M. L. Markelov, and S. A. Lukyanov. 1999. Fluorescent proteins from nonbioluminescent Anthozoa species. Nat. Biotechnol. 17:969–973. [DOI] [PubMed] [Google Scholar]
- Miesenböck, G., D. A. De Angelis, and J. E. Rothman. 1998. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature (Lond.). 394:192–195. [DOI] [PubMed] [Google Scholar]
- Mizuno, H., A. Sawano, P. Eli, H. Hama, and A. Miyawaki. 2001. Red fluorescent protein from Discosoma as a fusion tag and a partner for fluorescence resonance energy transfer. Biochemistry. 40:2502–2510. [DOI] [PubMed] [Google Scholar]
- O'Connor, D. V., and D. Phillips. 1984. Time-Correlated Single Photon Counting. Academic Press, London.
- Ormö, M., A. B. Cubitt, K. Kallio, L. A. Gross, R. Y. Tsien, and S. J. Remington. 1996. Crystal structure of the Aequorea victoria green fluorescent protein. Science. 273:1392–1395. [DOI] [PubMed] [Google Scholar]
- Pierini, L., D. Holowka, and B. Baird. 1996. FcɛRI-mediated association of 6-μm beads with RBL-2H3 mast cells results in exclusion of signaling proteins from the forming phagosome and abrogation of normal downstream signaling. J. Cell Biol. 134:1427–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocheleau, J. V., M. Edidin, and D. W. Piston. 2003. Intrasequence GFP in class I MHC molecules, a rigid probe for fluorescence anisotropy measurements of the membrane environment. Biophys. J. 84:4078–4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacchetti, A., V. Subramaniam, T. M. Jovin, and S. Alberti. 2002. Oligomerization of DsRed is required for the generation of a functional red fluorescent chromophore. FEBS Lett. 525:13–19. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan, S., D. De Angelis, J. E. Rothman, and T. A. Ryan. 2000. The use of pHluorins for optical measurements of presynaptic activity. Biophys. J. 79:2199–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuttrigkeit, T. A., U. Zachariae, T. von Feilitzsch, J. Wiehler, J. von Hummel, B. Steipe, and M. E. Michel-Beyerle. 2001. Picosecond time-resolved FRET in the fluorescent protein from Discosoma Red (wt-DsRed). Chem. Phys. Chem. 2:325–328. [DOI] [PubMed] [Google Scholar]
- Schwille, P., S. Kummer, A. A. Heikal, W. E. Moerner, and W. W. Webb. 2000. Fluorescence correlation spectroscopy reveals fast optical excitation-driven intramolecular dynamics of yellow fluorescent proteins. Proc. Natl. Acad. Sci. USA. 97:151–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheets, E. D., D. Holowka, and B. Baird. 1999. Critical role for cholesterol in Lyn-mediated tyrosine phosphorylation of FcɛRI and their association with detergent-resistant membranes. J. Cell Biol. 145:877–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminathan, R., C. P. Hoang, and A. S. Verkman. 1997. Photobleaching recovery and anisotropy decay of green fluorescent protein GFP-S65T in solution and cells: cytoplasmic viscosity probed by green fluorescent protein translational and rotational diffusion. Biophys. J. 72:1900–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terskikh, A., A. Fradkov, G. Ermakova, A. Zaraisky, P. Tan, A. V. Kajava, X. N. Zhao, S. Lukyanov, M. Matz, S. Kim, I. Weissman, and P. Siebert. 2000. Fluorescent timer: protein that changes color with time. Science. 290:1585–1588. [DOI] [PubMed] [Google Scholar]
- Tsai, P. S., N. Nishimura, E. J. Yoder, E. M. Dolnick, G. A. White, and D. Kleinfeld. 2002. Principles, design, and construction of a two-photon laser scanning microscope for in vitro and in vivo brain imaging. In Methods for In Vivo Optical Imaging. R. Frostig, editor. CRC Press, Boca Raton. 113–171.
- Tsien, R. Y. 1998. The green fluorescent protein. Annu. Rev. Biochem. 67:509–544. [DOI] [PubMed] [Google Scholar]
- Verkhusha, V. V., H. Otsuna, T. Awasaki, H. Oda, S. Tsukita, and K. Ito. 2001. An enhanced mutant of red fluorescent protein DsRed for double labeling and developmental timer of neural fiber bundle formation. J. Biol. Chem. 276:29621–29624. [DOI] [PubMed] [Google Scholar]
- Volkmer, A., V. Subramaniam, D. J. S. Birch, and T. M. Jovin. 2000. One- and two-photon excited fluorescence lifetimes and anisotropy decays of green fluorescent proteins. Biophys. J. 78:1589–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonakis, B. M., H. X. Chen, H. Haleem-Smith, and H. Metzger. 1997. The unique domain as the site on Lyn kinase for its constitutive association with the high affinity receptor for IgE. J. Biol. Chem. 272:24072–24080. [DOI] [PubMed] [Google Scholar]
- Vrzheshch, P. V., N. A. Akovbian, S. D. Varfolomeyev, and V. V. Verkhusha. 2000. Denaturation and partial renaturation of a tightly tetramerized DsRed protein under mildly acidic conditions. FEBS Lett. 487:203–208. [DOI] [PubMed] [Google Scholar]
- Wahlfors, J., S. Loimas, T. Pasanen, and T. Hakkarainen. 2001. Green fluorescent protein (GFP) fusion constructs in gene therapy research. Histochem. Cell Biol. 115:59–65. [DOI] [PubMed] [Google Scholar]
- Wall, M. A., M. Socolich, and R. Ranganathan. 2000. The structural basis for red fluorescence in the tetrameric GFP homolog DsRed. Nat. Struct. Biol. 7:1133–1138. [DOI] [PubMed] [Google Scholar]
- Widengren, J., B. Terry, and R. Rigler. 1999. Protonation kinetics of GFP and FITC investigated by FCS: aspects of the use of fluorescent indicators for measuring pH. Chem. Phys. 249:259–271. [Google Scholar]
- Xu, C., R. M. Williams, W. Zipfel, and W. W. Webb. 1996. Multiphoton excitation cross-sections of molecular fluorophores. Bioimaging. 4:198–207. [Google Scholar]
- Yanushevich, Y. G., D. B. Staroverov, A. P. Savitsky, A. F. Fradkov, N. G. Gurskaya, M. E. Bulina, K. A. Lukyanov, and S. A. Lukyanov. 2002. A strategy for the generation of non-aggregating mutants of Anthozoa fluorescent proteins. FEBS Lett. 511:11–14. [DOI] [PubMed] [Google Scholar]
- Yarbrough, D., R. M. Wachter, K. Kallio, M. V. Matz, and S. J. Remington. 2001. Refined crystal structure of DsRed, a red fluorescent protein from coral, at 2.0-angstrom resolution. Proc. Natl. Acad. Sci. USA. 98:462–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer, M. 2002. Green fluorescent protein (GFP): applications, structure, and related photophysical behavior. Chem. Rev. 102:759–781. [DOI] [PubMed] [Google Scholar]