

Abstract
We have investigated the pH dependence of the dynamics of conformational fluctuations of green fluorescent protein mutants EGFP (F64L/S65T) and GFP-S65T in small ensembles of molecules in solution by using fluorescence correlation spectroscopy (FCS). FCS utilizes time-resolved measurements of fluctuations in the molecular fluorescence emission for determination of the intrinsic dynamics and thermodynamics of all processes that affect the fluorescence. Fluorescence excitation of a bulk solution of EGFP decreases to zero at low pH (pKa = 5.8) paralleled by a decrease of the absorption at 488 nm and an increase at 400 nm. Protonation of the hydroxyl group of Tyr-66, which is part of the chromophore, induces these changes. When FCS is used the fluctuations in the protonation state of the chromophore are time resolved. The autocorrelation function of fluorescence emission shows contributions from two chemical relaxation processes as well as diffusional concentration fluctuations. The time constant of the fast, pH-dependent chemical process decreases with pH from 300 μs at pH 7 to 45 μs at pH 5, while the time-average fraction of molecules in a nonfluorescent state increases to 80% in the same range. A second, pH-independent, process with a time constant of 340 μs and an associated fraction of 13% nonfluorescent molecules is observed between pH 8 and 11, possibly representing an internal proton transfer process and associated conformational rearrangements. The FCS data provide direct measures of the dynamics and the equilibrium properties of the protonation processes. Thus FCS is a convenient, intrinsically calibrated method for pH measurements in subfemtoliter volumes with nanomolar concentrations of EGFP.
Because of the rapidly increasing number of applications of green fluorescent protein (GFP) and its mutants as noninvasive fluorescent markers in molecular biology (1, 2), considerable interest has developed in its biochemical and optical properties. Understanding its photophysics on the basis of recently solved crystal structures (3, 4) should allow the design of mutants tailored to specific needs. The chromophore of wild-type GFP is formed autocatalytically from Ser-65, Tyr-66, and Gly-67 (5) and has two major absorption maxima, at ≈390 nm and ≈480 nm. Structural analysis (6, 7) and quantum mechanical calculations (8) suggest that they represent chromophores with protonated and deprotonated tyrosyl hydroxyl groups, respectively. The hydroxyl group is part of an intricate network of hydrogen bonds that, depending on the specific local arrangement, favors either the protonated form (wild-type) or the deprotonated form [mutants GFP-S65T and GFP-F64L/S65T(≡ EGFP)], with both forms present in a fixed ratio over a certain pH range (about 8 to 11 for EGFP). Interestingly, excitation at either 390 nm or 490 nm leads to similar emission spectra, peaking around 505 nm. This photophysical pattern has been rationalized by an excited-state proton transfer from the hydroxyl group to the hydrogen bond network, leading to a deprotonated chromophore from which actual emission occurs, even when excitation is at 390 nm (9, 10). At lower pH values, however, GFP fluorescence is very sensitive to proton concentration, decreasing to zero below pH 4 (11–14). An objective of this paper is to report observations of the dynamics of the molecular fluorescence fluctuations, which reveal the underlying molecular processes.
The protonation state of individual molecules, however, is not fixed over time, but changes rapidly due to the intrinsic dynamics of the molecular system, leading to fluctuations of their photophysical properties. As the concept of protein substates suggests (15), dynamic transitions among conformational and/or chemical substates are a general phenomenon of polymers and are indispensable for protein function (16). Even in an equilibrium state, each molecule samples for a various lengths of time all accessible substates, which may differ in their properties. Bulk measurements, however, yield only average values of the ensemble, missing interesting dynamical aspects of distributions of properties. This limitation may be overcome by single-molecule detection (see, for example, refs. 17–20). A more analytical method utilizes observations of the fluorescence of small ensembles of molecules that fluctuate around a mean.
Fluorescence correlation spectroscopy (FCS) (21) was created to measure these molecular dynamics; it is sensitive to fluctuations in fluorescence intensity observed from a small open volume element (on the order of 10−15 liter) containing only a few molecules. Fluctuations may be due to molecules diffusing in and out of the volume or to chemical transitions between fluorescent and nonfluorescent states. The method of FCS extracts kinetic and thermodynamic information on these processes from the temporal autocorrelation of intensity fluctuations. The original technique (18) has become readily applicable for various dynamical systems having suitable fluorescent indicators by utilization of modern computer and detector technology (22, 23).
We applied FCS to study the dynamics of the fluctuations of the protonation state of GFP and find that at low pH protonation of the chromophore from the bulk solution occurs, whereas at high pH an internal protonation process takes over. These processes are distinct from the above-mentioned excited-state proton transfer and might provide a molecular explanation for GFP “blinking,” recently observed in different mutants of GFP on much longer time scales (24, 25). A convenient discovery is the applicability of the FCS measurements on EGFP to provide intrinsically calibrated values of pH in subfemtoliter volumes with only nanomolar EGFP concentrations. This feature appears particularly useful for pH measurement inside living cells.
MATERIALS AND METHODS
GFP-S65T and GFP-F64L/S65T (EGFP) were obtained from CLONTECH (Palo Alto, CA). The quadruple mutant F64L/Y66W/I167W/K238N (GFP-Y66W) was kindly provided by G. Palm (National Cancer Institute–Frederick Cancer and Research Development Center, Frederick, Maryland). Chemicals (highest purity) and water (HPLC-grade) were obtained from Sigma.
Absorption spectra were recorded on a single-beam HP-8451-A diode array spectrophotometer. GFP was diluted to 2 μM in CP buffer (10 mM citric acid/100 mM potassium phosphate) at various pH values, and spectra were taken within 1 min. Fluorescence spectra were taken on a PTI-QM-1 fluorescence spectrometer (PTI, South Brunswick, NJ). Samples were diluted to 10 to 20 nM in CP buffer and spectra were recorded within 1 min. Slits were set to 5 nm.
An FCS measurement apparatus was constructed. Briefly, the 488-nm line of a 10-mW Ar+ ion laser (Ion Laser Technology, Salt Lake City) is selected by a 485 ± 11 nm interference filter (Omega Optics, Brattleboro, VT), reflected by a dichroic mirror (495 nm longpass; Chroma Technology, Brattleboro, VT) and focused by an Olympus 1.15 NA/40× water-immersion objective. The beam waist at the back aperture of the objective was 3 mm. Fluorescence light is collected through the objective, passes the dichroic mirror and an interference filter (525 ± 25 nm, Chroma) and is focused by a 150-mm lens onto an optical fiber (25, 50, or 100 μm in diameter) coupled to an avalanche photodiode (SPCM-AQ-141-FC, EG & G Vaudreuil, Canada). The fiber entrance diameter represents the confocal aperture, which defines the detection focal volume. The signal of the photodiode is processed by a correlator card (ALV, Langen, Germany) that calculates the correlation curve with a time resolution of 200 ns. Data manipulation and fitting were performed with origin software (Microcal, Northampton, MA). Performance of the instrument was checked routinely with rhodamine green. The alternative filter set used for measurement of GFP-Y66W was 450 nm longpass dichroic, 450 ± 22 nm excitation, and 512 ± 15 nm emission. Similar measurements were carried out with two-photon fluorescence excitation (26) with appropriate femtosecond pulse excitation (27) focusing the fluorescence onto the 200-μm diameter active area of an avalanche photodiode (these less extensive data are not reported).
Temperature was adjusted by using a heated blower with temperature control or by flushing precooled nitrogen gas over the sample. The temperature was measured in the immersion fluid at the sample with a thermocouple. GFP solutions (1–5 nM in CP buffers) in a deep-well slide were covered with a coverslip and placed in front of the objective. For measurements in deuterium oxide (D2O) the buffers were prepared taking into account that the actual pD value is 0.41 higher than the reading of a pH meter using a standard glass electrode. GFP was diluted into deuterated buffer and concentrated again by using a Centricon spin concentrator. This operation was repeated three times, which results in better than 99% exchange.
The reduction of fluorescence intensity at very low pH was not fully reversible, depending on the time spent at low pH. Correlation curves obtained at different times after lowering the pH were indistinguishable, as were curves obtained before and after a pH excursion from pH 8.0 to pH 5.0 and back to pH 8.0. This observation shows that our results are not compromised by irreversibility.
RESULTS
pH Dependence of Fluorescence and Absorption.
Fluorescence emission of GFP is very pH sensitive, dropping to zero at very low and high pH values (11, 12, 28, 29). The emission intensity of EGFP as a function of pH is shown in the Inset of Fig. 1. The emission maximum lies at 509 nm with a pronounced shoulder at about 540 nm. Shapes and maxima of the emission spectra are essentially pH independent. A fit of the emission intensity F versus pH according to the equation F = x + y/(1 + 10z), where z is (pKa − pH) and x and y are related to the offset and dynamic range of the data, yields a pKa of 5.8 ± 0.1, indicating a single protonation step to be primarily responsible for the decreased fluorescence. From the equation ΔrG° = −RTlnK the standard free reaction energy may be calculated to be about 33 ± 1 kJ/mol at room temperature. At pH > 12 there is a drastic loss of fluorescence. Interestingly, fluorescence of the mutant GFP-Y66W first increases by a factor of 2 going from pH 8 to 5.5 but then also decays to zero at even lower pH (data not shown).
Figure 1.

Fluorescence excitation spectra of EGFP in 100 mM potassium phosphate/10 mM citrate at various pH values (down from the top): 9.0, 8.0, 11.0, 7.0, 6.5, 6.0, 5.5, and 5.0. (Inset) Fluorescence emission intensity at 510 nm (■), fluorescence excitation intensity at 490 nm (•), and absorption at 488 nm (♦) versus pH. The solid line is fit to the emission intensity from pH 4.0 to 9.0 yielding a pKa of 5.8 ± 0.1.
Excitation spectra of EGFP peak at 488 nm with a shoulder at about 465 nm and an additional ≈13% contribution at 400 nm. The pH dependence of the spectra is shown in Fig. 1. The shapes of the spectra at pH 9.0 and 5.0 are very similar except for a slightly higher excitation in the range of 400 nm at pH 9.0. Intensity of excitation versus pH (Fig. 1 Inset) is in good agreement with that of emission with a fitted pKa of 5.9 ± 0.1.
Like emission and excitation, absorption of EGFP is strongly pH dependent (Fig. 2). At pH 9.0 absorption and excitation spectra are basically congruent. Lowering the pH leads to an absorption decrease at 488 nm and a concomitant increase at 398 nm with a clear isosbestic point (425 nm), as one would expect for a single protonation reaction. Absorption at 488 nm versus pH is included in the Inset of Fig. 1 and shows the same behavior as fluorescence emission and excitation. This behavior implies that the reduced emission at low pH is due to a decreased molar absorption coefficient rather than a lower quantum efficiency. Given a pKa of 5.8 for the protonation equilibrium, one can calculate the molar absorption coefficient at 398 nm of the low-pH species to be about 38% of the high-pH species at 488 nm. Since the excitation spectra at low pH do not show an increased contribution around 400 nm, the species produced at low pH does not contribute to the fluorescence around 500 nm.
Figure 2.

Absorption spectra of EGFP in 100 mM potassium phosphate/10 mM citrate at various pH values (down from the top at 490 nm): 10.0, 9.0. 8.0. 7.0. 6.5, 6.0, 5.5, and 5.0.
FCS.
In Fig. 3 autocorrelations (blue curves) of EGFP at pH values from 5.0 to 11.0 are presented. The autocorrelation function G(t) is defined as G(t) = 〈δF(t)δF(0)〉/〈F(t)〉2, where F(t) is the fluorescence obtained from the volume at time t, angled brackets denote time averages, and δF(t) = F(t) − 〈F(t)〉. Above pH 8.0 the curves are basically indistinguishable. With increasingly acidic pH a new fast component grows with increasing amplitude and decreasing time constant; this might represent the expected protonation fluctuation of the chromophore. The correlation shoulder in the millisecond time range is known to be due to diffusional fluctuations of the number of EGFP molecules in the optically established focal volume comprising the grand canonical ensemble. The correlation function describing the fluctuations around equilibrium of a simple AH ⇌ A− + H+ reaction coupled with diffusion is given by
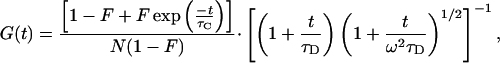 |
1 |
where N is the average number of molecules in the probe volume, F is the average fraction of molecules in the nonfluorescent state (AH), ω describes the length-to-diameter ratio of the three-dimensional gaussian volume element, and τC and τD are the chemical and diffusional relaxation times, respectively.
Figure 3.
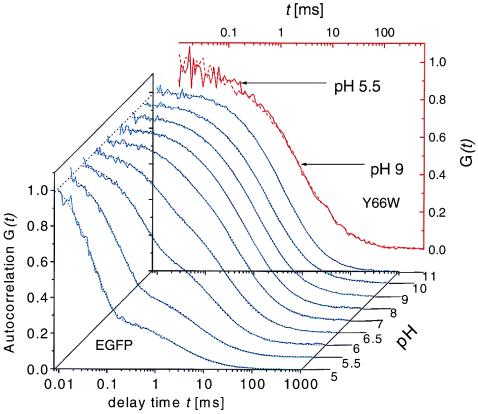
Autocorrelations G(t) for EGFP (blue curves) at different pH values (normalized to 1 at 10 μs). Fits of Eq. 2 to the data are shown as dotted lines. The decay of G(t) at high pH is dominated by diffusional relaxation, whereas upon decreasing pH, chemical relaxation at shorter time is growing in amplitude and speed. (Inset) Autocorrelations of GFP-Y66W (red curves) at pH 9.0 (solid line) and 5.5 (broken line) showing absence of pH-dependent fast fluorescence flicker in this mutant, which lacks a protonatable hydroxyl on the chromophore.
Fitting Eq. 1 to the data yields a decreasing τC (from 450 ± 65 μs to 50 ± 4 μs) and an increasing fraction F (up to 80% at pH 5.0) with decreasing pH. However, from pH 8.0 to 11.0 there is a reproducible, pH-independent, dark fraction of 13% ± 2% with an associated time constant of ≈450 μs. Finding this constant fraction over a wide pH range indicates that AH ⇌ A− + H+ does not describe the system completely. To account for the high-pH fraction we invoke a pH-independent internal protonation process—i.e., the proton fluctuates between the chromophore hydroxyl group and an internal proton binding site B−, as has been suggested on the basis of structural studies (6, 7). At low pH a second proton from the bulk solution can protonate the chromophore as hypothesized above, inducing a nonfluorescent state: 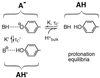
Species A− is the only one contributing to fluorescence. Assuming for simplicity that protonation from the solution is substantially faster than the internal process (τC < τ′C) and neglecting reactions between species AH and AH′ (molar ratio of A− to AH′ ≈ 8:1), the autocorrelation function is approximated by
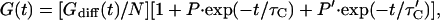 |
2 |
where Gdiff(t) = [(1 + t/τD)(1 + t/ω2τD)0.5]−1 describes the diffusional part, the pre-exponential factors are P = [K[H+]{1 + K′(1 + K[H+])}]/[K′(1 + K[H+])] and P′ = 1/[K′(1 + K[H+])], and [H+] denotes the hydrogen ion concentration. The equilibrium constants are K = [AH]/[A−][H+] and K′ = [A−]/[AH′], and the time constants τC and τ′C are associated with external and internal protonation, respectively. Between pH 9 and 11 Eq. 2 was fitted to the data, using the approximation P ≈ 0; P′ ≈ 1/K′. This yields an average diffusion time τD of 4.2 ± 0.3 ms, a K′ of 7.55 ± 0.60 corresponding to a 12% ± 1% fraction of nonfluorescent molecules, and τC′ = 341 ± 48 μs, which is somewhat faster than the results obtained with the single-exponential fit (450 μs). Data taken at 1/3 the usual excitation intensity show a 30% increase in both K′ and τ′C, suggesting the possible involvement of photoinduced processes at high pH.
The results of fitting the low-pH data with K′ and τ′C fixed at the average values (above) are shown in Fig. 4. As expected, the apparent rate constant kapp (= 1/τC) increases with decreasing pH. However, the equilibrium constant K appears to increase by a factor of 2.8 from pH 5.0 to 7.0, which may be due to the approximation τC ≪ τ′C ≪ τD assumed in the autocorrelation function 2. The rate constants for protonation and deprotonation at low pH can be obtained from a fit of kapp = 1/τC = kprot([H+] + [A−]) + kdeprot ≈ kprot[H+] + kdeprot because the concentration of deprotonated protein [A−] (in the nanomolar range) is negligible compared with [H+] below pH 7.0 (Fig. 4). This fit yields kprot = (1.53 ± 0.06) × 109 M−1⋅s−1 and kdeprot = (4.48 ± 0.27) × 103 s−1, from which a pKa [= log(kprot/kdeprot)] of 5.5 ± 0.3 can be calculated, in close agreement within the uncertainties to the value obtained from bulk measurements (see above). The mutant S65T behaves very similarly, although the chemical relaxation at low pH is about twice as fast as for EGFP (Fig. 4). The same analysis yields kprot = (3.45 ± 0.32) × 109 M−1⋅s−1, kdeprot = (9.05 ± 1.52) × 103 s−1, and a pKa of 5.6 ± 0.4 for GFP-S65T.
Figure 4.

Results for the apparent rate constants of chemical relaxation as obtained from fits of the correlation curves shown in Fig. 3, using Eq. 2 with the high-pH approximation for pH 8 to 11. For pH 5.0 to 7.0 the values for K′ and τ′C were fixed at the average values obtained from pH 8.0 to 11.0. Open symbols, GFP-S65T; closed symbols, EGFP. Solid lines, fits of the equation kapp = kprot([H+] + [A−]) + kdeprot to the data points from pH 5.0 to 7.0.
Control experiments were performed to exclude other possible origins of the fast decay time of the correlation curve at low pH—e.g., a light-induced process such as triplet formation or diffusion of a small contaminating molecule. In case of triplet formation the associated time constant is expected to depend on light intensity. Measurements taken at 8- and 80-μW laser power (Fig. 5B) demonstrate that the time constant for the fast (protonation) process does not change within experimental error (208 ± 8 μs at 8 μW and 219 ± 5 μs at 80 μW), excluding a photoreaction for the low-pH process. The curve at higher power, however, shows an additional contribution at even shorter times (≈30 μs), which is entirely consistent with triplet formation but merits further investigation.
Figure 5.

Influence of light intensity and detection volume on time constants. (A) At pH 5.5 fiber diameters of 25, 50, and 100 μm (left to right) were used to vary spot size. Fits are shown as dotted lines. (B) Autocorrelation curves at 8 and 80 μW.
The possibility of a fast-diffusing contaminant can be checked by changing the focal volume size by using different fiber diameters of 25, 50, and 100 μm (Fig. 5A). Whereas the diffusion time τD increases by a factor of 3 as expected, again the chemical relaxation time τC is unchanged within experimental error (124 ± 2 μs, 121 ± 4 μs, and 130 ± 3 μs). This result excludes a diffusional relaxation or photobleaching for the fast process and confirms the assignment of the slow decay time (4.2 ms) to EGFP diffusion.
Most convincing evidence for a protonation fluctuation as the basis for the chemical kinetics is gained from measurements of the mutant Y66W, which does not posses a protonatable hydroxyl group on the chromophore. In Fig. 3 the red curves show autocorrelations obtained at pH 9.0 and 5.5. Evidently, there is no additional kinetic process at low pH as in the case of EGFP and GFP-S65T.
From the above results we conclude that the pH-dependent kinetic component at low pH is based on a protonation/deprotonation reaction of the chromophore. Since one might expect an isotope effect on the kinetics of proton movement we investigated EGFP in D2O at different pD values. Although the scatter is much higher in the D2O data, there seems to be an increase by a factor of 1.25 in τ′C at high pH. This result might suggest that proton transfer is the rate-limiting step; however, D2O also has a 1.23 times higher viscosity than does H2O, which might influence kinetics.
To distinguish between viscosity and isotope effect we increased viscosity of the solution by adding glycerol. Diffusion time τD (pH 5.5 and pH 10) and the chemical relaxation kinetics τC at pH 5.5 as well as τ′C at pH 10 increase linearly with viscosity by factors of approximately 6.6 ± 0.3, 0.11 ± 0.01, and 0.61 ± 0.14, respectively (data not shown). Correspondingly, diffusion in D2O is slowed down by an expected factor of ≈1.25 due to the 1.23 times higher viscosity of D2O compared with H2O. This finding suggests that the increase in chemical relaxation times in D2O is a viscosity rather than an isotope effect and that proton transfer is not limiting. The high-pH process was ascribed to an internal protonation fluctuation. If such a proton movement is coupled to protein conformational changes it would be expected that its kinetics is influenced by solvent viscosity.
The kinetics of protonation reactions might be expected to be influenced by buffer concentration and/or ionic strength of the solution. In Fig. 6 we show that the ionic strength does not change the kinetics of the low-pH protonation process at constant buffer concentration. Inversely, a strong decrease of τC is seen with increased buffer concentration at constant ionic strength. This result seems to implicate buffer molecules in proton transfer reactions into the EGFP protein.
Figure 6.

Effect of ionic strength on the chemical relaxation time τC at pH 5.5 with constant phosphate buffer concentration of 10 mM, and effect of phosphate buffer concentrations with the ionic strength kept constant by adding appropriate amounts of KCl.
To obtain more information on the thermodynamics of the protonation equilibrium we performed a series of experiments at pH 6.5 in the temperature range from 15°C to 50°C. The temperature dependence of the equilibrium constant K allows the calculation of the standard reaction enthalpy ΔrH° according to the van’t Hoff equation dlnK/d(1/T) = −ΔrH°/R. From the slope of a linear fit to the graph lnK versus 1/T (Fig. 7) we obtain ΔrH° = −11.5 ± 0.7 kJ/mol, and using ΔrG° = ΔrH° − TΔrS° we obtain a standard reaction entropy at room temperature of ΔrS° = −150 ± 7 J/mol⋅K.
Figure 7.

Temperature dependence of equilibrium constant K at pH 6.5. Dotted line, linear fit to the data points.
DISCUSSION
The absorption spectra of GFPs typically show two maxima around 400 nm and 488 nm which have been assigned to chromophores with the hydroxyl group of Tyr-66 either protonated (AH′) or deprotonated (A−) (6–8). Absorption cross section at 488 nm as well as fluorescence emission intensity drops with decreasing pH, yielding a pKa of about 5.8 ± 0.1. This result shows that the transition is due to protonation of a single group and that the decreased emission is not due to fluorescence quenching at low pH but instead to decreased absorption at 488 nm, which agrees very well with similar results (12, 14, 29). The absorption decrease at 488 nm is paralleled by an increase at 400 nm, strongly suggesting that the species formed at low pH (AH) is GFP with a protonated chromophore. In wild-type GFP excitation at 400 nm leads to an excited-state proton transfer from the hydroxyl group to the hydrogen-bonded network, allowing fluorescence emission around 505 nm (9, 10). Interestingly, the excitation spectra at low pH (Fig. 1) do not show an increase in excitation cross section at 400 nm, demonstrating that species AH is nonfluorescent. This conclusion is consistent with proton acceptor site (B) protonation from the solvent at low pH, thereby blocking the excited-state proton transfer. Glu-222 has been suggested as the proton acceptor in the excited (AH′* → A−*) as well as ground state (AH′ → A−) on the basis of crystal structures (6, 7). In that case in the mutant E222G the protonated chromophore should be stabilized; however, the spectrum is very similar to that of S65T with a long-wavelength peak (30). Another possible proton acceptor site is His-148, which is within hydrogen bonding distance, or the proton might be delocalized in the hydrogen-bonded network, which also involves water molecules.
Recognizing that protein molecules continuously fluctuate between all conformational and/or chemical substates energetically accessible to them at a given temperature, we aim to utilize the fluorescence fluctuations to recognize the accessible states and the dynamics of the transitions by using FCS. The chromophore of EGFP is in an internal protonation equilibrium between pH 8 and 11 (6, 7) and is protonated from the solvent at low pH (12, 14, 28). Since the 488-nm laser excites only the deprotonated form of the GFP chromophore, one should expect the fluorescence from each individual molecule to fluctuate as it continuously transforms between states A−, AH, and AH′ in an equilibrium process—i.e., the protein “flickers.” Indeed, using FCS, we could identify two relaxation times (besides diffusional relaxation), one being pH dependent (at pH < 7.0) the other pH independent (pH 8–11). We have presented evidence that the pH dependence of the time constant of the low-pH process, as well as the fraction of molecules in the nonfluorescent state, agrees well with what is expected for a protonation of the chromophore from the solution. Strong support for this interpretation comes from the measurements of the mutant Y66W (Fig. 3), which does not have a protonatable hydroxyl group in its chromophore and correspondingly does not show the pH dependence of the correlation curves. For the mutant S65T, however, we measured almost doubled rate constants, indicating a strong influence of even small structural perturbations on the kinetics.
The high-pH process, on the other hand, might represent the internal protonation conformational equilibrium (6, 7), which is not directly influenced by bulk pH. It also exhibits a slight dependence on light intensity (30% increases of τ′C and K′ at 1/3 the light intensity), which could indicate some photo-assisted transition from state A− to AH′. The light dependence of the reverse process has long been recognized (31) but has also been suggested for the forward process in a recent study of “GFP-blinking” (24). Using a GFP-S65T/S72A/T203F triple mutant, these authors find fluorescence from individual GFP molecules to disappear and reappear slowly on the seconds time scale. Considering the strong influence of mutations (e.g., EGFP vs. S65T) and experimental conditions (buffer solution vs. gel matrix) on the kinetics, it is possible that the same phenomenon was observed in their study, but further investigation is needed to delineate the exact nature of the high-pH process.
It is generally accepted that the chromophore in GFP is very well shielded by the rigid β-can, since it is insensitive to fluorescence quenchers. How than can the chromophore “feel” the bulk proton concentration? An inspection of the structure reveals that the Tyr-66 hydroxyl group points toward the surface of the β-can (almost perpendicular to the cylinder axis) and is in direct hydrogen bonding with one water molecule (w316). A second ordered water molecule (w327) on the surface of the protein is located within 4.16 Å of w316, which is too far for a permanent hydrogen bond; however, it seems feasible that slight dynamical rearrangements on the order of 1 Å would allow communication of the Tyr-66 hydroxyl group with the bulk proton concentration via these two water molecules. This would require thermal fluctuations of the β-sheet structure and indeed, when NMR methods were used, locally collective conformational fluctuations in exactly the same time range (32–175 μs) were found for the all β-sheet fibronectin type III domain and were interpreted as a breathing motion of the β-sheet structure (32). In an approach using random mutagenesis to obtain pH-sensitive GFP mutants, Miesenböck et al. (29) identified amino acids 147, 149, 164, 165–168, and 220, all of which are located in three adjacent β-strands just above the Tyr-66 hydroxyl group. It is tempting to suggest that these mutations influence the conformational fluctuations and thereby the dependence of GFP fluorescence on bulk pH. However, it should be noted that the pH dependence of their mutant is inverse to what is expected for a simple protonation of the chromophore. Also, the internal protonation change of the chromophore (AH′ ⇌ A−) is coupled to structural rearrangements (6, 7). These structural fluctuations might be the key to understand why bulk viscosity influences the observed kinetics at low as well as at high pH. It might be interesting to measure the viscosity dependence of the NMR β-sheet domain signal and to compare it with our viscosity-dependent dynamics.
Reliable values for intracellular parameters such as viscosity, pH, or buffer capacity are difficult to obtain in vivo. We would like to point out that (FCS) measurements of intracellular GFP allow determination of these numbers with subcellular resolution as GFP may be directed to various compartments within the cell by using appropriate leader sequences. The fraction F of molecules in the nonfluorescent state is a robust number despite the environmental effects on the rate constants and is determined intrinsically by the FCS technique. It is an absolute indicator for pH. For this purpose calibration measurements with a simple analysis using Eq. 1 would be sufficient. This method does not suffer from the requirement to determine absolute or ratiometric intensities as used in other approaches (14, 28, 29, 33); it is noninvasive [it does not need subsequent calibration as in the approach used by Kneen et al. (14)]; and the fluorophore may be present at very low concentrations (nanomolar), minimizing the risk of interference with cellular processes (P.S. and W.W.W., unpublished work). Note that the fraction F and thus the pH is determined by the statistical thermodynamics of the protonation and is independent of the influence of viscosity and buffer concentrations that affect the kinetics. Thus its pH indication can be expected to be entirely independent of environment.
The diffusion time of GFP is a function of the viscosity of the medium and is easily determined from the slow factor of the autocorrelation function, allowing determination of medium viscosity in cell compartments, including membrane viscosity, by using GFP with a lipid anchor (P.S. and W.W.W., unpublished work). At known pH and viscosity, the relaxation rate depends on the buffer concentration, so that careful calibration should even allow an estimate for the intracellular buffer capacity. Unexpectedly, the kinetics was not affected by ionic strength at a given buffer concentration. Although the GFP surface has a many positively and negatively charged residues, water molecule w327, suggested to be the “entry point” for the proton, lies within a more hydrophobic patch, which might result in the independence of ionic strength.
Acknowledgments
We thank D. W. Piston and M. Nichols for help and advice and G. Palm for kindly providing the mutant GFP-Y66W. This research was carried out in the Developmental Resource for Biophysics Imaging and Opto-electronics with funding provided by the National Science Foundation and the National Institutes of Health. U.H. was supported by an Otto Hahn Grant from the Max Planck Society, and P.S. was supported by the Alexander-von-Humboldt Fellowship.
ABBREVIATIONS
- GFP
green fluorescent protein
- EGFP
GFP-F64L/S65T
- FCS
fluorescence correlation spectroscopy
References
- 1.Misteli T, Spector D L. Nat Biotechnol. 1997;15:961–963. doi: 10.1038/nbt1097-961. [DOI] [PubMed] [Google Scholar]
- 2.Welsh S, Kay S A. Curr Opin Biotechnol. 1997;8:617–622. doi: 10.1016/s0958-1669(97)80038-9. [DOI] [PubMed] [Google Scholar]
- 3.Ormö M, Cubitt A B, Kallio K, Gross L A, Tsien R Y, Remington S J. Science. 1996;273:1392–1395. doi: 10.1126/science.273.5280.1392. [DOI] [PubMed] [Google Scholar]
- 4.Yang F, Moss L G, Phillips G N., Jr Nat Biotechnol. 1997;14:1246–1251. doi: 10.1038/nbt1096-1246. [DOI] [PubMed] [Google Scholar]
- 5.Heim R, Prasher D C, Tsien R Y. Proc Natl Acad Sci USA. 1994;91:12501–12504. doi: 10.1073/pnas.91.26.12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brejc J, Sixma T, Kitts P A, Kain S R, Tsien R Y, Ormö M, Remington J S. Proc Natl Acad Sci USA. 1997;94:2306–2311. doi: 10.1073/pnas.94.6.2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palm G, Zdanov A, Gaitanaris G A, Stauber R, Pavlakis G N, Wlodawer A. Nat Struct Biol. 1997;4:361–365. doi: 10.1038/nsb0597-361. [DOI] [PubMed] [Google Scholar]
- 8.Voityuk A A, Michel-Beyerle M E, Rösch N. Chem Phys. 1997;272:162–167. [Google Scholar]
- 9.Chattoraj M, Kong B A, Bublitz G U, Boxer S G. Proc Natl Acad Sci USA. 1996;93:8362–8367. doi: 10.1073/pnas.93.16.8362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lossau H, Kummer A, Heinecke R, Pöllinger-Dammer F, Kompa C, Bieser G, Jonsson T, Silva C M, Yang M M, Youvan D C, Michel-Beyerle M E. Chem Phys. 1996;213:1–16. [Google Scholar]
- 11.Ward W W, Bokman S H. Biochemistry. 1982;21:4535–4540. doi: 10.1021/bi00262a003. [DOI] [PubMed] [Google Scholar]
- 12.Patterson G H, Knobel S M, Sharif W S, Kain S R, Piston D W. Biophys J. 1997;73:2782–2790. doi: 10.1016/S0006-3495(97)78307-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wachter M R, King B A, Heim R, Kallio K, Tsien R Y, Boxer S G, Remington S J. Biochemistry. 1997;36:9759–9765. doi: 10.1021/bi970563w. [DOI] [PubMed] [Google Scholar]
- 14.Kneen M, Farinas J, Li Y, Verkam A S. Biophys J. 1998;74:1591–1599. doi: 10.1016/S0006-3495(98)77870-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frauenfelder H, Parak F, Young R D. Annu Rev Biophys Biophys Chem. 1988;17:451–480. doi: 10.1146/annurev.bb.17.060188.002315. [DOI] [PubMed] [Google Scholar]
- 16.Varley G P, Pain R H. J Mol Biol. 1991;220:531–538. doi: 10.1016/0022-2836(91)90028-5. [DOI] [PubMed] [Google Scholar]
- 17.(1996) Acc. Chem. Res.29 (12).
- 18.Moerner W E. Science. 1997;277:1059–1060. [Google Scholar]
- 19.Edman L, Mets Ü, Rigler R. Proc Natl Acad Sci USA. 1996;93:6710–6715. doi: 10.1073/pnas.93.13.6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eggeling C, Fries J R, Brand L, Günther R, Seidel C A M. Proc Natl Acad Sci USA. 1998;95:1556–1561. doi: 10.1073/pnas.95.4.1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Magde D, Elson E, Webb W W. Phys Rev Lett. 1972;29:705–708. [Google Scholar]
- 22.Eigen M, Rigler R. Proc Natl Acad Sci USA. 1994;91:5470–5757. doi: 10.1073/pnas.91.13.5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maiti S, Haupts U, Webb W W. Proc Natl Acad Sci USA. 1997;94:11753–11757. doi: 10.1073/pnas.94.22.11753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dickson R M, Cubitt A B, Tsien R Y, Moerner W E. Nature (London) 1997;388:355–358. doi: 10.1038/41048. [DOI] [PubMed] [Google Scholar]
- 25.Pierce D W, Hom-Booher N, Vale R D. Nature (London) 1997;388:338. doi: 10.1038/41009. [DOI] [PubMed] [Google Scholar]
- 26.Denk W, Strickler J H, Webb W W. Science. 1990;248:73–76. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- 27.Xu C, Williams R M, Zipfel W, Webb W W. Bioimaging. 1996;3:198–207. [Google Scholar]
- 28.Llopis J, McCaffery M J, Miyawaki A, Farquhar M G, Tsien R Y. Proc Natl Acad Sci USA. 1998;95:6803–6808. doi: 10.1073/pnas.95.12.6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miesenböck G, DeAngelis D A, Rothman J E. Nature (London) 1998;394:192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- 30.Ehrig T, O’Kane D J, Prendergrast F G. FEBS Lett. 1995;367:163–166. doi: 10.1016/0014-5793(95)00557-p. [DOI] [PubMed] [Google Scholar]
- 31.Cubitt A B, Heim R, Adams S R, Boyd A E, Gross L A, Tsien R Y. Trends Biochem Sci. 1995;20:448–445. doi: 10.1016/s0968-0004(00)89099-4. [DOI] [PubMed] [Google Scholar]
- 32.Akke M, Liu J, Cavanagh J, Erickson H P, Palmer A G., III Nat Struct Biol. 1998;5:55–59. doi: 10.1038/nsb0198-55. [DOI] [PubMed] [Google Scholar]
- 33.Brooks Robey R, Riuz O, Santos A V P, Ma J, Kear F, Wang L-W, Li C-J, Bernardo A A, Arruda J A L. Biochemistry. 1998;37:9894–9901. doi: 10.1021/bi980857x. [DOI] [PubMed] [Google Scholar]