

Abstract
Mitogen-activated protein kinases are regulated by occupancy at two phosphorylation sites near the active site cleft. Previous studies using hydrogen exchange to investigate the canonical mitogen-activated protein kinase, extracellular signal-regulated protein kinase-2, have shown that phosphorylation alters backbone conformational mobility >10 Å distal to the site of phosphorylation, including decreased mobility within amino acids 102–105 and increased mobility within 108–109. To further describe changes after enzyme activation, site-directed spin labeling at amino acids 101, 105–109, 111, 112 and electron paramagnetic resonance spectroscopy were used to investigate this region. The anisotropic hyperfine splitting of the spin labels in glassy samples was unchanged by phosphorylation, consistent with previous crystallographic studies that indicate no structural change in this region. At positions 101, 111, and 112, the mobility of the spin label was unchanged by diphosphorylation, consistent with little or no conformational change. However, diphosphorylation caused small but significant changes in rotational diffusion rates at positions 105–108 and altered proportions of probe in a motionally constrained state at positions 105, 107, and 109. Thus, electron paramagnetic resonance indicates reproducible changes in nanosecond side-chain mobilities at specific residues within the interdomain region, far from the site of phosphorylation and conformational change.
INTRODUCTION
Protein kinases play a central role in transducing messages from the plasma membrane to the nucleus that ultimately alter key cellular responses. By catalyzing phosphoryl transfer to cytosolic and nuclear targets, protein kinases regulate the function of effector molecules that control cellular architecture, gene transcription, cell metabolism, and cell cycle. Thus, kinases play fundamental roles in all processes that define cellular behavior including motility, differentiation, proliferation, and growth.
Several kinase superfamilies have been examined by a variety of biophysical approaches including x-ray crystallography (Hu et al., 1994; Knighton et al., 1991; Narayana et al., 1997a,b; Madhusudan et al., 2002; Canagarajah et al., 1997; Fox et al., 1998; Zhang et al., 1994; Wilson et al., 1996), neutron diffraction (Olah et al., 1993), fluorescence anisotropy (Li et al., 2002), molecular dynamics (Emrick et al., 2001; Tsigelny et al., 1999; Young et al., 2001), and hydrogen exchange (Resing and Ahn, 1998; Resing et al., 1999; Hoofnagle et al., 2001; Anand et al., 2002). These studies have resulted in a general understanding of the canonical kinase catalytic domain structure, as well as insights into the dynamic conformational changes of kinases upon substrate and product binding. Many kinases in these superfamilies are regulated by posttranslational phosphorylation, which results in increased specific activity, homodimerization, and altered subcellular localization (Khokhlatchev et al., 1998; Pearson et al., 2001).
Members of the mitogen-activated protein kinase (MAPK) superfamily are activated by diphosphorylation in a region near the active site cleft, termed the activation lip (Robbins et al., 1993; Pearson et al., 2001; Johnson et al., 1996). Diphosphorylation of the MAPK, extracellular signal-regulated protein kinase-2 (ERK2), occurs at Thr-183 and Tyr-185, where modification of both residues is catalyzed by dual specificity kinases called MAP kinase kinases 1 and 2. Upon phosphorylation, the specific activity of ERK2 is increased by more than 1000-fold, which has been ascribed to an increase in the rate of phosphoryl transfer (Robbins et al., 1993; Prowse and Lew, 2001).
Structural insight into the mechanism of activation of ERK2 has been obtained from x-ray crystallographic analyses of unphosphorylated (ERK) and phosphorylated (ppERK) forms (Zhang et al., 1994; Canagarajah et al., 1997). The largest changes in structure occurred at the activation lip, involving reconfiguration of the lip through salt bridge interactions between the phosphothreonine and residues within the N-terminal αC-helix and catalytic cleft as well as between phosphotyrosine and arginine residues in the C-terminal substrate binding domain. From these studies, it was proposed that ERK2 activation results from increased affinity for protein substrate due to new phosphotyrosine contacts that reshape the substrate binding site to accommodate the Ser/Thr-Pro specificity motif (Canagarajah et al., 1997). However, it is clear from steady-state kinetic analysis that substrate binding accounts for only part of the activation mechanism (Prowse and Lew, 2001). Other structural changes that may contribute to activation include a slight closure of the active site cleft due to a 5° interdomain rotation of the N- and C-terminal lobes with respect to one another, and subtle realignments of active site residues. In addition to structural changes at the active site, diphosphorylation of ERK2 induces structural changes in the small N-terminal lobe of the kinase that forms a homodimerization interface (Canagarajah et al., 1997; Hoofnagle et al., 2001). At high concentrations, such as those used in this study, both ERK and ppERK exist as dimers in solution (Khokhlatchev et al., 1998).
The contribution of protein dynamics to kinase regulation is still poorly understood. Recently, hydrogen exchange mass spectral studies of ERK and ppERK revealed changes in conformational flexibility accompanying enzyme activation (Hoofnagle et al., 2001). Specifically, phosphorylation altered hydrogen exchange rates in the hinge region of the molecule (residues 101–112), which includes a region that is important for nucleotide binding as well as the pivot point for domain closure. X-ray crystallography indicates that this region is structurally identical between phosphorylated and unphosphorylated forms, suggesting that differences in hydrogen exchange rates reflect altered mobility, without major conformational change. Furthermore, the hinge region is >10 Å from the site of phosphorylation and major conformational change, indicating that there are long-range impacts on dynamic motion.
To more specifically describe changes in flexibility due to enzyme activation, we utilized the complementary approach of site-directed spin labeling–electron paramagnetic resonance spectroscopy (SDSL-EPR; Hubbell et al., 1996). EPR spectroscopy measures the absorption of microwave radiation by unpaired electrons. Attachment of a spin-label probe to engineered cysteine residues permits measurement of dynamic side-chain motions in the nanosecond (ns) timescale at specific sites in proteins. This technique has been used to study a wide variety of processes including folding and conformational changes in proteins, assembly of subunits, and insertion of proteins into membranes (Mchaourab and Perozo, 2000; Hubbell et al., 2000; Victor and Cafiso, 2001; Columbus and Hubbell, 2002). An important advance in interpreting slow motional EPR has been the development and release of software for simulation and nonlinear least squares fitting of various models to experimental spectra (Schneider and Freed, 1989; Earle et al., 1993; Budil et al., 1996), which has facilitated EPR studies to assess conformational changes and dynamics of systems not amenable to nuclear magnetic resonance spectroscopy (Alonso et al., 2001; Barnakov et al., 2002; Barnes et al., 1999; Glasgow et al., 1999; Liu et al., 2001).
Herein, we report the analysis of phosphorylation-dependent changes in protein dynamics in ERK2 by SDSL-EPR. We have focused on the hinge region of the molecule, which covalently links the N- and C-terminal lobes and facilitates interdomain closure, a dynamic conformational event important for catalysis. Spin-label probes incorporated into the hinge region were monitored by EPR and used to reveal changes in side-chain conformation and motion at several residues, upon ERK2 phosphorylation. The spectral changes demonstrate that phosphorylation changes the mobility of side chains at certain positions within the hinge on the ns timescale. We propose that these changes reflect a small conformational change that is important for interdomain closure and may be related to altered backbone motions observable by hydrogen exchange mass spectrometry (HX-MS). This study complements x-ray crystallography and hydrogen exchange measurements. The results are consistent with phosphorylation-induced interdomain closure as a mechanism for kinase regulation, and set an important precedent for the use of SDSL-EPR in studying dynamical responses to protein posttranslational modifications.
METHODS
Site-directed mutagenesis
A cysteineless mutant was constructed by incorporating mutations C38S, C63S, C125S, C159S, C164S, C214A, and C252S into ERK2, using the QuickChange PCR mutagenesis kit (Stratagene, La Jolla, CA). Within this background, native side chains I101, V102, Q103, D104, L105, M106, E107, T108, D109, L110, Y111, and K112 (numbered as in Hoofnagle et al., 2001) were individually mutated to cysteine. Mutants V102C, Q103C, D104C, and L110C were insoluble and were not included in the study. The parent plasmid used in this study was NpT7-5-His6-rERK2, kindly provided by Melanie Cobb (University of Texas, Southwestern Medical Center, Dallas, TX; described in Robbins et al., 1993). Primers were designed according to the manufacturer's specifications and included: 5′-GGCGCCTACGGCATGGTTTCTTCTGCTTATG-3′ (C38S), 5′-GAGCACCAGACCTACTCTCAGAGAACCCTGAG-3′ (C63S), 5′-CACCTCAGCAATGATCATATCTCCTATTTTCTTTATCAGATCC-3′ (C125S), 5′-CTGAACACCACTTCTGATCTCAAGATCTCTGACTTTGGCCTTG-3′ (C159S/C164S), 5′-GGTCTGTGGGCGCGATCCTGGCAGAGATGCTATCC-3′ (C214A), 5′-GATCTCCATCACAGGAAGATCTGAATTCTATAATAAATTTAAAAGC-3′ (C252S), 5′-CCAACCATTGAGCAGATGAAAGATGTATATTGCGTGCAGGACCTCATG-3′ (I101C), 5′-GATGTATATATAGTACAGGACTGCATGGAGACAGATCTTTACAAGC-3′ (L105C), 5′-GTACAGGACCTCTGCGAGACCGATCTTTACAAGCTCTTGAAGAC-3′ (M106C), 5′-GGACCTCATGTGCACCGATCTTTACAAGCTCTTGAAGACACAGC-3′ (E107C), 5′-CCTCATGGAGTGCGATCTTTACAAGCTCTTGAAGACACAGC-3′ (T108C), 5′-CCTCATGGAGACATGCCTTTACAAGCTCTTGAAGACACAGC-3′ (D109C), 5′-CCTCATGGAGACAGATCTTTGCAAGCTCTTGAAGACAC-3′ (Y111C), 5′-GGAGACAGATCTTTACTGCCTCTTGAAGACACAGCACCTC-3′ (K112C), as well as the complementary reverse primers. Protocols were carried out as described by the manufacturer except that 25 ng of plasmid, and 125 ng of forward and 125 ng of reverse primer were used in each reaction. The resulting plasmids were transformed into the XL-1 Blue strain of Escherichia coli, amplified, and purified using QIAgen miniprep spin columns (QIAGEN, Valencia, CA) according to the manufacturer's protocol. The purified plasmids were stored in water at −20°C at 60 ng/μL.
Protein preparation
Plasmids were expressed in BL21-Star(DE3)pLysS E. coli (Invitrogen, Carlsbad, CA). Overnight starter cultures were grown at 30°C and used to inoculate 3 L of growth medium (LB amp) to OD600 = 0.1. Cultures were grown at 30°C until OD600 = 0.6, when they were induced with 400 μM isopropyl-β-D-thiogalactoside (IPTG). After 4 h, the cells were pelleted and frozen at −20°C. Pellets were thawed and sonicated individually in 30 mL sonication buffer (50 mM KPi, pH 7.5, 300 mM NaCl, 0.2% 2-mercaptoethanol). After centrifugation for 30 min at 35,000 × g at 4°C, the supernatant protein was bound by batch absorption to 1 mL bed volume Ni2+-NTA agarose, washed with 30 mL 25 mM imidazole in sonication buffer, and eluted with 300 mM imidazole in sonication buffer. Peak fractions were pooled and desalted using PD-10 columns (Amersham Pharmacia, Piscataway, NJ).
ERK (350 μg/mL) was phosphorylated to stoichiometry with 70 μg/mL MKK1-G7B (ΔN4/S218D/M219D/N221D/S222D; Mansour et al., 1996) in reactions containing phosphorylation buffer (10 mM HEPES, pH 7.5, 100 mM NaCl, 0.01% Triton X-100, 0.1% 2-mercaptoethanol), with 10 mM MgCl2 and 4 mM ATP, run at 30°C for 2 h. Unphosphorylated samples were incubated in parallel without ATP and MKK1-G7B. The ppERK was purified away from MKK1-G7B using Ni2+-NTA agarose as described above, except that the columns were washed with 5 mL phosphorylation buffer before elution. Peak fractions were pooled and desalted.
Protein in 50 mM potassium phosphate, pH 7.5, 50 mM NaCl was purified by Mono Q (HR 5/5) anion exchange fast protein liquid chromatography at 4°C. Protein was loaded at 0.5 mL min−1 in buffer A (50 mM KPi, pH 7.5) and eluted with a 30-min gradient of 0–300 mM KCl in 50 mM KPi, pH 7.5. Peak fractions containing 0.4–1.0 mg ERK2 were pooled and spin-labeled with 40–100 molar excess (0.16–1.3 mM) of (1-oxy-2,2,5,5-tetramethyl-δ3-pyrroline-3-yl)methyl methanethiosulfonate (MTSL; Toronto Research Chemicals, North York, Ont.) for 4 h at room temperature, followed by 8–10 h at 4°C. Protein was then desalted with PD-10 columns into 50 mM NaPi, pH 8.0, 200 mM NaCl, 5 mM MgCl2, 5% glycerol. VivaSpin-4 polyethersulfone 10,000 Da MWCO spin concentrators (Vivascience, Carlsbad, CA) were used to concentrate the protein to 100–200 μM. At these concentrations, ERK and ppERK are dimeric (Khokhlatchev et al., 1998).
Electron paramagnetic resonance spectroscopy
For room temperature solution studies, samples were drawn into 1 mm i.d. capillary tubes (sample height of 1.7–2.5 cm) and sealed with vacuum grease (Dow Corning, Midland, MI). The capillary tubes were then placed into 4 mm i.d. quartz EPR tubes. EPR spectra were acquired with a PC using DOS in-house software (CWSCAN.exe; Quine et al., 1986) controlling a Varian (Palo Alto, CA) E9 X-band spectrometer with a TE102 rectangular resonator containing a 5-mm or 6-mm quartz dewar insert. The temperature of the samples was ∼21°C. Continuous wave spectra were collected at 9.1 GHz, 200 G sweep width, 1024 steps, 180 s sweep time, 100 kHz modulation frequency, 1.0 G modulation amplitude, and 1 mW bridge power. The signal was amplified using a low-noise gallium-arsenide (GaAsFET) amplifier. The quantity of MTSL probe in each sample was determined from the double integral of the first-derivative EPR spectra. The double integral was compared to that of 1 mM MTSL in water, and the percent labeling was determined by dividing the concentration of label by the concentration of protein determined by Bradford assay. In some cases, this percent labeling was corrected for the concentration of free probe, which was determined by subtracting variable fractions of spectra for a known concentration of MTSL. Samples for low temperature spectroscopy were prepared by adding 100 μL glycerol to 100 μL protein solution and freezing in liquid nitrogen in 4 mm i.d. quartz tubes. The spectra were collected similar to above except that the temperature of the sample was held at −170°C with a Varian temperature controller, and the microwave power was 20 μW. The magnetic field was calibrated with DPPH (g = 2.0036).
Liquid chromatography/mass spectrometry (LC/MS)
We used mass spectrometry to evaluate the stoichiometry of ERK2 phosphorylation as described (Resing and Ahn, 1997). Briefly, 10–20 μg of protein was denatured in 6 M urea and incubated at room temperature for 15–20 min. The denatured protein was digested with 1 μg trypsin (100 μL total reaction volume) in trypsin digestion buffer (200 mM Tris, pH 8.0, 1 mM CaCl2, 1.6 M urea) at 37°C for 2 h. The resulting peptides were then reduced with 4 mM dithiothreitol (DTT) for 30 min at 50°C, and alkylated with 15 mM iodoacetamide for 30 min at room temperature. The peptides were separated using a hand-packed capillary reversed-phase column (POROS 20 R1, 15 cm × 500 μm i.d.) and analyzed by mass using an API-III+ triple quadrupole mass spectrometer (PE Sciex, Foster City, CA).
Kinase assays for enzyme activity
Kinase assays were carried out in 25 μL reaction volumes with 0.5–1 μg ERK2 in kinase assay buffer (25 mM Tris, pH 8.0, 100 μM ATP, 0.5–2.0 μCi [γ-32P] ATP, 10 mM MgCl2) with 200 μg/mL myelin basic protein (MBP; Sigma, St. Louis, MO) as substrate. ERK2 was phosphorylated and activated with 0.05 μg MKK1-G7B for 30 min before initiation of the kinase assay with MBP. Reactions were carried out for 10 min at 30°C, quenched with 8 μL 5X Laemmli sample loading buffer, separated by SDS-polyacrylamide gel electrophoresis, and visualized by autoradiography. Specific activities were calculated by normalizing to the amount of phosphorylated ERK2 in each sample (assessed by 32P incorporation into ERK2).
Determination of EPR parameters
Spectra obtained at −170°C in glassy 1:1 buffer/glycerol were simulated using the program MONMER.exe, which is based on the equations in Toy et al. (1971) to determine the principal components of the anisotropic hyperfine interaction matrix (Azz, Axx, and Ayy) and the g-values. These parameters were used in the slow-motion simulations of room temperature spectra.
The signal from free MTSL (present in solution but not attached to cysteines in the protein) was simulated using ASYM.exe software (Amin et al., 2000), and fractions of the calculated spectrum were subtracted from the observed spectra. The resulting corrected spectra were then simulated using a nonlinear least-squares fitting algorithm, NLSL.exe (Budil et al., 1996; Schneider and Freed, 1989), kindly provided by David Budil (Northeastern University). The three components of the diffusion rate tensor, rxx, ryy, and rzz (units of log(sec−1)) were further adjusted to match the spectrum calculated using an anisotropic microscopic-order–macroscopic-disorder model of probe motion (Budil et al., 1996; Columbus et al., 2001; Columbus and Hubbell, 2002) to the experimental data. Individual parameters were then manually adjusted to higher and lower values to estimate uncertainties in parameters and to test the sensitivity of the simulated spectra to varying parameter values. For positions 105–108, the differences in values of Rbar between the phosphorylated and unphosphorylated samples were greater than the uncertainties in the parameters. Differences between the three principal components of R (diffusion rate tensor) up to an order of magnitude signified anisotropic motion of the spin label at some locations. The three components of the tensors were averaged to report the overall rate of diffusion or tumbling (Rbar; Table 1). The size of the cone in which the probe diffuses was modeled with the restoring force of the diffusion tensor, c20. The simulations included separate populations of probe in higher or lower mobility conformations (referred to as site 1 or site 2, respectively). The tumbling correlation time for the lower mobility conformation is comparable to the overall tumbling rate of dimeric ERK2 (22 ns).
TABLE 1.
Parameters from spectral simulation of ERK versus ppERK*
| Position | Phos. state† | Azz (G)‡ | Rbar (log [sec−1])§ | c20¶ | Site 2| (% total area) | Rbar site 2 (log [sec−1]) |
|---|---|---|---|---|---|---|
| 101 | U | 37.0 | 6.97** | 2.1 | ||
| P | 6.97 | |||||
| 105 | U | 37.0 | 7.70 | 1.1 | 37 | 7.25 |
| P | 7.63 | 47 | ||||
| 106 | U | 36.5 | 6.88** | 2.5 | ||
| P | 7.24 | |||||
| 107 | U | 37.0 | 7.75** | 2.5 | 8 | 6.80 |
| P | 7.37 | 36 | ||||
| 108 | U | 37.3 | 7.20 | 1.1 | ||
| P | 7.37 | |||||
| 109 | U | 37.0 | 7.70 | 0.5 | 19 | 7.09 |
| P | 7.70 | 25 | ||||
| 111 | U | 36.5 | 8.00 | 1.9 | ||
| P | 8.00 | |||||
| 112 | U | 37.5 | 7.93 | 2.1 | 5 | 7.08 |
| P | 7.93 | 7 |
The parameters were determined by simulating the slow motional spectra of spin labels at indicated positions in the hinge region of ERK2. In all simulations, gxx = 2.00918, gyy = 2.00635, and gzz = 2.00298; Axx and Ayy were permitted to float between 4.0 and 7.0 G.
Parameters are shown for unphosphorylated (U) and diphosphorylated (P) forms of the kinase.
Azz is the largest component of the anisotropic hyperfine interaction matrix and is determined from spectra of glassy samples at −170°C. The best fits to the room temperature spectra were selected from simulations with Azz values varying between 36.0 and 37.5.
Rbar is the log10 of the diffusion rate constant calculated from the average of the logarithms of the three principal components of the tensor (rxx, ryy, and rzz).
c20 is the restoring force describing a cone within which the spin label diffuses; the higher the restoring force, the smaller the angle of the cone.
Site 2 is the percentage of bound probe in a second, less mobile conformation. Site 2 was simulated with isotropic diffusion and with Azz = 37.0, Axx, Ayy = 4.0–7.0, and c20 = 1.0.
Markedly anisotropic rotational diffusion was seen at three sites. For residue 101, rxx = 7.31, ryy = 6.15, rzz = 7.46 (U,P). For residue 106, rxx = 6.25, ryy = 6.00, rzz = 8.19 (U); and rxx = 6.95, ryy = 6.37, rzz = 8.40 (P). For residue 107, rxx = 8.43, ryy = 7.30, rzz = 7.52 (U); and rxx = 8.30, ryy = 6.30, rzz = 7.50 (P).
RESULTS
Site-directed mutagenesis was used to modify seven endogenous cysteines in ERK2 (Fig. 1). Six native cysteine residues (C38, C63, C125, C159, C164, and C252) were all mutated to serine. Cysteine 214, which is deeply buried in the C-terminal lobe, was changed to alanine due to low solubility of the serine mutant (data not shown). In vitro assays showed that the cysteineless mutant retained 20% of wild-type kinase specific activity toward myelin basic protein.
FIGURE 1.

X-ray crystal structure of phosphorylated ERK2. Conserved features of the kinase catalytic core are present, including the N-terminal lobe, which contacts ATP, and the large C-terminal lobe, which contacts protein substrate (PDB code 2ERK). The activation lip is the site of phosphoThr-183 and phosphoTyr-185 (displayed as ball-and-stick). Phosphorylation at these sites increases the specific activity of ERK2. The backbone positions of seven endogenous cysteines that were removed are shown as black spheres, and the positions of eight hinge residues that were individually engineered to cysteine are shown in gray. N and C refer respectively to the N- and C-termini.
Within the background of cysteineless ERK2, eight cysteine side chains were individually engineered into the hinge region, spanning residues 101–112, which includes β5, αD, and the intervening loop. The mutants were phosphorylated in vitro with active MKK1, yielding products that were >80% diphosphorylated at Thr-183 and Tyr-185, and the rest monophosphorylated, as determined by LC/MS. Phosphorylated and unphosphorylated ERK2 were then covalently coupled to the MTSL spin label. The specific activity of each engineered cysteine mutant, before and after MTSL coupling, was the same as that of the cysteineless mutant, within error uncertainty. Because labeling does not change enzymatic activity, it is unlikely that modification alters the protein structure. Therefore, differences between phosphorylated and unphosphorylated forms of the spin-labeled proteins can be interpreted as molecular responses of ERK2 to activation. Furthermore, the level of ERK2 activation due to phosphorylation (i.e., the ratio of phosphorylated to unphosphorylated enzyme specific activity) was 1000-fold, which is identical to that of wild-type enzyme (data not shown). Overall, ERK2 provided a stable system in which to use SDSL-EPR to measure changes in flexibility due to phosphorylation.
Continuous wave room-temperature EPR spectra of the spin-labeled mutants were acquired at X-band (9.1 GHz). The spin-label g-values were approximately the same for all of the samples. The anisotropic hyperfine interaction depends strongly on the polarity of the local environment of the probe. To measure A-values, samples were diluted with equal volumes of glycerol and frozen in liquid nitrogen, producing glassy samples in which motions of the probe were attenuated. Simulation of spectra of the glassy samples provided values of Azz, the largest component of the A-matrix (Table 1). Azz was identical, within error, between phosphorylated and unphosphorylated forms of each mutant, indicating that the polarity of the local environment near the spin probe was unperturbed by phosphorylation. This finding is consistent with x-ray information, which shows complete structural overlap between ERK and ppERK in the hinge region.
The method of protein preparation yielded samples contaminated by probe that had not reacted with protein cysteine sulfhydryls, which is designated as “free probe.” Most of the free probe had dimerized via disulfide bond cross-linking, yielding a spectrum with five lines and alternating linewidths. The free probe contributed 0–30% of the total EPR signal for each sample. Room temperature spectra were corrected by subtracting the simulated 5-line spectrum of free probe (Fig. 2). Room-temperature spectra corrected for free probe were examined for effects of ERK2 phosphorylation. Visual inspection revealed significant and reproducible differences in the separation between the outermost peaks, or extrema, which is sensitive to changes in motion (Fig. 3). For simplicity in the qualitative descriptions, we use the distance between the downfield (left) and central peaks as a gauge of mobility.
FIGURE 2.

Correction of spectra for free probe. The room-temperature spectra of unphosphorylated ERK2, modified at position 106 with MTSL spin label, are shown as an example. (A) The uncorrected spectrum of labeled side chain 106 includes signals assigned to free probe, with alternating sharp (*) and broad (**) lines as expected for a dimer with fluctuating electron-electron interaction. The simulated spectrum shown in B was subtracted from the spectrum in A, to obtain the corrected spectrum shown in C.
FIGURE 3.

EPR spectra of spin-labeled ERK2. Spectra corrected for free probe are shown, corresponding to sites labeled at positions (A) 101, (B) 105, (C) 106, (D) 107, (E) 108, (F) 109, (G) 111, and (H) 112 of unphosphorylated (black) and diphosphorylated (red) ERK2. Spectra obtained from independent protein preparations and labeling reactions are shown to illustrate the reproducibility of the experiment.
A more detailed analysis of the spectra was achieved using a simulation algorithm based on the microscopic-order–macroscopic-disorder model described in Budil et al. (1996) (see also Columbus et al., 2001). In this model, motion of the spin-label probe is described in terms of a restoring force (c20) that defines a cone within which the probe diffuses. A larger restoring force means that the probe explores less space in a given amount of time. The rate at which the probe explores this space is defined by the diffusion rate tensor R, from which a correlation time for tumbling of the probe can be calculated 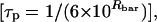 where Rbar is the average of the three components of the diffusion rate tensor. The rotational diffusion of the probe may be as slow as the overall tumbling of the protein molecule, or faster due to rotations around bonds in the side chain or motion of local backbone structure. The spectral simulations of unphosphorylated and phosphorylated pairs of ERK2 mutants confirmed qualitative conclusions based on visual inspection (Figs. 3 and 4; Table 1).
where Rbar is the average of the three components of the diffusion rate tensor. The rotational diffusion of the probe may be as slow as the overall tumbling of the protein molecule, or faster due to rotations around bonds in the side chain or motion of local backbone structure. The spectral simulations of unphosphorylated and phosphorylated pairs of ERK2 mutants confirmed qualitative conclusions based on visual inspection (Figs. 3 and 4; Table 1).
FIGURE 4.

Simulation of EPR spectra. Observed spectra of spin-labeled cysteine 107 from unphosphorylated (left) and diphosphorylated (right) ERK2 are shown in black. The simulated spectra, shown in gray, were derived using the NLSL algorithm of Budil et al. (1996), which accounts for the rate and extent of rotational diffusion of the probe. In this example, two different conformations of probe are present, one less mobile than the other. The proportion of the less mobile conformation (site 2) increases in the phosphorylated state. The arrows mark the most distinctive region of the spectrum for site 2.
For variants 106 and 108, phosphorylation of ERK2 caused the peak separation to decrease, which is indicative of increasing motion (Fig. 3, C and E). This qualitative judgment was confirmed by the simulations, which showed that the rates of rotational diffusion for probes at positions 106 and 108 increased by 2.3- and 1.5-fold, respectively, upon phosphorylation. The spin label on cysteine 106 moved anisotropically (rxx = 6.25, ryy = 6.00, rzz = 8.19; Table 1), probably because it is lying in a cleft on the surface of the protein (Figs. 1 and 5).
FIGURE 5.

Enlarged view of the ERK2 hinge region. (A) The two lobes of ERK2 are joined by the hinge, which is shown from a perspective similar to Fig. 1. Leu-105 and Glu-107 interact with surface features in conserved strands β3 and β7, whereas Thr-108 has more freedom to move above the surface. (B) A different perspective, rotated ∼90° counterclockwise as viewed from the top of the molecule in A, provides a view of the Met-106 side chain, which is inserted into a surface cleft, explaining the anisotropic motion of the spin-label probe at this position. The figure shows the native side chains, which were replaced by spin-labeled cysteine for the EPR experiments.
Peak separations increased upon ERK2 phosphorylation at positions 105 and 107, indicative of decreased probe mobility (Fig. 3, B and D). Correspondingly, diffusion rates decreased upon phosphorylation by 1.2-fold and 2.4-fold for variants 105 and 107, respectively. In addition, variants 105, 107, and to a smaller extent, 109, after phosphorylation, all had increased proportions of probe in a site-2 conformation with lower mobility (Fig. 3, B, D, and F; Table 1), which likely reflects shifts toward more crowded side-chain environments. The equilibrium constants describing the increase in the population of site 2 indicate energy differences of a few kJ/mole.
In contrast to the changes seen at positions 105–109, diphosphorylation of ERK2 induced no changes in spin-label mobility at positions 101, 111, and 112 in the hinge region (Fig. 3, A, G, and H; Table 1). In addition, cysteine residues engineered into the glycine-rich ATP binding loop within the cysteineless ERK2 mutant were compared by SDSL-EPR. The spectra revealed little or no change in mobility at eight sites in this region (T24, N25, S27, Y28, I29, A33, Y34, V37; data not shown). Thus the spectral changes observed in the hinge region report localized alterations in motions that are not observed in other regions of the molecule.
Finally, the average correlation time 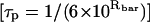 for probe in the least mobile (site 2) conformation is ∼26 ns, in excellent agreement with the correlation time (∼22 ns) predicted by the generalized Stokes-Einstein equation (Wand et al., 1998). Thus, the lower limit of probe motion is dominated by the tumbling of the protein, precluding assessment of the loss in side-chain mobility below this value.
for probe in the least mobile (site 2) conformation is ∼26 ns, in excellent agreement with the correlation time (∼22 ns) predicted by the generalized Stokes-Einstein equation (Wand et al., 1998). Thus, the lower limit of probe motion is dominated by the tumbling of the protein, precluding assessment of the loss in side-chain mobility below this value.
DISCUSSION
This study demonstrates the use of SDSL-EPR to assess the effects of posttranslational modifications on protein dynamics. The results show that upon phosphorylation and activation, ERK2 undergoes small changes within the hinge, a region of the kinase that covalently joins the N-terminal and C-terminal lobes, providing a pivot point for the closure of the active site cleft, and which makes important hydrogen bonding interactions with nucleotide (Narayana et al., 1997a; Zhang et al., 1994). We find no evidence for large changes in side-chain motions throughout the hinge after enzyme activation. However, small but reproducible changes in side-chain mobilities on the nanosecond timescale are observed at specific residues. Spin labels attached to residues 106 and 108 move more rapidly after phosphorylation. On the other hand, spin labels on 105 and 107 move more slowly, and in the company of the probe attached to residue 109, spend a greater amount of time in a more restrained conformation. These changes in dynamics upon activation may reflect changes in tertiary contacts between side chains within the interdomain region.
The increased proportion of the site-2 conformation at residues 105 and 107 indicates significantly more time spent in a less mobile conformation upon phosphorylation. Such changes may be explained by the proximity of the side chains to nearby surface features as shown in Fig. 5, A and B. We propose that the less mobile conformation may correspond to the spin label interacting with conserved strands β3 and β7. It seems possible that if increased flexibility in the hinge postulated from HX-MS measurements facilitates interdomain closure in the phosphorylated molecule, then β3 and β7 would, on average, move closer to one another, crowding spin labels 105 and 107 and causing them to interact with β3 and β7 to a greater extent. Therefore, changes in the slower site-2 population are consistent with a hypothesis for more favorable interdomain closure after phosphorylation, occurring on a slower timescale than measurable in the EPR experiment.
The observed effects on Rbar in the hinge region are more readily ascribed to changes in side-chain mobility, rather than major conformational change, because x-ray crystallography shows complete overlay between static structures of phosphorylated and unphosphorylated forms. This interpretation is consistent with the observation that at each residue monitored, within experimental uncertainty, the hyperfine component Azz and the restoring force (c20) were unchanged by phosphorylation.
It was of interest to integrate the results from SDSL-EPR with measurements previously made by HX-MS, to conceptualize perturbations in the hinge region upon enzyme activation. HX-MS measures the accessibility of backbone amide hydrogens to solvent deuterium, which is determined by protein flexibility as well as conformation (reviewed by Hoofnagle et al., 2003). HX-MS studies with ERK2 indicated decreased backbone flexibility within residues 102–105, which was attributed to residues 104 and/or 105, and increased flexibility within residues 108–109, most likely occurring at residue 108 (Hoofnagle et al., 2001). These changes were attributed to regulated protein flexibility at the hinge, based on comparison with x-ray data, which showed no difference in average structure between unphosphorylated and phosphorylated forms.
HX-MS and SDSL-EPR do not necessarily report similar motions, because EPR monitors labels attached to side chains, whereas HX-MS measures chemical exchange rates from backbone atoms, and the methods report nonoverlapping timescales of side-chain (ns) and backbone (ms, or microsecond) motions. Spin labels attached to surface sites on helices can reflect backbone motions (Langen et al., 2000; Columbus et al., 2001; Columbus and Hubbell, 2002); however, the relationship between side-chain and backbone motion may be more complicated for buried side chains. Interestingly, the decreased backbone flexibility reported by HX-MS within residues 104 and/or 105, and the increased flexibility at residue 108, correlate with results from EPR in which Rbar of spin-label probes at positions 105 and 108 decrease and increase, respectively. The situation at residues 106 and 107 is more complicated. EPR shows increased side-chain motion at 106 and decreased motion at 107, whereas no change was observed by HX-MS. One possibility is that the HX-MS experiment reported only the average of a small increase and a small decrease at the two residues. Alternatively, the side-chain motions at these sites may be dominated by interactions that do not have a significant impact on backbone motion. Conceivably, both the EPR and HX-MS measurements may report changes in motions between domains in this region upon enzyme activation, which cause changes in side-chain contacts as well as backbone mobility.
In conclusion, SDSL-EPR measurements provide additional characterization of the effects of phosphorylation on residues within the hinge, demonstrating small but significant changes in side-chain mobility in this region, located far from the site of phosphorylation and major conformational change. These occur against a background of slower (ms) backbone mobility changes observed by HX-MS. The measurements complement conclusions from HX-MS, that motional information responsive to posttranslational modifications can be communicated over long distances in protein kinases, from the activation lip to the hinge of the molecule. The data provide insight complementary to x-ray structural information, by suggesting that the population of conformers in solution is enriched for a closed state necessary for catalysis (Cox et al., 1994; Narayana et al., 1997b). Thus the combination of HX-MS and EPR provides insight not easily achieved by other methods, in dissecting regulated dynamics in a large multidomain protein.
Acknowledgments
We are indebted to Arthur Pardi, Deborah Wuttke, Joseph Falke, Gareth Eaton, and David Budil for innumerable helpful discussions and advice. We also thank David Budil, who provided software for the slow motional simulation of room-temperature samples, and John Hardin, who prepared several of the mutant plasmids used in this work.
This work was supported by National Institutes of Health grants GM48521 (N.G.A.) and GM21156 (S.S.E.).
References
- Alonso, A., W. P. dos Santos, S. J. Leonor, J. G. dos Santos, and M. Tabak. 2001. Stratum corneum protein dynamics as evaluated by a spin-label maleimide derivative: Effect of urea. Biophys. J. 81:3566–3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin, S. S., K. Cryer, B. Y. Zhang, S. K. Dutta, S. S. Eaton, O. P. Anderson, S. M. Miller, B. A. Reul, S. M. Brichard, and D. C. Crans. 2000. Chemistry and insulin-mimetic properties of bis(acetylacetonate)oxovanadium(IV) and derivatives. Inorg. Chem. 39:406–416. [DOI] [PubMed] [Google Scholar]
- Anand, G. S., C. A. Hughes, J. M. Jones, S. S. Taylor, and E. A. Komives. 2002. Amide H/H-2 exchange reveals communication between the cAMP and catalytic subunit-binding sites in the R-I alpha subunit of protein kinase A. J. Mol. Biol. 323:377–386. [DOI] [PubMed] [Google Scholar]
- Barnakov, A., C. Altenbach, L. Barnakova, W. L. Hubbell, and G. L. Hazelbauer. 2002. Site-directed spin labeling of a bacterial chemoreceptor reveals a dynamic, loosely packed transmembrane domain. Protein Sci. 11:1472–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes, J. P., Z. C. Liang, H. S. McHaourab, J. H. Freed, and W. L. Hubbell. 1999. A multifrequency electron spin resonance study of T4 lysozyme dynamics. Biophys. J. 76:3298–3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budil, D. E., S. Lee, S. Saxena, and J. H. Freed. 1996. Nonlinear-least-squares analysis of slow-motion EPR spectra in one and two dimensions using a modified Levenberg-Marquardt algorithm. J. Magn. Reson. 120:155–189. [Google Scholar]
- Canagarajah, B. J., A. Khokhlatchev, M. H. Cobb, and E. J. Goldsmith. 1997. Activation mechanism of the MAP kinase ERK2 by dual phosphorylation. Cell. 90:859–869. [DOI] [PubMed] [Google Scholar]
- Columbus, L., and W. L. Hubbell. 2002. A new spin on protein dynamics. Trends Biochem. Sci. 27:288–295. [DOI] [PubMed] [Google Scholar]
- Columbus, L., T. Kalai, J. Jeko, K. Hideg, and W. L. Hubbell. 2001. Molecular motion of spin labeled side chains in alpha-helices: analysis by variation of side chain structure. Biochemistry. 40:3828–3846. [DOI] [PubMed] [Google Scholar]
- Cox, S., E. Radzio-Andzelm, and S. S. Taylor. 1994. Domain movements in protein kinases. Curr. Opin. Struct. Biol. 4:893–901. [DOI] [PubMed] [Google Scholar]
- Earle, K. A., D. E. Budil, and J. H. Freed. 1993. 250-GHz EPR of nitroxides in the slow-motional regime: models of rotational diffusion. J. Phys. Chem. 97:13289–13297. [Google Scholar]
- Emrick, M. A., A. N. Hoofnagle, A. S. Miller, L. F. Ten Eyck, and N. G. Ahn. 2001. Constitutive activation of extracellular signal-regulated kinase 2 by synergistic point mutations. J. Biol. Chem. 276:46469–46479. [DOI] [PubMed] [Google Scholar]
- Fox, T., J. T. Coll, X. L. Xie, P. J. Ford, U. A. Germann, M. D. Porter, S. Pazhanisamy, M. A. Fleming, V. Galullo, M. S. S. Su, and K. P. Wilson. 1998. A single amino acid substitution makes ERK2 susceptible to pyridinyl imidazole inhibitors of p38 MAP kinase. Protein Sci. 7:2249–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasgow, B. J., O. K. Gasymov, A. R. Abduragimov, T. N. Yusifov, C. Altenbach, and W. L. Hubbell. 1999. Side chain mobility and ligand interactions of the G strand of tear lipocalins by site-directed spin labeling. Biochemistry. 38:13707–13716. [DOI] [PubMed] [Google Scholar]
- Hoofnagle, A. N., K. A. Resing, and N. G. Ahn. 2003. Protein analysis by hydrogen exchange mass spectrometry. Annu. Rev. Biophys. Biomol. Struct. 32:1–25. [DOI] [PubMed] [Google Scholar]
- Hoofnagle, A. N., K. A. Resing, E. J. Goldsmith, and N. G. Ahn. 2001. Changes in protein conformational mobility upon activation of extracellular regulated protein kinase-2 as detected by hydrogen exchange. Proc. Natl. Acad. Sci. USA. 98:956–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, S. H., M. W. Parker, J. Y. Lei, M. C. J. Wilce, G. M. Benian, and B. E. Kemp. 1994. Insights into autoregulation from the crystal-structure of twitchin kinase. Nature. 369:581–584. [DOI] [PubMed] [Google Scholar]
- Hubbell, W. L., D. S. Cafiso, and C. Altenbach. 2000. Identifying conformational changes with site-directed spin labeling. Nat. Struct. Biol. 7:735–739. [DOI] [PubMed] [Google Scholar]
- Hubbell, W. L., H. S. McHaourab, C. Altenbach, and M. A. Lietzow. 1996. Watching proteins move using site-directed spin labeling. Structure. 4:779–783. [DOI] [PubMed] [Google Scholar]
- Johnson, L. N., M. E. M. Noble, and D. J. Owen. 1996. Active and inactive protein kinases: structural basis for regulation. Cell. 85:149–158. [DOI] [PubMed] [Google Scholar]
- Khokhlatchev, A. V., B. Canagarajah, J. Wilsbacher, M. Robinson, M. Atkinson, E. Goldsmith, and M. H. Cobb. 1998. Phosphorylation of the MAP kinase ERK2 promotes its homodimerization and nuclear translocation. Cell. 93:605–615. [DOI] [PubMed] [Google Scholar]
- Knighton, D. R., J. H. Zheng, L. F. Teneyck, N. H. Xuong, S. S. Taylor, and J. M. Sowadski. 1991. Structure of a peptide inhibitor bound to the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science. 253:414–420. [DOI] [PubMed] [Google Scholar]
- Langen, R., K. J. Oh, D. Cascio, and W. L. Hubbell. 2000. Crystal structures of spin labeled T4 lysozyme mutants: implications for the interpretation of EPR spectra in terms of structure. Biochemistry. 39:8396–8405. [DOI] [PubMed] [Google Scholar]
- Li, F., M. Gangal, C. Juliano, E. Gorfain, S. S. Taylor, and D. A. Johnson. 2002. Evidence for an internal entropy contribution to phosphoryl transfer: a study of domain closure, backbone flexibility, and the catalytic cycle of cAMP-dependent protein kinase. J. Mol. Biol. 315:459–469. [DOI] [PubMed] [Google Scholar]
- Liu, Y. S., P. Sompornpisut, and E. Perozo. 2001. Structure of the KcsA channel intracellular gate in the open state. Nat. Struct. Biol. 8:883–887. [DOI] [PubMed] [Google Scholar]
- Madhusudan, P. Akamine, N. H. Xuong, and S. S. Taylor. 2002. Crystal structure of a transition state mimic of the catalytic subunit of cAMP-dependent protein kinase. Nat. Struct. Biol. 9:273–277. [DOI] [PubMed] [Google Scholar]
- Mansour, S. J., J. M. Candia, J. E. Matsuura, M. C. Manning, and N. G. Ahn. 1996. Interdependent domains controlling the enzymatic activity of mitogen-activated protein kinase kinase 1. Biochemistry. 35:15529–15536. [DOI] [PubMed] [Google Scholar]
- Mchaourab, H. S., and E. Perozo. 2000. Determination of protein folds and conformational dynamics using spin-labeling EPR spectroscopy. Biol. Magn. Reson. 19:185–247. [Google Scholar]
- Narayana, N., S. Cox, N. H. Xuong, L. F. TenEyck, and S. S. Taylor. 1997a. A binary complex of the catalytic subunit of cAMP-dependent protein kinase and adenosine further defines conformational flexibility. Structure. 5:921–935. [DOI] [PubMed] [Google Scholar]
- Narayana, N., S. Cox, S. Shaltiel, S. S. Taylor, and N. H. Xuong. 1997b. Crystal structure of a polyhistidine-tagged recombinant catalytic subunit of cAMP-dependent protein kinase complexed with the peptide inhibitor PKI(5–24) and adenosine. Biochemistry. 36:4438–4448. [DOI] [PubMed] [Google Scholar]
- Olah, G. A., R. D. Mitchell, T. R. Sosnick, D. A. Walsh, and J. Trewhella. 1993. Solution structure of the cAMP-dependent protein kinase catalytic subunit and its contraction upon binding the protein kinase inhibitor peptide. Biochemistry. 32:3649–3657. [DOI] [PubMed] [Google Scholar]
- Pearson, G., F. Robinson, T. B. Gibson, B. E. Xu, M. Karandikar, K. Berman, and M. H. Cobb. 2001. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr. Rev. 22:153–183. [DOI] [PubMed] [Google Scholar]
- Prowse, C. N., and J. Lew. 2001. Mechanism of activation of ERK2 by dual phosphorylation. J. Biol. Chem. 276:99–103. [DOI] [PubMed] [Google Scholar]
- Quine, R. W., G. R. Eaton, and S. S. Eaton. 1986. Versatile computer interface for a Varian E9 EPR spectrometer. J. Magn. Reson. 66:164–167. [Google Scholar]
- Resing, K. A., and N. G. Ahn. 1997. Protein phosphorylation analysis by electrospray ionization mass spectrometry. Methods Enzymol. 283:29–44. [DOI] [PubMed] [Google Scholar]
- Resing, K. A., and N. G. Ahn. 1998. Deuterium exchange mass spectrometry as a probe of protein kinase activation. Analysis of wild-type and constitutively active mutants of MAP kinase kinase-1. Biochemistry. 37:463–475. [DOI] [PubMed] [Google Scholar]
- Resing, K. A., A. N. Hoofnagle, and N. G. Ahn. 1999. Modeling deuterium exchange behavior of ERK2 using pepsin mapping to probe secondary structure. J. Am. Soc. Mass Spectrom. 10:685–702. [DOI] [PubMed] [Google Scholar]
- Robbins, D. J., E. Z. Zhen, H. Owaki, C. A. Vanderbilt, D. Ebert, T. D. Geppert, and M. H. Cobb. 1993. Regulation and properties of extracellular signal-regulated protein kinases 1 and 2 in vitro. J. Biol. Chem. 268:5097–5106. [PubMed] [Google Scholar]
- Schneider, D. J., and J. H. Freed. 1989. Calculating slow motional magnetic resonance spectra. Biol. Magn. Reson. 8:1–76. [Google Scholar]
- Toy, A. D., S. H. H. Chaston, J. R. Pilbrow, and T. D. Smith. 1971. Electron spin resonance study of copper(II) chelates of certain monothio-beta-diketones and diethyldithiocarbamate. Inorg. Chem. 10:2219–2225. [Google Scholar]
- Tsigelny, I., J. P. Greenberg, S. Cox, W. L. Nichols, S. S. Taylor, and L. F. Ten Eyck. 1999. 600 ps molecular dynamics reveals stable substructures and flexible hinge points in cAMP dependent protein kinase. Biopolymers. 50:513–524. [DOI] [PubMed] [Google Scholar]
- Victor, K. G., and D. S. Cafiso. 2001. Location and dynamics of basic peptides at the membrane interface: electron paramagnetic resonance spectroscopy of tetramethyl-piperidine-N-oxyl-4-amino-4-carboxylic acid-labeled peptides. Biophys. J. 81:2241–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wand, A. J., M. R. Ehrhardt, and P. F. Flynn. 1998. High-resolution NMR of encapsulated proteins dissolved in low-viscosity fluids. Proc. Natl. Acad. Sci. USA. 95:15299–15302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson, K. P., M. J. Fitzgibbon, P. N. Caron, J. P. Griffith, W. Y. Chen, P. G. McCaffrey, S. P. Chambers, and M. S. S. Su. 1996. Crystal structure of p38 mitogen-activated protein kinase. J. Biol. Chem. 271:27696–27700. [DOI] [PubMed] [Google Scholar]
- Young, M. A., S. Gonfloni, G. Superti-Furga, B. Roux, and J. Kuriyan. 2001. Dynamic coupling between the SH2 and SH3 domains of c-Src and Hck underlies their inactivation by C-terminal tyrosine phosphorylation. Cell. 105:115–126. [DOI] [PubMed] [Google Scholar]
- Zhang, F. M., A. Strand, D. Robbins, M. H. Cobb, and E. J. Goldsmith. 1994. Atomic structure of the MAP kinase ERK2 at 2.3 A resolution. Nature. 367:704–711. [DOI] [PubMed] [Google Scholar]