

Abstract
The conformation of mononucleosome complexes reconstituted with recombinant core histones on a 614-basepair-long DNA fragment containing the Xenopus borealis 5S rRNA nucleosome positioning sequence was studied by scanning/atomic force microscopy in the absence or presence of linker histone H1. Imaging without prior fixation was conducted with air-dried samples and with mononucleosomes that were injected directly into the scanning force microscopy fluid cell and visualized in buffer. From a quantitative analysis of ∼1,700 complexes, the following results were obtained: i), In the absence of H1, a preferred location of the nucleosome at the X. borealis 5S rRNA sequence in the center of the DNA was detected. From the distribution of nucleosome positions, an energy difference of binding to the 5S rRNA sequence of ΔΔG ≈ 3 kcal mol−1 as compared to a random sequence was estimated. Upon addition of H1, a significantly reduced preference of nucleosome binding to this sequence was observed. ii), The measured entry-exit angles of the DNA at the nucleosome in the absence of H1 showed two maxima at 81 ± 29° and 136 ± 18° (air-dried samples), and 78 ± 25° and 137 ± 25° (samples imaged in buffer solution). In the presence of H1, the species with the smaller entry-exit angle was stabilized, yielding average values of 88 ± 34° for complexes in air and 85 ± 10° in buffer solution. iii), The apparent contour length of the nucleosome complexes was shortened by 34 ± 13 nm as compared to the free DNA due to wrapping of the DNA around the histone octamer complex. Considering an 11 nm diameter of the nucleosome core complex, this corresponds to a total of 145 ± 34 basepairs that are wound around the nucleosome.
INTRODUCTION
In eukaryotes, the DNA is packaged by histone proteins into a chain of nucleosomes, in which 146 or 147 basepairs of DNA are wrapped in 1.67 turns of a left-handed superhelix around a histone octamer complex (Davey et al., 2002; Hansen, 2002; Luger et al., 1997a; Ramakrishnan, 1997b; Richmond and Widom, 2000; van Holde, 1989). This protein core consists of two copies each of the four histones, H2A, H2B, H3, and H4. The nucleosome is the basic building block of the chromatin fiber and changes of its conformation are likely to modulate the organization of the fiber. The structure of the free histone octamer and that of the nucleosome complex has been determined by high resolution x-ray diffraction (Arents et al., 1991; Davey et al., 2002; Harp et al., 2000; Luger et al., 1997a). The nucleosome has a cylindrical shape with a diameter of ∼11 nm and a height of 5.5 nm.
A fifth histone, the so-called linker histone H1 or H5, binds to the nucleosome core complex (Graziano et al., 1994; Ramakrishnan, 1997a; Zhou et al., 1998; Zlatanova and van Holde, 1996). Although its exact binding site and orientation are still unknown, it seems to affect the trajectory of the DNA at the nucleosome. It has been proposed that H1 binding constrains the entry-exit angle of the DNA at the nucleosome and thus contributes to the formation of higher order chromatin structure (Bednar et al., 1998; Furrer et al., 1995; Hamiche et al., 1996; Leuba et al., 1998a,b; Ramakrishnan, 1997a; Tóth et al., 2001; Travers, 1999; Zlatanova et al., 1998). The linker histones can modulate gene transcription (Zlatanova et al., 2000). For example, it has been shown that histone H1 can selectively repress the transcription of oocytic 5S rDNA in Xenopus borealis (Buttinelli et al., 1999). Linker histones can also act as general transcription regulators as they play a role in inhibition of histone acetylation (Herrera et al., 2000).
We have applied scanning force microscopy (SFM, also termed atomic force microscopy) to determine the structural parameters of unfixed mononucleosomes reconstituted with recombinant core histone proteins DNA and linker histone H1. The recombinant histones allow the analysis of highly purified and well-defined nucleosomes in the absence of any posttranslational modifications or contaminations with other chromosomal proteins (Luger et al., 1997a,b). SFM can be used for imaging DNA and protein-DNA complexes without further fixing or staining of the sample, and imaging can be conducted in physiological buffer solutions. Accordingly, the technique is especially suited to study the native conformation of nucleosome complexes with minimal disturbance of the sample. Over the past years, SFM has been applied successfully in various studies of chromatin samples including oligonucleosomes, chromatin fibers, and whole metaphase chromosomes (Allen et al., 1993; Bash et al., 2003, 2001; Fritzsche and Henderson, 1996; Fritzsche et al., 1994; Karymov et al., 2001; Leuba et al., 1998a,b, 1994; Martin et al., 1995; Wang et al., 2002; Yodh et al., 1999, 2002; Zlatanova et al., 1998). Results from the SFM chromatin analysis as well as technical aspects of these studies have been described in a number of reviews (Bustamante et al., 1997; Fritzsche et al., 1997, 1995; Leuba and Bustamante, 1999).
As proposed previously, essential determinants of the chromatin fiber structure are the entry-exit angle of the nucleosomal DNA and the rotational orientation and distance of neighboring nucleosomes, which depend on the linker DNA length (Leuba et al., 1994; Woodcock et al., 1993; Yang et al., 1994). In a series of articles, these parameters have been defined by SFM imaging and electron cryomicroscopy of chromatin fibers isolated from chicken erythrocytes (Bednar et al., 1998; Horowitz et al., 1994; Leuba et al., 1998a,b, 1994; Woodcock et al., 1993; Yang et al., 1994; Zlatanova et al., 1998). Here, we present an SFM study of unfixed recombinant nucleosome core particles with special focus on the effect of linker histone H1 binding. The samples were imaged either after air-drying or in a buffer solution that preserves the native hydration state, and a quantitative analysis of different conformational states was conducted. We find that binding of H1 stabilizes a nucleosome conformation with a smaller entry-exit angle and reduces the binding preference to the high affinity 5S rRNA sequence from X. borealis.
MATERIALS AND METHODS
Expression and purification of recombinant histone proteins
Overexpression and purification of recombinant histone proteins as well as reconstitution of histone octamers and mononucleosomes were essentially conducted as described (Luger et al., 1997b, 1999) using a slightly different chromatography procedure scheme on a fast protein liquid chromatography system (Amersham Biosciences, Freiburg, Germany). The supernatant from the resolubilized histone protein inclusion bodies was filtered and loaded on a 5 × 60-cm Sephacryl S-200 high resolution gel filtration column equilibrated with 1.5 column volumes of SAU-1000 buffer (7 M deionized urea, 20 mM sodium acetate, pH 5.2, 1 mM EDTA, 1 M KCl, 5 mM 2-mercaptoethanol). The column was run at room temperature at a flow rate of 3 ml/min. Elution of proteins was monitored by the absorbance at 280 nm. Fractions were analyzed on an 18% SDS-acrylamide gel and by ultraviolet spectroscopy. Pooled fractions were dialyzed against water containing 5 mM 2-mercaptoethanol overnight at 4°C using 6000–8000 MWCO dialysis bags (Spectrum Laboratories, Rancho Dominguez, CA). Proteins were lyophilized and stored at −20°C. For further purification, the probes were redissolved in SAU-50 buffer (7 M deionized urea, 20 mM sodium acetate, pH 5.2, 1 mM EDTA, 50 mM KCl, 5 mM 2-mercaptoethanol) and the protein solution was applied to a Mono S HR 5/5 cation exchange column equilibrated with the same buffer. Histones were eluted by linearly increasing the KCl concentration in 20 column volumes to 0.6 M.
Reconstitution of histone octamers was done as described previously (Luger et al., 1997b, 1999). The final purification of the octamers was conducted with a Superdex 200 HR 10/10 column equilibrated with refolding buffer (2 M KCl, 10 mM Tris-HCl, pH 7.5, 0.1 mM EDTA, 5 mM 2-mercaptoethanol). The flow rate was 0.6 ml/min. The stoichiometry of the octamer complex was checked on an 18% SDS-acrylamide gel, and fractions with equimolar amounts of the four histones were pooled and stored at −20°C in refolding buffer supplemented with glycerol to a final concentration of 50%. Octamer concentrations were determined using an extinction coefficient at 276 nm ɛ276 = 38 400 M−1·cm−1 (Luger et al., 1999).
Preparation of linker histone H1
Full-length histone protein H1 from calf thymus was obtained from Roche Diagnostics (Mannheim, Germany) and further purified. 5 mg of lyophilized protein were dissolved in 1 ml 1× TE buffer overnight. The protein was loaded on a 1 ml Sepharose SP-Fast Flow column and washed with 1× TE buffer supplemented with 0.3 M NaCl and 0.5 mM PMSF. Then the protein was eluted in a step gradient of 1 ml 1 × TE, 0.5 M NaCl, 0.5 mM PMSF followed by 1.4 ml of 1 × TE, 2 M NaCl, 0.5 mM PMSF. The last fractions of ∼0.8 ml that showed the highest absorption at 280 nm were pooled. The protein was desalted on a NAP-5 column (Amersham Biosciences, Freiburg, Germany) previously equilibrated with H2O. The H1-containing fraction was concentrated with Vivaspin 500 centrifugal concentrators (10000 MWCO, Sartorius, Göttingen, Germany) to a volume of 0.5 ml. This fraction was loaded on a second NAP-5 column equilibrated with 1× TE and eluted with 1 ml 1× TE. The DNA-binding activity of the purified H1 preparation was determined by analytical ultracentrifugation and fluorescence anisotropy measurements of H1 binding to a 20 bp DNA duplex under conditions of stoichiometric binding.
Reconstitution of nucleosomes
For reconstitution of nucleosomes, 100 nM of DNA fragment and 100–130 nM histone octamer were incubated for 30 min at room temperature in a buffer containing 10 mM Tris-HCl pH 7.5 and 2 M KCl. Two different DNA fragments were used: a 146-bp-long palindromic sequence derived from human α-satellite DNA studied previously (Luger et al., 1997a) was assembled by ligating the HPLC-purified synthetic oligonucleotides ATC AAT ATC CAC CTG CAG ATT CTA CCA AAA GTG TAT TTG GAA ACT GCT CCA TCA AAA GGC ATG TTC AGC T (N1), ATT CAG CTG AAC ATG CCT TTT GAT GGA GCA GTT TCC AAA TAC ACT TTT GGT AGA ATC TGC AGG TGG ATA TTG AT (N2), GAA TTC AGC TGA ACA TGC CTT TTG ATG GAG CAG TTT CCA AAT ACA CTT TTG GTA GAA TCT GCA GGT GGA TAT TGA T (N3) and ATC AAT ATC CAC CTG CAG ATT CTA CCA AAA GTG TAT TTG GAA ACT GCT CCA TCA AAA GGC ATG TTC AGC TGA (N4) (PE-Applied Biosystems, Weiterstadt, Germany) under standard conditions. The ligation product of the N1·N2 and the N3·N4 duplex was purified by HPLC chromatography using an ion-exchange Gen-Pak Fax column (Millipore, Milford, MA). The gradient was 0–0.5 NaCl (10 min) followed by 0.5–1.0 M NaCl (20 min) at 0.75 ml/min flow rate. The buffer contained 20 mM Tris-HCl, pH 7.6 and 0.1 mM EDTA. For the SFM imaging, mononucleosomes were reconstituted with a 614-bp-long DNA fragment, which was amplified by polymerase chain reaction from the plasmid pXP-10. It contains a nucleosome positioning sequence from the X. borealis 5S rRNA gene in the center. The polymerase chain reaction product was purified from a 1% TBE agarose gel using a gel extraction kit according to the protocol provided by the manufacturer (Qiagen, Hilden, Germany).
Formation of the octamer-DNA complex was done by salt dialysis with two different methods that yielded equivalent results. According to the first protocol, the KCl concentration was decreased from 2 M to zero in 10 mM Tris-HCl pH 7.5 buffer in a continuous gradient overnight at 4°C. The second protocol involved a stepwise reduction of the salt concentration at 4°C (Gottesfeld et al., 2001). Six different buffers with 10 mM Tris-HCl pH 7.5 and 1.5, 1, 0.8, 0.67, 0.2 M, or no KCl were used. In each dialysis step, the sample was incubated for at least 1 h. In the reconstitutions with linker histone H1, the protein was added at a ratio of 1.1 molecule H1 per nucleosome in the dialysis step with 0.67 M KCl.
After dialysis, the samples were incubated for ∼2 h at 37° to equilibrate the distribution of binding positions (Luger et al., 1999), and then stored at 4°C. Reconstituted mononucleosomes were analyzed on an 8% polyacrylamide gel (29:1) or on a 1% agarose gel in 1× TBE buffer. Bands were visualized by ethidium bromide staining. To compare the amount of free DNA relative to mononucleosomes, it should be noted that the intensity of ethidium bromide fluorescence is ∼1.4 times higher for free DNA than for nucleosomal DNA (McMurray et al., 1991).
Analytical ultracentrifugation
Analytical ultracentrifugation experiments were carried out on a Beckmann Coulter (Palo Alto, CA) Optima XL-A with absorbance optics. Mononucleosome solutions of A260 ≈ 0.4 were centrifuged at 20 C°. The buffer solution contained 20 mM Tris-HCl, pH 7.5, 0.1 mM EDTA, 0.1 mM DTT. For the histone octamer, a molecular mass M = 108 kDa and a partial specific volume 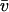 = 0.744 ml·g−1 at 20°C was derived from the sequence with the program SEDNTERP V1.05 by J. Philo, D. Hayes, and T. Laue. The same program was used to calculate a buffer density ρ = 0.999 g·ml−1 and a viscosity η = 1.008 mPa s at 20°C. For the 146 bp DNA, a partial specific volume
= 0.744 ml·g−1 at 20°C was derived from the sequence with the program SEDNTERP V1.05 by J. Philo, D. Hayes, and T. Laue. The same program was used to calculate a buffer density ρ = 0.999 g·ml−1 and a viscosity η = 1.008 mPa s at 20°C. For the 146 bp DNA, a partial specific volume 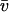 = 0.55 ml·g−1 at 20°C (Durchschlag, 1986) and a calculated molecular mass M = 116 kDa (Dorigo et al., 2003) was used so that nucleosome values of M = 224 kDa and
= 0.55 ml·g−1 at 20°C (Durchschlag, 1986) and a calculated molecular mass M = 116 kDa (Dorigo et al., 2003) was used so that nucleosome values of M = 224 kDa and 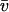 = 0.655 ml·g−1 were derived. The sedimentation velocity data were recorded at 260 nm with 42,000 rpm using a spacing of 0.003 cm in the continuous scan mode. Data were analyzed by computing the g (s*) distribution (Stafford 1992, 1997) with the program DCDT+ version 1.13 by John Philo according to the algorithm described (Philo, 2000).
= 0.655 ml·g−1 were derived. The sedimentation velocity data were recorded at 260 nm with 42,000 rpm using a spacing of 0.003 cm in the continuous scan mode. Data were analyzed by computing the g (s*) distribution (Stafford 1992, 1997) with the program DCDT+ version 1.13 by John Philo according to the algorithm described (Philo, 2000).
SFM imaging
SFM images in air and in fluid were obtained with a Multimode Scanning Probe Microscope and a Nanoscope IIIa controller from Veeco Instruments (Woodbury, NY) operating in the “tapping mode”. The SFM samples were prepared according to the following protocols:
Imaging of air dried samples
10 μl of a solution of 2–10 nM nucleosomes in 5 mM Hepes-KOH pH 7.5, 10 mM Mg-acetate, and 50 mM K-acetate was deposited onto freshly cleaved mica (Plano, Wetzlar, Germany) and immediately washed with distilled water. The mica was then dried with a constant stream of nitrogen. Images were recorded in air at ambient humidity using etched Si-probes type “Nanosensor” (LOT Oriel, Darmstadt, Germany) with a force constant 17–64 N/m, a thickness of 3.5–5.0 μm, a resonance frequency between 250 and 400 kHz, and a tip curvature radius of ≈10 nm (specifications given by the manufacturer).
Imaging in buffer solutions
A solution of 60–80 μl of a buffer containing 10 mM Hepes-KOH 8.0, 30 mM NaCl, 10 mM MgCl2, and nucleosomes at a concentration between 0.4 and 0.8 nM was pipetted onto a freshly cleaved mica disk. In this buffer, complexes were barely visible, most likely due to weak adhesion to the surface as observed previously, e.g., Rippe et al. (1997a,b). To increase the binding affinity of the complexes to the mica surface, NiCl2 was added to a final Ni2+ concentration of ∼10 mM (Hansma and Laney, 1996; Schulz et al., 1998). Images were recorded with NP-S Tips (Veeco Instruments) with a force constant of 0.32 N/m, a nominal tip radius curvature of 5–40 nm, and a resonance frequency ∼8.7 kHz.
Image analysis
Only molecules with a single nucleosome not obstructed by other DNA fragments or protein molecules were analyzed. Complexes with the protein being bound at the end of the DNA were not included into the analysis. DNA contour length, DNA entry-exit angles, and nucleosome positions were measured with the installed Nanoscope Software and with the program Image SXM version 1.67 by Steve Barrett based on NIH Image Software from the National Institutes of Health (Bethesda, MD). The data were plotted and fitted with the program Kaleidagraph (Synergy Software, Reading, PA). The error bars correspond to the square root of the number of samples within each bin. At least four independent experiments were analyzed for the complexes studied in fluid and in air.
Analysis of nucleosome binding sites
The location of the bound nucleosome was determined by measuring the DNA contour length from the middle of the nucleosome complex to the free end of the DNA fragment that was nearer. This length was divided by the total contour length of the complex leading to values of the ratio r between 0 and 0.5. Since the X. borealis 5S rRNA sequence was located in the middle of the fragment, a value of r = 0.5 would correspond to a nucleosome at this position.
Measurement of DNA entry-exit angles
The DNA entry-exit angles at the octamer were measured with Image SXM by drawing lines through the DNA axes on both sides of the nucleosome and measuring the angle at their intersection.
Measurements of apparent contour length
The contour length was determined by drawing a curved line through the middle of the DNA contour from one end of the molecule to the other end. For the protein-DNA complexes, the curve was drawn through the center of the protein from the DNA entry and exit points.
RESULTS
Reconstitution of mononucleosomes
Recombinant histone proteins were overexpressed and purified as described (Luger et al., 1997b, 1999). Fig. 1 A shows a Coomassie-stained SDS gel with the purified single histones H2A, H2B, H3, and H4 and the reconstituted histone octamer. The SDS gel demonstrates that the single proteins are highly purified except for a minor shorter contaminant in the H4 preparations that was removed during further purification of the octamer. The recombinant histones H2A und H2B from Xenopus laevis have a very similar mobility under standard electrophoretic conditions and are hardly separated in the octamer sample (Luger et al., 1997b).
FIGURE 1.

Gel-electrophoretic analysis of mononucleosomes reconstituted with recombinant histone proteins. (A) SDS gel of purified recombinant histones H2A, H2B, H3, and H4, and reconstituted histone octamers (lane Oct). (B) Nondenaturing polyacrylamide gel of mononucleosomes reconstituted with a 146 bp DNA fragment used previously for crystallography (Luger et al., 1997a). As compared to the free DNA, the octamer-DNA complex (Nuc) showed a strongly reduced mobility (Luger et al., 1999). (C) Agarose gel of mononucleosomes reconstituted with a 614 bp DNA fragment.
With the octamer preparation shown in Fig. 1 A, mononucleosomes were reconstituted by salt dialysis. Fig. 1 B displays a nondenaturing polyacrylamide gel of mononucleosomes assembled on a 146-bp-long palindromic sequence derived from human α-satellite DNA. This complex corresponds to the one used in previous crystallographic studies (Luger et al., 1997a, 1999). As compared to the free DNA, the octamer-DNA complex shows a strongly reduced mobility in the gel. An analysis of purified mononucleosomes on the 146 bp DNA by analytical ultracentrifugation yielded a sedimentation coefficient of s = 11.7 ± 0.4 S (20°C, water) and a diffusion coefficient of D = 3.5 × 10−7 cm2 s−1 corresponding to a molecular mass of M = 240 ± 30 kDa. The expected sedimentation coefficient calculated from the crystal structure coordinates (Davey et al., 2002) using the program HYDROPRO (Garcia de la Torre et al., 2000) is s = 11.1 ± 0.3 S.
For the SFM imaging, mononucleosomes were reconstituted on a 614 bp DNA. A longer fragment was chosen to visualize the DNA entering and leaving the nucleosome so that the entry-exit angle and contour length could be measured. The reconstituted complexes were analyzed on a 1% agarose gel. The sample typically contained a small fraction of free DNA (10–20%) and most of the complexes carried only a single nucleosome. Formation of the nucleosome resulted in the expected shift to lower gel-electrophoretic mobility. However, in contrast to the preparation obtained with the 146 bp DNA (Fig. 1 B), no distinct bands were observed, but the mobility was reduced over a rather broad range (Fig. 1 C). This reflects binding of the nucleosome complex to various positions on the DNA-fragment (Hamiche et al., 1996).
Images of mononucleosomes
Representative images obtained by SFM with nucleosomes assembled on the 614 bp fragment are displayed in Fig. 2. The DNA and protein complexes imaged in air (Fig. 2, A and B) and fluid (Fig. 2 C) had typical dimensions of 6 nm width and 0.5 nm height (DNA) and 16 nm width and 2 nm height (nucleosome). The true dimensions of the DNA are a diameter of 2.4 nm whereas the nucleosome has a cylindrical shape of 11 nm diameter and 5.5 nm height. The observed broadening of the sample can be attributed to the finite dimensions of the scanning tip. Conversely, the measured height is somewhat lower than the expected dimensions of 2.4 nm (DNA) and 5.5 nm (nucleosome) most likely due to interaction of the tip and the sample as discussed previously (e. g., Rippe et al. (1997b); Schulz et al. (1998)).
FIGURE 2.

Scanning force microscopy images of mononucleosomes reconstituted with a 614 bp DNA fragment. (A) Image of a 2 × 2 μm area of a sample without H1 scanned in air. (B) A 1 × 1 μm scan of air-dried mononucleosomes reconstituted with linker histone H1. (C) SFM scan in buffer of a 400 × 400 nm area of complexes without H1.
The magnification (600 × 400 nm) of single complexes given in Fig. 3 demonstrates that the protein-complex and the DNA were clearly visible so that a detailed quantitative analysis of the conformation of the complexes could be conducted as described below.
FIGURE 3.

Magnified view of single mononucleosome complexes. An area of 600 × 400 nm from an SFM image of mononucleosomes acquired in air is shown in a three-dimensional representation.
Nucleosome binding positions
The measured distribution of the nucleosome binding position as expressed by the value of r is given in Fig. 4. The analysis of air-dried samples (Fig. 4, A and B) and those imaged in buffer yielded similar results (Fig. 4, C and D). The contour length measurements of the complexes showed a standard deviation of ∼10% in air and in fluid. Given this accuracy for the contour length determination, a Gaussian distributions of r = 0.50 ± 0.07 would be expected for a sample that had all the nucleosomes bound at the 5S rRNA sequence. However, in the absence of H1, the experimentally determined values for r of 0.49 ± 0.18 (Fig. 4 A, air) and 0.44 ± 0.17 (Fig. 4 C, fluid) displayed a larger standard deviation. This indicates that a significant number of nucleosomes were also positioned at other sites of the DNA template. The difference between the measured width of the distribution and that expected for a sample with all nucleosomes located at the 5S rRNA sequence indicated that both in air and in fluid, ∼40% of the nucleosomes are bound to the 5S rRNA sequence.
FIGURE 4.

Histogram of the nucleosome binding position on the 614 bp DNA fragment. Images were recorded in air and in buffer in the absence or presence of linker histone H1. (A) Air, without H1; (B) air, with H1; (C) fluid, without H1; and (D) fluid, with H1.
According to the Boltzmann equation, the probability Pi to find a complex with energy Ei is described by
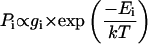 |
(1) |
with gi being the number of states with energy Ei. The octamer binding site covers 147 basepairs of DNA, so that on a 614-basepair-long DNA, there are 614 − 146 = 468 binding sites. Only nucleosomes were analyzed in which both free DNA ends were detectable, i.e., r ≥ 0.11. Accordingly, complexes bound at the terminal 70 basepairs were not included in the analysis. This reduces the effective value of the unspecific binding sites by 140 and gun = 328. From Eq. 1, the energy difference ΔE between specific and unspecific complexes can be calculated from the experimental ratio of unspecific to specific complexes Pun/Psp = 1.5 given in Eq. 2:
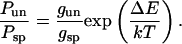 |
(2) |
The resulting energy difference for nucleosome binding to the specific positioning sequence in the X. borealis 5S rRNA as compared to a random sequence is ∼5 kT. The corresponding free energy ΔΔG is 3 kcal mol−1.
Upon addition of the linker histone H1, the distribution profile changed significantly (Fig. 4, B and D). The distribution was rather broad and little preference of nucleosome binding to the X. borealis 5S rRNA sequence was observed. Again, the samples with H1 imaged in air (Fig. 4 B) and in buffer (Fig. 4 D) were undistinguishable within the accuracy of the measurement.
DNA contour length
The apparent DNA contour length of the mononucleosomes was determined in air and in fluid (Fig. 5) as described previously (Schulz et al., 1998). For the free DNA fragments, a length of 190 ± 9 nm was measured (air-dried samples, 691 fragments analyzed). This corresponds to a value of 0.31 ± 0.015 nm/bp, which is slightly smaller than the canonical value for DNA of 0.34 nm per basepair. As discussed previously, the DNA contour length determined by SFM is frequently found to be somewhat shorter than that expected for B-DNA (Rivetti and Codeluppi, 2001). The contour length of mononucleosomes without H1 was 156 ± 14 nm in air (Fig. 5 A) and 156 ± 22 nm in fluid (Fig. 5 C). Mononucleosomes with H1 yielded a contour length of 154 ± 11 nm in air (Fig. 5 B) and 157 ± 16 nm in fluid (Fig. 5 D).
FIGURE 5.

Histogram of contour length measurements. The free 614 bp DNA fragment displayed a 190 ± 9 nm contour length. (A) Air, without H1; (B) air, with H1; (C) fluid, without H1; and (D) fluid, with H1.
No significant differences between measurements in air (Fig. 5, A and B) and in buffer (Fig. 5, C and D) were apparent. In addition, no effect of the addition of linker histone H1 was detected except for a slightly smaller standard deviation of the distribution (Fig. 5). In the complexes studied, the apparent DNA contour length was reduced on an average by 34 ± 13 nm as compared to the free DNA. This shortening would correspond to ∼110 bp of DNA with a contour length of 0.31nm/bp. To determine the amount of DNA that is wrapped around the nucleosome, one has to take into account that the part of the measured contour length of the mononucleosomes traced through the nucleosome complex itself should be subtracted from the total contour length (Hamiche et al., 1996). Since the true dimensions of this part could not be determined directly on the SFM images, a value of 11 nm for the diameter of the nucleosome was taken (Davey et al., 2002; Luger et al., 1997a). This leads to a total of 45 nm or 145 ± 34 bp that are wrapped around the histone octamer complex.
DNA entry-exit angle at the nucleosome
The histogram of DNA entry-exit angles at the nucleosome complexes displayed a bimodal distribution for complexes without H1 (Fig. 6, A and C). In air, two maxima at 81 ± 29° and 136 ± 18° were observed (Fig. 6 A). For measurements in fluid, the values were similar with 78 ± 25° and 137 ± 25° (Fig. 6 C). These data were derived by fitting the distributions with a sum of two Gaussian functions. Addition of the linker histone H1 led to a significant increase in the amount of complexes that displayed smaller DNA entry-exit angles (Fig. 6, B and D). The population centered around 136° was reduced whereas a corresponding increase of the species with the smaller DNA entry-exit angle was evident. A mean value of 88 ± 34° was measured for the air-dried samples (Fig. 6 B). In fluid, a maximum at 85 ± 10° and a rather small amount of complexes at 132 ± 22° were detected (Fig. 6 D).
FIGURE 6.

Histogram of DNA entry-exit angles at the octamer. (A) Air, without H1; (B) air, with H1; (C) fluid, without H1; and (D) fluid, with H1.
DISCUSSION
To our knowledge, we present in this report the first quantitative microscopy analysis of nucleosomes containing recombinant core histone proteins. These histone proteins lack any posttranslational modifications and are completely free of other chromosomal proteins. Therefore, they are particularly suitable to examine the nucleosome conformation under defined conditions and the effect of linker histone H1 binding. Recently, glutaraldehyde fixed nucleosome arrays were visualized successfully by SFM imaging in water with good resolution, and conformation changes due to the DNA sequence or the solution conditions were revealed (Bash et al., 2003; Wang et al., 2002). Furthermore, it has been shown by SFM that air-dried protein-DNA complexes may adopt a conformation that is different from that observed in buffer (Schulz et al., 1998). Thus, SFM provides a valuable contribution to the imaging of biological samples and their conformation changes under conditions where the native hydration state is preserved. Our quantitative comparison of unfixed nucleosomes imaged in air and in buffer yielded essentially the same results. Although the composition of the two deposition buffers was somewhat different (see Materials and Methods), both contained millimolar concentrations of divalent cations and the total ionic strength was very similar. It corresponded to nearly physiological concentrations (84 and 97 mM). Thus, the results obtained here indicate that with respect to the structural features quantitated (binding position of nucleosome, apparent contour length, and DNA entry-exit angle), drying of the samples did not induce changes of the nucleosome conformation, and the differences in buffer composition had no effect. This is in agreement with previous studies of mono- and oligonucleosomes, in which no structural changes have been observed for an ionic strength between 40 and 100 mM (reviewed in van Holde and Zlatanova (1996)).
Reconstitution with the 146 bp DNA according to the published protocols (Luger et al., 1997b, 1999) produced a homogenous population of mononucleosomes and a well-defined band on the polyacrylamide gel (Fig. 1 B). The sedimentation velocity analysis by analytical ultracentrifugation yielded an s value of 11.7 ± 0.4 S that is in good agreement with the value of 12.0 S for mononucleosomes from chicken erythrocytes (Butler and Thomas, 1998) and a theoretically determined value of s = 11.1 ± 0.3 S from the crystal structure. The measured molecular mass of 240 ± 30 kDa in the velocity sedimentation analysis compares favorably with the calculated value of 224 kDa for the complex and demonstrates the integrity of our mononucleosome preparations.
After mononucleosome reconstitution on a 614 bp DNA, the gel electrophoretic mobility of the complex varied over a relatively large range on a 1% agarose gel (Fig. 1 C). This reflects the distribution of different nucleosome binding positions on the DNA fragment. A similar gel mobility of mononucleosomes has been observed previously with a shorter DNA fragment (Hamiche et al., 1996). Since the DNA entry-exit angles at the nucleosome are significantly smaller than 180°, the electrophoretic mobility will be lowest for nucleosomes bound at the center of the DNA. The effect of gel retardation due to DNA bending is reduced the more the bent region is located at the end of the fragment (Koo and Crothers, 1988). Accordingly, both the distribution of binding sites and the magnitude of the DNA entry-exit angle will determine the electrophoretic mobility. These effects cannot be distinguished in the comparison of the samples with and without linker histone H1 by gel-electrophoresis (Fig. 1 C). However, the SFM image analysis conducted here clearly demonstrates that both parameters are changed by H1 binding. This effect of H1 is due to interaction of the protein with the nucleosome since nonspecific H1 binding to free DNA can be excluded under the conditions of the SFM imaging. As determined by fluorescence anisotropy measurements, H1 binds to a 20 basepair DNA with a dissociation constant of Kd ≈ 10−7 M in SFM buffer (F. Kepert and K. Rippe, unpublished). Thus, at nanomolar DNA and H1 concentrations as in the SFM experiments, the fraction of H1 complexed with free DNA is neglectable.
Position of the bound nucleosomes
In the absence of linker histone H1, ∼40% of the nucleosomes were bound at the X. borealis 5S rRNA positioning sequence, which is located in the center of the 614 bp DNA. In agreement with other studies, a significant amount of nucleosomes was also located at other positions (Dong et al., 1990; Karymov et al., 2001; Meersseman et al., 1991; Panetta et al., 1998). It has been shown that the related 208 bp 5 S rRNA sequence from the sear urchin Lytechinus variegatus contains additional minor binding sites so that ≈50% of the nucleosomes are bound at a unique position (Dong et al., 1990; Karymov et al., 2001; Meersseman et al., 1991). Previously, values of 0.84–2.2 kT or 0.7–1.3 kT have been estimated for the energy difference of this five S rRNA sequence considering either four or more (Dong et al., 1990) or only two alternative (Karymov et al., 2001) nucleosome binding positions. However, it is usually assumed for the analysis of unspecific binding of a ligand to DNA that every basepair constitutes a possible start of a binding site so that a sequence of length L has L–146 possible locations for the histone octamer (McGhee and von Hippel, 1974; Widom, 2001). Accordingly, a 208 bp sequence contains 208 − 146 = 62 binding positions. In the analysis presented above (Eq. 2) this would correspond to an energy difference of 4 kT or ΔΔG = 2.4 kcal mol−1 for binding to the sea urchin 5 S rRNA high affinity site within the 208 bp DNA fragment. This energy difference is similar to the value of ΔΔG ≈ 3 kcal mol−1 measured here for the corresponding sequence from X. borealis. Numerous experiments in which nucleosomes are successfully reconstituted with regular spacing on 12 tandemly arranged 208-bp fragments of the 5 S rRNA gene of L. variegatus (reviewed in Hansen (2002)) also support the view that this sequence has a binding energy that is several kT higher than a random sequence.
Remarkably, the unfavorable cost of DNA bending around the nucleosome has been calculated to be as high as ≈76 kcal mol−1 (Widom, 2001). Thus, the histone-DNA interaction energy has to be quite large and a free energy difference of ≈3 kcal mol−1 between different sites is easily conceivable. It can be estimated from the data given by Cotton and Hamkalo (1981) that at approximately physiological salt concentrations (150 mM NaCl), the average dissociation constant of DNA in mononucleosomes isolated from a mouse cell line is 2 nM (ΔG ≈ 12 kcal mol−1). For chicken erythrocytes, a value of 3.6 nM (ΔG = 11.4 kcal mol−1) at 100 mM NaCl has been measured (Ausio et al., 1984). Nucleosome core particle reconstituted on 146 bp sequences containing the human α-satellite DNA or the L. variegatus 5S DNA showed Kds of 0.03 and 0.06 nM, respectively, at 50 mM NaCl, indicating that complexes with these sequences were ∼2 kcal mol−1 more stable than those formed with bulk DNA (Gottesfeld and Luger, 2001). An up to 1000-fold range in relative affinities between certain sequences has been demonstrated that corresponds to a >4 kcal mol−1 range in ΔΔG (Lowary and Widom, 1997; Widom, 2001). However, in the later studies, a comparison of the L. variegatus 5S rRNA with bulk chicken genomic DNA showed only a relatively modest free-energy difference of 0.5 kcal mol−1. This value is similar in magnitude to that of the thermal fluctuations RT ≈ 0.6 kcal mol−1, and therefore too small to explain the preferred binding observed here. According to Eq. 2, <1% of the complexes would be bound to the central 5S rRNA sequence of the template studied here if the binding affinity difference to this site would only be 0.5 kcal mol−1. Thus, to explain the experimental results presented in Fig. 4, A and C, as well as the previously observed binding preference to the related L. variegatus 5 S rRNA sequence (Dong et al., 1990; Karymov et al., 2001; Meersseman et al., 1991) a higher binding energy difference of ΔΔG = 2–3 kcal mol−1 as compared to random sites is required. This ΔΔG value is also consistent with a thermodynamic analysis of nucleosome dissociation constants (Gottesfeld and Luger, 2001).
The addition of linker histone H1 led to a distribution of binding positions that displayed little preference for the 5S rRNA positioning sequence (Fig. 4, B and D). This suggests that in the presence of H1, the free-energy difference between binding positions is reduced to values of ∼2 RT (1.2 kcal mol−1) or lower, which would not result in a significant increase of occupancy at the central 5 S rRNA sequence considering the accuracy of the measurement. And alternative explanation would be that the effect of H1 addition is mostly on the kinetics of nucleosome movement. Although various reports indicate that in the absence of H1, considerable rearrangement of histone octamers can occur on the DNA, it appears that H1 inhibits the short-range mobility of histone octamers on DNA and traps the nucleosomes at a certain position (Panetta et al., 1998; Pennings et al., 1994). If this is the case, a broader distribution of binding sites might be observed despite a significant energy difference to the 5 S rRNA positioning sequence if an equilibrium is not reached.
DNA contour length
From x-ray crystallography studies it is known that 146 bp (Luger et al., 1997a) or 147 bp (Davey et al., 2002) of DNA are wrapped around the histone octamer core complex. Our results yield a similar value of 145 ± 34 bp from the SFM contour length measurements of the same recombinant histone samples but reconstituted on a longer DNA fragment (Fig. 5). However, the value for the amount of DNA wrapping determined from SFM images displayed a rather large standard deviation. Although the contour length measurements of the free DNA had a standard deviation of ≈5%, the corresponding value for the nucleosome complexes was twice as high. This indicates that some heterogeneity with respect to the amount of DNA wrapped into the nucleosome is present. It is also possible that the binding of the nucleosome to the surface introduces some broadening of the measured contour length distributions. The addition of linker histone H1 has little effect on the apparent DNA contour length except for a small narrowing of the distribution, the significance of which is unclear (Fig. 5). Binding of H1 has been reported to protect another 20 bp in addition to the 146/147 bp wrapped around the octamer (Travers, 1999). This would correspond to a length of only 6 nm or 4% of the total contour length. It is likely that an additional shortening of this magnitude or less would not be detected in our analysis as a significant reduction.
DNA entry-exit angles at the nucleosome
For mononucleosomes without linker histone H1, two maxima in the distribution of bending angles at ∼80° and at 136° were identified, indicating that two species with a different DNA geometry were present (Fig. 6, A and C). Upon binding of H1 to the nucleosome, the complexes with angles ≥120° mostly disappeared (Fig. 6, B and D), and the first maximum ∼85° became more pronounced. Thus, the linker histone seems to stabilize the conformation of nucleosomes in which the DNA entry-exit angle is ∼85°. A reduction of the entry-exit angles has also been observed by SFM, electron microscopy (EM), and electron cryomicroscopy (cryo-EM) studies of native nucleosome preparations (Bednar et al., 1998; Hamiche et al., 1996; Leuba et al., 1998a,b; Zlatanova et al., 1998). Thus, in support of previous results, we conclude from our experiments that histone H1 stabilizes a certain DNA geometry at the nucleosome, in which the DNA entry-exit angle is significantly reduced as compared to the linker histone free nucleosome.
Studies of chromatin isolated mostly from chicken erythrocytes but also from other sources indicate that the DNA flanking the nucleosome core and the linker histone can interact to form a so called stem motif (Bednar et al., 1998; Hamiche et al., 1996; Zlatanova et al., 1998). In this structure, the two DNA segments leaving the nucleosome core particle associate ∼8 nm from the nucleosome center and remain in contact for 3–5 nm before diverging (Bednar et al., 1998). On the SFM images in the presence of H1, we did not observe any complexes that had a nucleosome stem motif. This could be partly attributed to an insufficient resolution to examine the trajectory of the DNA directly at the nucleosome. Obviously, detection of a stem motif would require that the region in which the two DNA segments associate is separated well from the nucleosome core. However, also the small DNA entry-exit angle characteristic for a nucleosome stem (Bednar et al., 1998; Hamiche et al., 1996) was not apparent upon quantitative inspection of the samples. Only a very small fraction of the complexes had angles <45° (Fig. 6, B and D). Thus, it appears also conceivable that the formation of a stem structure with recombinant core histone proteins might require additional components/modifications besides the simple addition of linker histone H1 that are not present in our in vitro system.
By cryo-EM imaging of native chromatin fibers, mean DNA entry-exit angles between ≈30° (80 mM salt) to ≈90° (5 mM salt) have been estimated (Bednar et al., 1995, 1998; Woodcock et al., 1993; Woodcock and Horowitz, 1995). In another cryo-EM study on reconstituted chicken erythrocyte mononucleosomes without H1, average values of 58 ± 42° were determined but there was also a significant population of complexes with angles ≥120° (Furrer et al., 1995). In general, the values for the DNA entry-exit angle determined by cryo-EM appear to be significantly lower than those measured by SFM (Leuba et al., 1998a,b; Zlatanova et al., 1998). Since resolution and contrast are different for the two imaging techniques, the relative location of the measured bending angle with respect to the nucleosome core might vary. Effects of sample preparation and effective salt concentration have also to be considered. For the SFM imaging, the samples are bound to a surface. It has been previously argued that no systematic error is introduced in the mean DNA bending angle that exists in solution under the conditions of deposition used here (Erie et al., 1994; Rees et al., 1993; Rivetti et al., 1996). Furthermore, the conventional EM imaging of surface bound mononucleosomes also shows small entry-exit angles in the presence of linker histone, although no quantitative analysis is given (Hamiche et al., 1996). However, an effect of binding the complexes on the surface cannot be excluded, and the absolute DNA entry-exit angles determined by SFM might not be directly comparable to those obtained by true solution methods such as analysis of DNA labeled mononucleosomes by fluorescence resonance energy transfer (Tóth et al., 2001). Nevertheless, the relative changes of the angle distribution observed here upon addition of linker histone H1 provide reliable evidence that H1 stabilizes a smaller entry-exit angle.
Previous SFM studies of fixed chromatin fibers yielded average values of 100 ± 40° in the presence and 130 ± 40° in the absence of linker histones (Leuba et al., 1998a,b; Zlatanova et al., 1998). These data are consistent with the distribution of angles between 70 and 150° determined here for nucleosome core particles and DNA entry-exit angles of 85 ± 10° (air) and 88 ± 34° (buffer) for nucleosome complexes with H1 (Fig. 6). The agreement suggests that the entry-exit angle is predominantly determined on the level of a single nucleosome by the binding or dissociation of the linker histone, which in turn is expected to affect the compaction of the whole chromatin fiber.
We emphasize that the experiments described here were conducted in a well-defined system with highly purified protein components and recombinant proteins. Thus, the approach can be extended to study the effect of posttranslational histone modifications as well as the binding of other chromosomal proteins. Furthermore, it could be demonstrated that unfixed mononucleosomes can be studied in their native hydration state by SFM. This is a specific advantage of the technique that will prove to be useful in studies of the higher order chromatin organization.
Acknowledgments
We gratefully acknowledge the support of Tom Jovin and Peter Lichter, and thank Gudrun Heim and Nathalie Brun for their help, Karolin Luger for providing the histone expression vectors, and Jacek Mazurkiewicz for critical reading of the manuscript. Part of the work by J.F.K., K.F.T., and K.R., and the work of M.C., was done at the Division Biophysics of Macromolecules of the Deutsches Krebsforschungszentrum. J.F.K. did some of the SFM imaging at the Department of Molecular Biology at the Max-Planck-Institut für biophysikalische Chemie in Göttingen.
The project was supported by the Volkswagen Foundation in the program “Junior Research Groups at German Universities”.
References
- Allen, M. J., X. F. Dong, T. E. O'Neill, P. Yau, S. C. Kowalczykowski, J. Gatewood, R. Balhorn, and E. M. Bradbury. 1993. Atomic force microscope measurements of nucleosome cores assembled along defined DNA sequences. Biochemistry. 32:8390–8396. [DOI] [PubMed] [Google Scholar]
- Arents, G., R. W. Burlingame, B.-C. Wang, W. E. Love, and E. N. Moudrianakis. 1991. The nucleosomal core histone octamer at 3.1 Å resolution: a triparatite protein assembly and a left-handed superhelix. Proc. Natl. Acad. Sci. USA. 88:10148–10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausio, J., D. Seger, and H. Eisenberg. 1984. Nucleosome core particle stability and conformational change. Effect of temperature, particle and NaCl concentrations, and crosslinking of histone H3 sulfhydryl groups. J. Mol. Biol. 176:77–104. [DOI] [PubMed] [Google Scholar]
- Bash, R., H. Wang, J. Yodh, G. Hager, S. M. Lindsay, and D. Lohr. 2003. Nucleosomal arrays can be salt-reconstituted on a single-copy MMTV promoter DNA template: their properties differ in several ways from those of comparable 5S concatameric arrays. Biochemistry. 42:4681–4690. [DOI] [PubMed] [Google Scholar]
- Bash, R. C., J. Yodh, Y. Lyubchenko, N. Woodbury, and D. Lohr. 2001. Population analysis of subsaturated 172-12 nucleosomal arrays by atomic force microscopy detects nonrandom behavior that is favored by histone acetylation and short repeat length. J. Biol. Chem. 276:48362–48370. [DOI] [PubMed] [Google Scholar]
- Bednar, J., R. A. Horowitz, J. Dubochet, and C. L. Woodcock. 1995. Chromatin conformation and salt-induced compaction: Three-dimensional structural information from cryoelectron microscopy. J. Cell Biol. 131:1365–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bednar, J., R. A. Horowitz, S. A. Grigoryev, L. M. Carruthers, J. C. Hansen, A. J. Koster, and C. L. Woodcock. 1998. Nucleosomes, linker DNA, and linker histone form a unique structural motif that directs the higher-order folding and compaction of chromatin. Proc. Natl. Acad. Sci. USA. 95:14173–14178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustamante, C., G. Zuccheri, S. H. Leuba, G. L. Yang, and B. Samori. 1997. Visualization and analysis of chromatin by scanning force microscopy. Methods. 12:73–83. [DOI] [PubMed] [Google Scholar]
- Butler, P. J., and J. O. Thomas. 1998. Dinucleosomes show compaction by ionic strength, consistent with bending of linker DNA. J. Mol. Biol. 281:401–407. [DOI] [PubMed] [Google Scholar]
- Buttinelli, M., G. Panetta, D. Rhodes, and A. Travers. 1999. The role of histone H1 in chromatin condensation and transcriptional repression. Genetica. 106:117–124. [DOI] [PubMed] [Google Scholar]
- Cotton, R. W., and B. A. Hamkalo. 1981. Nucleosome dissociation at physiological ionic strengths. Nucleic Acids Res. 9:445–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey, C. A., D. F. Sargent, K. Luger, A. W. Maeder, and T. J. Richmond. 2002. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 Å resolution. J. Mol. Biol. 319:1097–1113. [DOI] [PubMed] [Google Scholar]
- Dong, F., J. C. Hansen, and K. E. van Holde. 1990. DNA and protein determinants of nucleosome positioning on sea urchin 5S rRNA gene sequences in vitro. Proc. Natl. Acad. Sci. USA. 87:5724–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorigo, B., T. Schalch, K. Bystricky, and T. J. Richmond. 2003. Chromatin fiber folding: requirement for the histone H4 N-terminal tail. J. Mol. Biol. 327:85–96. [DOI] [PubMed] [Google Scholar]
- Durchschlag, H. 1986. Specific volumes of biological macromolecules and some other molecules of biological interest. In Thermodynamic Data for Biochemistry and Biotechnology. H.-J. Hinz, editor. Springer-Verlag, Berlin. 45–128.
- Erie, D. A., G. Yang, H. C. Schultz, and C. Bustamante. 1994. DNA bending by Cro protein in specific and nonspecific complexes: implications for protein site recognition and specificity. Science. 266:1562–1566. [DOI] [PubMed] [Google Scholar]
- Fritzsche, W., and E. Henderson. 1996. Scanning force microscopy reveals ellipsoid shape of chicken erythrocyte nucleosomes. Biophys. J. 71:2222–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritzsche, W., A. Schaper, and T. M. Jovin. 1994. Probing chromatin with the scanning force microscope. Chromosoma. 103:231–236. [DOI] [PubMed] [Google Scholar]
- Fritzsche, W., L. Takac, and E. Henderson. 1997. Application of atomic force microscopy to visualization of DNA, chromatin, and chromosomes. Crit. Rev. Eukaryot. Gene Expr. 7:231–240. [DOI] [PubMed] [Google Scholar]
- Fritzsche, W., J. Vesenka, and E. Henderson. 1995. Scanning force microscopy of chromatin. Scanning Microsc. 9:729–739. [PubMed] [Google Scholar]
- Furrer, P., J. Bednar, J. Dubochet, A. Hamiche, and A. Prunell. 1995. DNA at the entry-exit of the nucleosome observed by cryoelectron microscopy. J. Struct. Biol. 114:177–183. [DOI] [PubMed] [Google Scholar]
- Garcia de la Torre, J., M. L. Huertas, and B. Carrasco. 2000. Calculation of hydrodynamic properties of globular proteins from their atomic-level structure. Biophys. J. 78:719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesfeld, J. M., and K. Luger. 2001. Energetics and affinity of the histone octamer for defined DNA sequences. Biochemistry. 40:10927–10933. [DOI] [PubMed] [Google Scholar]
- Gottesfeld, J. M., C. Melander, R. K. Suto, H. Raviol, K. Luger, and P. B. Dervan. 2001. Sequence-specific recognition of DNA in the nucleosome by pyrrole-imidazole polyamides. J. Mol. Biol. 309:615–629. [DOI] [PubMed] [Google Scholar]
- Graziano, V., S. E. Gerchman, D. K. Schneider, and V. Ramakrishnan. 1994. Histone H1 is located in the interior of the chromatin 30-nm filament. Nature. 368:351–354. [DOI] [PubMed] [Google Scholar]
- Hamiche, A., P. Schultz, V. Ramakrishnan, P. Oudet, and A. Prunell. 1996. Linker histone-dependent DNA structure in linear mononucleosomes. J. Mol. Biol. 257:30–42. [DOI] [PubMed] [Google Scholar]
- Hansen, J. C. 2002. Conformational dynamics of the chromatin fiber in solution: determinants, mechanisms, and functions. Annu. Rev. Biophys. Biomol. Struct. 31:361–392. [DOI] [PubMed] [Google Scholar]
- Hansma, H. G., and D. E. Laney. 1996. DNA binding to mica correlates with cationic radius: assay by atomic force microscopy. Biophys. J. 70:1933–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harp, J. M., B. L. Hanson, D. E. Timm, and G. J. Bunick. 2000. Asymmetries in the nucleosome core particle at 2.5 Å resolution. Acta Crystallogr. D Biol. Crystallogr. 56:1513–1534. [DOI] [PubMed] [Google Scholar]
- Herrera, J. E., K. L. West, R. L. Schiltz, Y. Nakatani, and M. Bustin. 2000. Histone H1 is a specific repressor of core histone acetylation in chromatin. Mol. Cell. Biol. 20:523–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz, R. A., D. A. Agard, J. W. Sedat, and C. L. Woodcock. 1994. The three-dimensional architecture of chromatin in situ: electron tomography reveals fibers composed of a continuously variable zig-zag nucleosomal ribbon. J. Cell Biol. 125:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karymov, M. A., M. Tomschik, S. H. Leuba, P. Caiafa, and J. Zlatanova. 2001. DNA methylation-dependent chromatin fiber compaction in vivo and in vitro: requirement for linker histone. FASEB J. 15:2631–2641. [DOI] [PubMed] [Google Scholar]
- Koo, H.-S., and D. M. Crothers. 1988. Calibration of DNA curvature and a unified description of sequence-directed bending. Proc. Natl. Acad. Sci. USA. 85:1763–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuba, S. H., and C. Bustamante. 1999. Analysis of chromatin by scanning force microscopy. Methods Mol. Biol. 119:143–160. [DOI] [PubMed] [Google Scholar]
- Leuba, S. H., C. Bustamante, K. van Holde, and J. Zlatanova. 1998a. Linker histone tails and N-tails of histone H3 are redundant: scanning force microscopy studies of reconstituted fibers. Biophys. J. 74:2830–2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuba, S. H., C. Bustamante, J. Zlatanova, and K. van Holde. 1998b. Contributions of linker histones and histone H3 to chromatin structure: scanning force microscopy studies on trypsinized fibers. Biophys. J. 74:2823–2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuba, S. H., G. Yang, C. Robert, B. Samori, K. van Holde, J. Zlatanova, and C. Bustamante. 1994. Three-dimensional structure of extended chromatin fibers as revealed by tapping-mode scanning force microscopy. Proc. Natl. Acad. Sci. USA. 91:11621–11625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowary, P. T., and J. Widom. 1997. Nucleosome packaging and nucleosome positioning of genomic DNA. Proc. Natl. Acad. Sci. USA. 94:1183–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger, K., A. W. Mäder, R. K. Richmond, D. F. Sargent, and T. J. Richmond. 1997a. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 389:251–260. [DOI] [PubMed] [Google Scholar]
- Luger, K., T. J. Rechsteiner, A. J. Flaus, M. M. Waye, and T. J. Richmond. 1997b. Characterization of nucleosome core particles containing histone proteins made in bacteria. J. Mol. Biol. 272:301–311. [DOI] [PubMed] [Google Scholar]
- Luger, K., T. J. Rechsteiner, and T. J. Richmond. 1999. Preparation of nucleosome core particle from recombinant histones. Methods Enzymol. 304:3–19. [DOI] [PubMed] [Google Scholar]
- Martin, L. D., J. P. Vesenka, E. Henderson, and D. L. Dobbs. 1995. Visualization of nucleosomal substructure in native chromatin by atomic force microscopy. Biochemistry. 34:4610–4616. [DOI] [PubMed] [Google Scholar]
- McGhee, J. D., and P. H. von Hippel. 1974. Theoretical aspects of DNA-protein interactions: co-operative and non-co-operative binding of large ligands to a one-dimensional homogenous lattice. J. Mol. Biol. 86:469–489. [DOI] [PubMed] [Google Scholar]
- McMurray, C. T., E. W. Small, and K. E. van Holde. 1991. Binding of ethidium to the nucleosome core particle. Internal and external binding modes. Biochemistry. 30:5644–5652. [DOI] [PubMed] [Google Scholar]
- Meersseman, G., S. Pennings, and E. M. Bradbury. 1991. Chromatosome positioning on assembled long chromatin. Linker histones affect nucleosome placement on 5 S rDNA. J. Mol. Biol. 220:89–100. [DOI] [PubMed] [Google Scholar]
- Panetta, G., M. Buttinelli, A. Flaus, T. J. Richmond, and D. Rhodes. 1998. Differential nucleosome positioning on Xenopus oocyte and somatic 5 S RNA genes determines both TFIIIA and H1 binding: a mechanism for selective H1 repression. J. Mol. Biol. 282:683–697. [DOI] [PubMed] [Google Scholar]
- Pennings, S., G. Meersseman, and E. M. Bradbury. 1994. Linker histones H1 and H5 prevent the mobility of positioned nucleosomes. Proc. Natl. Acad. Sci. USA. 91:10275–10279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philo, J. S. 2000. A method for directly fitting the time derivative of sedimentation velocity data and an alternative algorithm for calculating sedimentation coefficient distribution functions. Anal. Biochem. 279:151–163. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan, V. 1997a. Histone H1 and chromatin higher-order structure. Crit. Rev. Eukaryot. Gene Expr. 7:215–230. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan, V. 1997b. Histone structure and the organization of the nucleosome. Annu. Rev. Biophys. Biomol. Struct. 26:83–112. [DOI] [PubMed] [Google Scholar]
- Rees, W. A., R. W. Keller, G. Y. Vesenka, and C. Bustamante. 1993. Evidence of DNA bending in transcription complexes imaged by scanning force microscopy. Science. 260:1646–1649. [DOI] [PubMed] [Google Scholar]
- Richmond, T. J., and J. Widom. 2000. Nucleosome and chromatin structure. In Chromatin Structure and Gene Expression, Frontiers in Molecular Biology. S. Elgin and J. L. Workman, editors. Oxford University Press, Oxford, UK. 328–351.
- Rippe, K., N. Mücke, and J. Langowski. 1997a. Molecules in motion: Imaging DNA with the scanning force microscope in aqueous solutions. Bioforum International. 1:42–44. [Google Scholar]
- Rippe, K., N. Mücke, and J. Langowski. 1997b. Superhelix dimensions of a 1868 base pair plasmid determined by scanning force microscopy in air and in aqueous solution. Nucleic Acids Res. 25:1736–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivetti, C., and S. Codeluppi. 2001. Accurate length determination of DNA molecules visualized by atomic force microscopy: evidence for a partial B- to A-form transition on mica. Ultramicroscopy. 87:55–66. [DOI] [PubMed] [Google Scholar]
- Rivetti, C., M. Guthold, and C. Bustamante. 1996. Scanning force microscopy of DNA deposited onto mica: equilibration versus molecular kinetic trapping studied by statistical polymer chain analysis. J. Mol. Biol. 264:919–932. [DOI] [PubMed] [Google Scholar]
- Schulz, A., N. Mücke, J. Langowski, and K. Rippe. 1998. Scanning force microscopy of E. coli RNA polymerase·σ54 holoenzyme complexes with DNA in buffer and in air. J. Mol. Biol. 283:821–836. [DOI] [PubMed] [Google Scholar]
- Stafford, W. F. 1992. Boundary analysis in sedimentation transport experiments: a procedure for obtaining sedimentation coefficient distributions using the time derivative of the concentration profile. Anal. Biochem. 203:295–301. [DOI] [PubMed] [Google Scholar]
- Stafford, W. F. 1997. Sedimentation velocity spins a new weave for an old fabric. Curr. Opin. Biotechnol. 8:14–24. [DOI] [PubMed] [Google Scholar]
- Tóth, K., N. Brun, and J. Langowski. 2001. Trajectory of nucleosomal linker DNA studied by fluorescence resonance energy transfer. Biochemistry. 40:6921–6928. [DOI] [PubMed] [Google Scholar]
- Travers, A. 1999. The location of the linker histone on the nucleosome. Trends Biochem. Sci. 24:4–7. [DOI] [PubMed] [Google Scholar]
- van Holde, K., and J. Zlatanova. 1996. What determines the folding of the chromatin fiber. Proc. Natl. Acad. Sci. USA. 93:10548–10555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Holde, K. E. 1989. Chromatin. Springer, Heidelberg.
- Wang, H., R. Bash, J. G. Yodh, G. L. Hager, D. Lohr, and S. M. Lindsay. 2002. Glutaraldehyde modified mica: a new surface for atomic force microscopy of chromatin. Biophys. J. 83:3619–3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widom, J. 2001. Role of DNA sequence in nucleosome stability and dynamics. Quart. Rev. Biophys. 34:269–324. [DOI] [PubMed] [Google Scholar]
- Woodcock, C. L., S. A. Grigoryev, R. A. Horowitz, and N. Whitaker. 1993. A chromatin folding model that incorporates linker variability generates fibers resembling the native structures. Proc. Natl. Acad. Sci. USA. 90:9021–9025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodcock, C. L., and R. A. Horowitz. 1995. Chromatin organization reviewed. Trends Cell Biol. 5:272–277. [DOI] [PubMed] [Google Scholar]
- Yang, G., S. H. Leuba, C. Bustamante, J. Zlatanova, and K. van Holde. 1994. Role of linker histones in extended chromatin fibre structure. Nat. Struct. Biol. 1:761–763. [DOI] [PubMed] [Google Scholar]
- Yodh, J. G., Y. L. Lyubchenko, L. S. Shlyakhtenko, N. Woodbury, and D. Lohr. 1999. Evidence for nonrandom behavior in 208-12 subsaturated nucleosomal array populations analyzed by AFM. Biochemistry. 38:15756–15763. [DOI] [PubMed] [Google Scholar]
- Yodh, J. G., N. Woodbury, L. S. Shlyakhtenko, Y. L. Lyubchenko, and D. Lohr. 2002. Mapping nucleosome locations on the 208-12 by AFM provides clear evidence for cooperativity in array occupation. Biochemistry. 41:3565–3574. [DOI] [PubMed] [Google Scholar]
- Zhou, Y. B., S. E. Gerchman, V. Ramakrishnan, A. Travers, and S. Muyldermans. 1998. Position and orientation of the globular domain of linker histone H5 on the nucleosome. Nature. 395:402–405. [DOI] [PubMed] [Google Scholar]
- Zlatanova, J., P. Caiafa, and K. Van Holde. 2000. Linker histone binding and displacement: versatile mechanism for transcriptional regulation. FASEB J. 14:1697–1704. [DOI] [PubMed] [Google Scholar]
- Zlatanova, J., S. H. Leuba, and K. van Holde. 1998. Chromatin fiber structure: morphology, molecular determinants, structural transitions. Biophys. J. 74:2554–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlatanova, J., and K. van Holde. 1996. The linker histones and chromatin structure: new twists. Prog. Nucleic Acid Res. Mol. Biol. 52:217–259. [DOI] [PubMed] [Google Scholar]