

Abstract
This report provides a detailed analysis of developmental changes in cytoplasmic free calcium (Ca2+) buffering and excitation-contraction coupling in embryonic chick ventricular myocytes. The peak magnitude of field-stimulated Ca2+ transients declined by 41% between embryonic day (ED) 5 and 15, with most of the decline occurring between ED5 and 11. This was due primarily to a decrease in Ca2+ currents. Sarcoplasmic reticulum (SR) Ca2+ content increased 14-fold from ED5 to 15. Ca2+ transients in voltage-clamped myocytes after blockade of SR function permitted computation of the fast Ca buffer power of the cytosol as expressed as generalized values of Bmax and KD. Bmax rose with development whereas KD did not change significantly. The computed SR Ca2+ contribution to the Ca2+ transient and gain factor for Ca2+-induced Ca2+ release increased markedly between ED5 and 11 and slightly thereafter. These results paralleled the maturation of SR and peripheral couplings reported by others and demonstrated a strong relationship between structure and function in development of excitation-contraction coupling. Modeling of buffer power from estimates of the major cytosolic Ca binding moieties yielded a Bmax and KD in reasonable agreement with experiment. From ED5 to 15, troponin C was the major Ca2+ binding moiety, followed by SR and calmodulin.
INTRODUCTION
Contraction of the myocardium is initiated by depolarization of the membrane, followed by net entry of Ca2+ through voltage-gated Ca2+ channels. In the adult heart, this Ca2+ causes a further release of Ca2+ from the sarcoplasmic reticulum (SR) via a mechanism termed Ca2+-induced Ca2+ release (CICR). The subsequent rise in intracellular Ca2+ causes cyclic interactions between the contractile proteins myosin and actin, which give rise to force production and shortening of the muscle cell. Relaxation of the cell is caused primarily by a decrease in the intracellular level of Ca2+ due to active uptake of Ca2+ back into the SR and extrusion via Na+/Ca2+ exchange. The processes linking depolarization of the membrane and the ensuing rise and fall in intracellular Ca2+ is termed excitation-contraction (EC) coupling (reviewed in Bers, 2001).
In mammals, it is often considered that SR content is sparse at birth and undergoes most of its development postnatally (Nakanishi and Jarmakani, 1984). This appears to be true in rabbit and rat hearts (Seguchi et al., 1986a; Nakanishi et al., 1992) but not true for guinea pigs, which have a fully developed SR at birth (Goldstein and Traeger, 1985). Thus, there is variation among mammalian species with respect to the degree of maturity of SR at birth. In the chick, SR morphology and function is well developed by hatching. Presumably because of technical difficulties, there are currently no published reports of detailed analysis of SR function and CICR in mammalian embryonic heart development.
The chick heart has long been the standard for the study of developmental physiology and morphology and remains the best described of any vertebrate model of development. The normal physiology of the developing chick heart has long been characterized (Sperelakis, 1982) and there is information available on altered cardiac function in a chick model of human heart disease (Creazzo et al., 1998). Moreover, a comprehensive study of the morphology of the formation and maturation of SR junctional complexes is available only for the chick embryo (Protasi et al., 1996).
CICR in chick develops early and is present well before hatching, probably by embryonic day (ED) 4–5, as ryanodine receptors are first detected at about this time (Dutro et al., 1993). The chick heart begins beating at ∼ED1.5 (Sperelakis, 1982). As the myocardium develops, SR and contractile proteins proliferate and myofibrils are formed. After ED5, the myofibrils are abundant and are aligned parallel to one another in the cell (Manasek, 1970). As the content of myofibrils increases, the force of contraction increases apace (Godt et al., 1991). In the early chick heart, relaxation is caused primarily by removal of Ca2+ from the cell by Na+/Ca2+ exchange proteins in the sarcolemma. With development, the SR begins to play a larger role in control of intracellular Ca2+, such that from around the time of hatching (ED22) and beyond, Na+/Ca2+ exchange is subsidiary to the SR (Vetter et al., 1986).
Recently, the elegant work of Franzini-Armstrong, Protasi, and colleagues has provided detailed information on the development of SR-plasma membrane junctional complexes involved in EC coupling in the chick heart (Sun et al., 1995; Protasi et al., 1996). In these studies, complete junctional complexes are described as having a junctional gap, which is fully zippered by closely spaced feet (RyRs). Dihydropyridine receptors (DHPRs) are clustered in the plasma membrane in close proximity to the RyRs at junctional complexes but as in adult hearts, lack the ordered arrays seen in skeletal muscle. The junctions are confined to the peripheral surface membrane as avian heart, like mammalian embryonic heart, lacks t-tubules, and extended junctional SR and corbular SR do not begin to appear until after hatching (Jewett et al., 1973; Sommer, 1995; Junker et al., 1994). Complete junctions or peripheral couplings are essentially absent at ED4 but increase rapidly and are nearly at the adult level by ED11. From these earlier findings, it is expected that both the efficiency or gain factor for CICR and the overall SR contribution to the Ca2+ transient will increase with development, and parallel the development of SR junctional complexes.
An important factor in considering the development of Ca2+ handling mechanisms in cardiac muscle is the concomitant developmental change in the rapid Ca2+ buffering capacity of the cytoplasm. In adult myocardium, cytoplasmic buffering capacity is quite robust. For every 80–100 Ca2+ entering during an action potential, only one Ca2+ can be detected with a fluorescent indicator. The rest are bound by rapid Ca2+ binding moieties in the cytoplasm (Berlin et al., 1994). The more prevalent Ca2+ binding moieties include troponin C (TnC), the SR, and negatively charged membranous structures (Bers, 2001). These and other Ca2+ binding moieties are expected to increase with development as the contractile elements increase and become more compact and the myocytes mature. Therefore the impact of “fast” Ca2+ buffers on cytosolic Ca2+ transients is expected to increase with development.
In this report we find that Ca2+ transients and Ca2+ currents decrease whereas the SR Ca2+ content and the Ca2+ binding capacity of fast Ca2+ buffers in the cytosol increases with development. Whereas TnC binds >50% of the entering Ca2+ throughout development, the proportion of Ca2+ bound by various other moieties during a Ca2+ transient change with development and largely reflects the increasing SR component. The Ca2+ buffering capacity (expressed as Bmax and KD) along with physiological measurements are used to calculate a gain factor for CICR. As expected, the gain increases with development. The relative increase in SR, as indicated by the increase in gain, paralleled closely the morphological maturation of SR/sarcolemmal junctional complexes previously reported in the elegant study by Protasi et al. (1996; see their Fig. 4). Our results indicate a strong relationship between structure and function in the development of cardiac EC coupling.
FIGURE 4.

Example of Ca2+ transients used to calculate buffer power. SR function was blocked with 1 μM thapsigargin and 100 μM ryanodine. Shown are Ca2+ transients from five consecutive 200-ms voltage-clamp pulses to +10 mV from a holding potential of −80 mV at 5-s intervals. In some instances the Ca2+ channel agonist Bay k 8644 was added to increase the size of the Ca2+ current as in the examples for ED11 and 15 shown here. These data were used to calculate [Ca2+]m as described in the text and illustrated in Fig. 5.
METHODS
Myocyte preparation
The culture method used in this study was as originally established by R. L. DeHaan ∼20 years ago specifically for the study of Ca2+ and Na+ currents in chick heart development (personal communication; Kawano and DeHaan, 1991; Fujii et al., 1988). After decapitation of the embryos, the hearts from one to five embryos were rapidly removed, trimmed of the atria and great vessels, and the ventricles (left and right) were cleaned of connective tissue and dissociated with brief, repeated exposures to collagenase and DNAase at 37°C as previous described (Brotto and Creazzo, 1996). The cultures were enriched for myocytes by 20-min incubations in tissue-culture flasks before plating the cells. During these short incubations other cell types attach to the flasks whereas myocytes do not. The myocytes were sparsely plated (∼5 × 105 cells/35-mm culture dish) in DeHaan's 21212 medium with 1.8 mM CaCl2, onto plastic petri dishes (Falcon, 1008, Becton Dickenson, San Jose, CA) containing a glass coverslip (25-mm diameter and 0.16-mm thickness). The cultures were incubated in a humidified 5% CO2-95% air atmosphere at 37°C. The myocytes were cultured overnight and used within 24 h of enzymatic dissociation to allow for attachment to glass coverslips and to recover from the cell isolation procedure. EC coupling does not appear to be measurably affected by enzymatic dispersion and overnight culture. This is evident when comparing reports of SR contributions to electrically stimulated Ca2+ transients in isolated myocytes (Brotto and Creazzo, 1996; Creazzo et al., 1995) with Ca2+ transients elicited in freshly dissected intact cardiac trabeculae (Nosek et al., 1997), which shows that the Ca2+ transients from isolated myocytes are virtually identical to those obtained in intact trabeculae whether or not SR function is blocked with ryanodine. Moreover, a high level of colocalization of RyRs and DHPRs is maintained even after enzymatic dissociation and overnight culture in which the embryonic myocytes, unlike adult myocytes, have typically lost their elongated appearance and have assumed a spherical shape (Creazzo et al., 2001). Thus, junctional complexes are quite stable and not disrupted under the conditions used in this study. All experiments were carried out at room temperature (22–24°C).
Calcium transient measurements
Myocytes were loaded with fura-2 AM and calibrations were carried out as previously determined in our laboratory and described in detail elsewhere (Brotto and Creazzo, 1996). Within 24 h of cell isolation, the coverslips were transferred to a perfusion chamber (Warner Instruments, Hamden, CT), with maximum volume of 1 ml, and the cells were gently washed five times with a Ringer solution containing: 142.5 mM NaCl, 4.0 mM KCl, 1.8 mM MgCl2, 1.8 mM CaCl2, 5.0 mM HEPES (n-2-hydroxyethylpiperazine-n′-2-ethanesulfonic acid), and 10.0 mM dextrose, adjusted to pH 7.4 with NaOH, at room temperature. After washing the myocytes were incubated in 1 ml of Ringer solution with 1.5–2.0 μM fura-2 AM for 8 min at 37°C in a rotating water bath. The myocytes were subsequently washed five times with the Ringer solution without the dye and allowed to stand at room temperature for 30 min to facilitate the de-esterification of the dye and rinsed again before use. The chamber was subsequently placed on the stage of a Olympus IX-70 inverted microscope (Olympus America, Inc., Melville, NY). We have found that after exposure of myocytes loaded with fura-2 to saponin (a skinning agent that selectively permeabilizes the sarcolemmal membrane), the remaining fluorescence counts were not significantly different from the background autofluorescent counts before loading. These results indicate that subcellular compartmentalization of fura-2 is not detectable when using our standardized fura-2 AM loading conditions. A DeltaScan microspectrofluorometer with dual monochrometers (Photon Technology International, Inc., Lawrenceville, NJ) was used to collect the fura-2 calcium transients (340:380-nm ratios).
Electrical field stimulation
Electrical field stimulation at 0.2Hz was accomplished by applying 5 V for 5–7 ms via a pulse stimulator (Model II, Hewlett-Packard Co., Palo Alto, CA) through two platinum electrodes placed on either side of a single myocyte. The stimulator was triggered by a digital timer (Winston Electronics, San Francisco, CA).
Patch-clamp
The myocytes loaded with fura-2 were voltage-clamped, using the perforated patch-clamp technique as previously described (Brotto and Creazzo, 1996). Before use the culture medium was replaced with extracellular solution comprised of 1.8 mM CaCl2, 20 mM CsCl, 120 mM TEA-Cl, 1.8 mM MgCl2, 10 mM HEPES, 3 μM TTX (∼500 × KI for the chick cardiac Na channel), and 5 mM dextrose, adjusted to pH 7.4 with NaOH. The perforated patch solution was comprised of 55 mM KCl, 70 mM Cs2SO4, 7 mM MgCl2, 10 mM HEPES, and 5 mM dextrose (pH 7.3, KOH). Nystatin was added to this solution from a 50 mg/ml stock in DMSO (1:500 dilutions). The high concentrations of Cs and TEA were used to limit interference from K+ currents.
For buffer power determinations Ca2+ transients were stimulated by activating Ca2+ current under voltage-clamp conditions by stepping to +10 mV from the holding potential of −80 mV. To block the SR contribution to the transient, SR Ca2+ release was inhibited with 100 μM ryanodine and SR Ca2+ uptake was blocked using 1 μM thapsigargin. It should be noted that computations involving the proportion of Ca2+ bound by the SR are subject to a potential error due to the use of thapsigargin in our experiments. Thapsigargin has been shown to reduce the Ca2+ binding affinity of the Ca2+-ATPase in competition assays where the molar ratio of inhibitor to ATPase approaches 1 (Witcome et al., 1995). However, this effect should be limited under our experimental conditions because the concentration of SR Ca2+-ATPase in the cytoplasm is much greater than the 1 μM thapsigargin used for the determination of Ca buffering. The molar ratios calculated from table 1 are 0.2:1, 0.08:1, and 0.06:1 for ED5, 11, and 15, respectively. Therefore, the error is expected to be small and probably not significant, particularly for ED11 and 15.
TABLE 1.
Binding constants for major fast-acting Ca binding moieties
| ED15
|
ED11
|
ED5
|
||||
|---|---|---|---|---|---|---|
| Embryonic day (ED) | Bmax (μM) | KD (μM) | Bmax (μM) | KD (μM) | Bmax (μM) | KD (μM) |
| TnC | 33.6* | 0.6† | 28.7* | 0.6† | 20.3* | 0.6† |
| SR | 16‡ | 0.6 | 12‡ | 0.6 | 4‡ | 0.6 |
| SL high§ | 0.25 | 0.3 | 0.25 | 0.3 | 0.25 | 0.3 |
| SL low§ | 0.7 | 13 | (Same all ages) | |||
| Calmodulin total¶ | 24 | 0.1–1 | (Same all ages) | |||
| ATP‖ | 5000 | 200 | (Same all ages) | |||
| Creatine phosphate | 12,000 | 71,073 | (Same all ages) | |||
All values for KD were taken from Table 10 in Bers (2001), except for those indicated by footnotes.
Estimates of TnC in the embryonic chick heart were obtained from SDS PAGE gels of ventricular strips from ED5, 11, 15, and 24 days posthatching, run together on the same gel, using the larger and more prominent myosin band and assuming a 1:1 correspondence between myosin and TnC. Bmax for TnC was assumed to be proportional to the myosin content normalized to total protein loading. We assumed that the TnC content at 24 days posthatching was the same as in adult rat myocytes, which Bers (2001) estimates is 70 μM.
We have no accurate estimate of the KD for TnC in embryonic chick myocytes and thus assume it is similar to adult rat myocytes (Bers, 2001).
To obtain an estimate of Bmax for SR Ca2+ binding in embryonic chick heart we assumed 1), that the Bmax was proportional to the oxalate-supported Ca2+ uptake in isolated chick SR reported by Vetter et al. (1986; their Fig. 2) and 2), that the Bmax thus estimated from adult chicken SR uptake was the same as that in adult rat myocytes given in Bers (2001). Bmax for ED5–15 were scaled proportionately from the adult value.
Estimates of Bmax for high- and low-affinity Ca2+ binding sites on SL were obtained using values for surface/volume ratio and the average volume of adult rat myocytes (Satoh et al., 1996). The chick myocytes used in this study were spherical with an average volume of ∼3 pl. From geometry we obtained the surface area of chick myocytes. Bmax for SL high- and low-affinity sites were scaled proportional to Bmax of adult rat SL sites according to the relative surface area of chick and rat myocytes.
Calmodulin has four classes of calcium binding sites at which Mg and K also bind. Calcium binding to calmodulin was computed from binding affinities given in Fabiato (1983), assuming [Mg2+] = 0.6 mM and [K+] = 140 mM.
Calcium binding to ATP was computed from binding affinities for Ca, Mg, and K (Fabiato, 1983), given the values of [Mg2+] and [K+] above.
Estimation of accessible cell volume in chick ventricle
Computation of intracellular Ca2+ buffer power requires an estimate of the cell volume in which the fura-2 is dispersed, termed accessible cell volume (Vacc). Vacc is roughly the intracellular volume outside of mitochondria, which is ∼65% of total cell volume in adult rat ventricular myocytes (Table 3 in Bers, 2001; Berlin et al., 1994). Similar values for extramitochondrial volume were obtained in adult avian heart (Table 3 in Bers, 2001). However, the volume of the sarcoplasmic reticulum (SR, 3.5% in rat ventricle; Bers, 2001) and that physically occupied by the concentrated protein in the cytosol (∼10–15%; Bers, 2001) must also be considered, leaving a Vacc of ∼47–52% for adult rat. Inasmuch as we were unaware of similar estimates in embryonic chick heart we devised a method to measure accessible cell volume in this tissue.
Small segments of embryonic left ventricle (8–12 mg wet weight) were dissected and split into strands connected centrally, like an octopus, to facilitate rapid diffusion into and out of the preparation. Dissection was performed at room temperature in oxygenated chick Ringer containing 30 mM BDM (2,3-butanedione 2-monoxime) to eliminate contractility and minimize cellular damage (Wiggins et al., 1980; Brotto et al., 1995). To estimate extracellular volume (Vextra), preparations were transferred to 0.5 ml of chick Ringer containing 0.024 μCi/μl-tritiated water (to label all cellular water) and 0.0024 μCi/μl of 14C-labeled mannitol (to label extracellular volume) for 30 min at 4°C. Samples were gently agitated to facilitate diffusion. The samples were then briefly blotted to remove excess fluid, placed in 5 ml of scintillation fluid, and gently agitated in a refrigerator for three days, after which the radioactivity was counted. The concentrations of tritiated water and 14C-mannitol and incubation times were determined by trial and error to give sufficient counts for accuracy. Along with the tissue samples, vials containing serial dilutions of tritiated water and 14C-mannitol were also counted, which established a linear relation to relate counts and marker concentrations.
The tissue volume outside of mitochondria, SR, and concentrated protein (Vmito) was estimated by bathing preparations for 30 min in a relaxing solution containing 0.45 mM Mg2+, 2.59 mM MgATP, 15 mM disodium phosphocreatine, 5 mM EGTA, and 84.5 mM potassium methanesulfonate, at pH 7.1, with 5% v/v Dextran T-500 and 50 μg/ml saponin. Dextran minimizes swelling of the preparation (Godt and Maughan, 1981) and saponin at this concentration permeabilizes the sarcolemma but not the SR (Nosek et al., 1997). Preparations were subsequently treated as in the extracellular volume determinations above. The accessible cell volume (Vacc) necessary for our calculations is thus Vmito − Vextra.
As a test of the methodology, we treated other preparations for 30 min with 1% Triton X-100, a nonionic detergent, in chick relaxing solution with 5% Dextran to remove all membranes. The tissues were loaded with radiolabels and processed as outlined above. In this case, we expected the tritium space should be identical to that measured with mannitol.
Although ventricular tissue from ED5 was too fragile to withstand the blotting and transfer between solutions, successful estimates were obtained from ED11 and 15. At both ages, regression analysis showed that the ratio of tritium and mannitol spaces was not significantly different from 1.0, thus validating the method. Estimates of Vextra and Vmito for ED11 and 15 were not significantly different, so data were pooled to obtain values of Vextra of 5.7% (mean ± 0.1% SE, n = 25) and Vmito of 60.6% (mean ± 2.5% SE, n = 26). Taken together, these give an estimate of Vacc of 54.9% (mean ± 1.8% SE), a value in reasonable agreement with that of adult rat myocytes.
Cytoplasmic calibration of the fura-2
At the end of the experiment the myocyte was exposed to 10 μM of ionomycin (Ca salt) for the determination of the parameters Rmax and βmax. The same cell was then exposed to 25 mM EGTA to determine Rmin and βmin. In most cases the new ratio after exposure of the cell to either ionomycin or EGTA was achieved in 5–10 min. The myocyte autofluorescence was determined before loading with fura-2. Fura-2 transients were then calibrated in terms of [Ca]i with the ratiometric procedure used by Grynkiewicz et al. (1985) with the equation
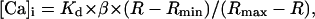 |
where KD is the dissociation constant for fura-2 (224 nM); β is the ratio of the fluorescence signal at 380 nm of a solution with high Ca2+ (βmax) and low Ca2+ (βmin); R is the ratio at 340:380 nm; Rmin is the ratio at 340:380 when the cell is in the presence of EGTA (low Ca2+); and Rmax is the ratio at 340:380 when the cell is in the presence of ionomycin (high Ca2+). The KD = 224 nM for fura-2 was taken from the estimate of Grynkiewicz et al. (1985) for a buffered high K+, low Na+, and Mg2+ solution.
Estimation of cytosolic fura-2 dye concentration
Dye concentration in myocytes is estimated by making fluorescence measurements of fura-2, of a known concentration, in an oil droplet submersed in a Ca2+-free high K+ solution as detailed by Moore et al. (1990) and as previously described (Brotto and Creazzo, 1996). Briefly, myocyte fura-2 concentration is calculated from
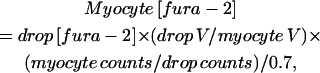 |
where V is volume and counts is photon counts at the isosbestic point. The counts are divided by 0.7, since light collection in oil is 70% as efficient as in water, as indicated by Moore and co-workers for a 40× objective.
Using our standardized loading conditions, [fura-2]i = 5.9 ± 2.4 μM (±SD; n = 23). The range varied from 3.3 to 10.9 μM [fura-2]i. All measurements of cytosolic Ca buffering were corrected for Ca binding by fura-2 by assuming a KD value of 224 μM.
Statistics
Statistical comparisons among the three embryonic ages were made using analysis of variance (ANOVA) for a single factor. Individual comparisons were made using either the student's t-test or the paired student's t-test when comparing paired data (i.e., before and after addition of ryanodine). P < 0.05 was considered significant. Bmax and KD were determined from experimental or theoretical data using Origin 6.1 software (OriginLab Corporation, Northampton, MA). Curve fitting was carried out using the nonlinear least-squares method and Origin 6.1 software.
RESULTS
For this study, embryonic ventricular myocytes were obtained from chick embryos at ED5, 11, and 15 and cultured overnight. The ages selected encompass nearly the full range of development for SR junctional complexes in the chick embryo, including the age when SR peripheral junctions are first detected to an age when the relative frequency of junctions is similar to an adult level (Protasi et al., 1996).
Ca2+ transients in ventricular development
The peak magnitude of Ca2+ transients, in the presence or absence of 100 μM ryanodine (to block SR Ca2+ release), was measured in field-stimulated ventricular myocytes at ED5, 11, and 15 (Fig. 1). Maximum reduction of the Ca2+ transient was achieved after a 20-min exposure to 100 μM ryanodine (Brotto and Creazzo, 1996). The magnitude of the transients was greatest at ED5 and declined with age. Most of the decline occurred between ED5 and 11 with the peak magnitude reduced by 33% over this time period. A further but much smaller decline occurred between ED11 and 15 (8.5%). Ryanodine (100 μM) reduced the magnitude by ∼45% at ED11 and 15, compared with only 6% at ED5. The reduction with ryanodine shown for ED11 was similar to a previously reported observation from this laboratory for this age (Brotto and Creazzo, 1996). The time to reach peak magnitude after electrical stimulation was not significantly different for any of the ages either with or without ryanodine exposure (197 ± 11 ms, all groups combined; p = 0.95, ANOVA). However, the time for the transient to decay by half the peak magnitude was significantly faster at ED5 in comparison with either ED11 or 15 in the absence of ryanodine (332 ± 38, 477 ± 54, and 499 ± 63 ms for ED5, 11, and 15, respectively; p = 0.02 ANOVA) or in the presence of ryanodine (346 ± 20, 480 ± 39, and 512 ± 29 ms; p = 0.001). There was no significant difference in the decay times when comparing ED11 and 15 with or without ryanodine. The faster decay time at ED5 reflects the faster decay for the Ca2+ current at this age (see below). Ryanodine treatment did not significantly affect the decay times. These results most likely indicated that the Na+/Ca2+ exchanger was sufficient to rapidly extrude Ca2+ in transients in which the magnitude had been reduced by the ryanodine. The diastolic Ca2+ level at each age was slightly decreased by the addition of ryanodine and this was more evident at the older ages. The results indicate that Ca2+-induced Ca2+ release (CICR) from the SR is very limited at ED5 but increases substantially by ED11. The increase in CICR appears to level off between ED11 and 15, as there is little difference in the effect of ryanodine between these ages. Finally, the small effect of ryanodine on the magnitude of the Ca2+ transient at ED5 indicates that most of the transient at this age is due to an influx of extracellular Ca2+.
FIGURE 1.

The effect of ryanodine (Ry) on electrically stimulated intracellular Ca2+ transients during development. (A) Examples of Ca2+ transients illustrating the effects before and after the addition of 100 μM ryanodine. Note that Ca2+ transients are larger at ED5 compared to ED11 and 15, whereas the effect of ryanodine is smaller. (B) Graphical representation illustrating that most of the decline in the magnitude of Ca2+ transients as well as the largest increase in the effect of ryanodine occurs between ED5 and 11. Peak indicates the peak magnitude of the Ca2+ transient and base indicates the baseline Ca2+ level after decay of the Ca2+ transient and just before the beginning of the next transient. N = 7 myocytes for each of the three ages. The data points represent 2–3 experiments per age with 2–4 myocytes per experiment. Error bars indicate mean ± SE. Single asterisk (*) indicates statistical significance when comparing either ED11 or 15 with ED5. Section symbol (§) indicates significant reduction in the peak after ryanodine treatment in paired comparisons with the student's t-test. Double asterisk (**) indicates that the baseline was significantly less after ryanodine treatment when compared using the paired student's t-test.
Ca2+ current decreases with development
The results in Fig. 1 indicate that there is sparse SR function at ED5. A plausible explanation for the larger electrically stimulated Ca2+ transients at ED5 is that the voltage-gated Ca2+ current is larger at this age. To test this hypothesis, the magnitude of the Ca2+ current was measured in voltage-clamped myocytes (Fig. 2). Embryonic ventricular myocytes have both L-and T-type Ca2+ currents (Kawano and DeHaan, 1991; Brotto and Creazzo, 1996; Cribbs et al., 2001; Kitchens et al., 2003). The current-voltage (I-V) relationships in Fig. 2 B illustrate that the peak magnitudes of Ca2+ currents from ED5 to ED15 qualitatively parallels the decrease in the magnitude of the electrically stimulated Ca2+ transients during development. The holding potential for these experiments was −80 mV and both L- and T-type Ca2+ currents were activated with this voltage-clamp protocol (Kawano and DeHaan, 1991). This was evident in the biphasic appearance of the I-V relationships evident at ED11 and 15 (Fig. 2 B). At ED5 the I-V relationships for both currents overlapped more than at later ages, reflecting a more positive activation threshold for ICa,T and a more negative activation for ICa,L at earlier ages in the chick (Kawano and DeHaan, 1991). To examine in greater detail the decline of Ca2+ currents with development we relied on the voltage-dependent differences between L- and T-type current (Kawano and DeHaan, 1991; Brotto and Creazzo, 1996). To obtain the I-V relationship for L-type current,T-type current was inactivated by a 500-ms prepulse to −40 mV (Fig. 2 C). The T-type current I-V was calculated from the difference in the current elicited from −80 mV and that following the prepulse to −40 mV. The peak magnitude of T-type current occurred at ∼−30 mV and L-type at 0 to +10 mV. The I-V relationship for T-type current at ED5 showed proportionately greater current at positive potentials. This may be due to differences in voltage-dependent properties observed at earlier embryonic ages (Kawano and DeHaan, 1991). These data indicated that both L- and T-type Ca2+ currents decline with development. Most of the decline inL-type current occurred between ED5 and 11, whereasT-type current appeared to decline steadily over the entire period from ED5 to 15. A similar developmental decline of L- and T-type currents was observed in an earlier study that employed a strategy of blocking L-type current with nifedipine when holding at −80 mV (Kitchens et al., 2003). These results indicate that the relatively large Ca2+ transients observed at ED5 were most likely due to a larger voltage-gated Ca2+ current although we cannot rule out a possible contribution from reverse Na+/Ca2+ exchange.
FIGURE 2.

Examples of L- and T-type Ca2+ currents and current-voltage (I-V) relationships from ED5, 11, and 15. (A) Example of current traces illustrating T-type Ca2+ current elicited from steps to −30 mV from the holding potential of −80 mV and L-type current from a step to +10 mV after a prepulse to −40 mV to inactivate the T-current. Note that the Ca2+ currents are largest at ED5. The overlying smooth curves illustrate the single-exponential fits to the decaying phase of the currents. (B) I-V curves elicited by voltage steps from a −80-mV holding potential showing the peak values for inward Ca2+ currents. The biphasic I-V curve most noticeable at ED11 is due to the presence of T-type Ca2+ current which activates at a lower threshold compared to L-type current (see text). (C) I-V relationships for L-type Ca2+ current elicited after inactivation of T-type current with a 500-ms prepulse to −40 mV. (D) The I-V relationship for T-type current was determined by subtracting the values illustrated in C from those in B. N = 15, 10, and 15 for ED5, 11, and 15, respectively, and 3–5 cells per experiment. Error bars indicate mean ± SE.
From visual examination of the Ca2+ current records such as the examples in Fig. 2 A it appeared that the current decay was slower at ED11 and 15 compared to ED5. To verify this observation we fitted the decaying phase of the Ca2+ current with a single exponential curve. We used the current records from ∼15 to 150 ms after the voltage step to −30 mV from the −80 mV holding potential for T-type current and after the voltage step to +10 mV following the prepulse to −40 mV for L-type current. These data were well fitted by a single exponential with no detectable improvement in the fit when fitted to a double-exponential curve (not shown). The exponential time constant for decay of the current at −30 mV was not significantly different among the three ages (combined τ = 24.5 ± 1.7 ms; p = 0.74, ANOVA). However at +10 mV, the decay times were significantly different (τ = 32.4 ± 3.5, 51.9 ± 4.6, and 54.4 ± 4.5 ms for ED5, 11, and 15, respectively; p = 0.02, ANOVA). As anticipated, the rate of decay at ED11 and 15 was significantly slower than at ED5 (p < 0.01 for both ages, Student's t-test). There was no difference between ED11 and 15 (p = 0.41). In summary, the rate of decay of L-type Ca2+ current is faster at ED5 than at the later ages.
SR Ca2+ content increases with development
The other major source of cytosolic Ca2+ for EC coupling is the SR. The Ca2+ content of the SR was estimated by releasing SR Ca2+ with a rapid application of a high concentration of caffeine (50 mM) and integrating the resultant Na+/Ca2+ exchange current in voltage-clamped myocytes (Diaz et al., 1996). As expected, the SR Ca2+ content increased markedly in a near-linear fashion with development (Fig. 3). These data are consistent with observations by others showing a developmental increase in SR Ca2+ release units and an accumulation of electron-dense material in SR vesicles (presumably calsequestrin) over a similar developmental period in chick heart (Protasi et al., 1996).
FIGURE 3.

SR Ca2+ content determined from integration of the Na+/Ca2+ exchange current after caffeine application. A shows an example of the exchanger current after caffeine application to a ventricular myocyte from ED5, 11, and 15 (left to right, respectively). The noisy baseline before the caffeine-induced current was due to the experimenter placing his hand and arm within the Faraday cage and close to the patch-amplifier head stage during the caffeine addition. The seal remained stable throughout this process. N = 5, 6, and 6, for ED5, 11, and 15, respectively, and representing two experiments per age and 2–3 cells per experiment. (B) Graphical representation of SR Ca2+ content. Single asterisk (*) indicates that ED11 SR content is significantly greater compared to ED5. Double asterisk (**) indicates significance when comparing ED15 with either ED5 or 11. Bars indicate mean ± SE.
To assess the functional ability of the Na+/Ca2+ exchanger to extrude Ca2+ with development, the rate of decay of the caffeine-induced transient was determined by exponential curve-fitting. The data were well-fitted by a single exponential (not shown) and were not significantly different among the three ages (τ = 77 ± 16, 102 ± 11, and 82 ± 5 ms for ED5, 11, and 15, respectively; p = 0.20, ANOVA). These results indicate that the exchanger density is not the rate-limiting step in the extrusion of Ca2+ in the absence of SR function.
Ca2+ buffer power during development
The approach of Berlin et al. (1994) was used to estimate the fast Ca2+ buffering active during the rapid rising phase of the Ca2+ transient. For these experiments, SR function was blocked by application of 1 μM thapsigargin (to prevent SR Ca2+ uptake) and 100 μM ryanodine (to block SR Ca2+ release) to the extracellular solution and Ca2+ currents were elicited by voltage-clamp steps to +10 mV from a holding potential of −80 mV which elicits both T- and L-type Ca2+ currents (see Fig. 2). In some instances the Ca2+ channel agonist Bay k 8644 was added to increase the size of the current and facilitate obtaining measurable transients. Examples of Ca2+ transients elicited under these voltage-clamp conditions from ED5 to ED15 are shown in Fig. 4. Data such as those shown in Fig. 4 were used to calculate the cytosolic buffer power (BP) following the method of Berlin et al. (1994; reviewed briefly here). To determine BP, the integrated Ca2+ current (∫ICa) was compared to the simultaneous change in free Ca2+. The ∫ICa gives the total moles of Ca2+ entering the cytosol which must be converted into moles/liter to calculate BP. This is done using the cytosolic volume into which the Ca2+ is diluted (see Methods). The Michaelis-Menten relationship was used to compare free and bound Ca2+,
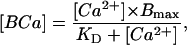 |
(1) |
where Bmax is the sum of the capacities of all cytosolic buffers, KD is the lumped dissociation constant for all buffers, and [BCa] is the concentration of bound Ca2+ calculated as total Ca2+ minus free Ca2+. To compute fast Ca2+ BP where the change in free Ca2+ (d[Ca2+]) parallels the change in bound Ca (d[BCa]), Eq. 1 can be differentiated with respect to Ca2+ such that
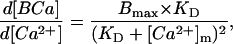 |
(2) |
where [Ca2+]m is the mean level of Ca2+ during a step increase in Ca2+. [Ca2+]m was calculated by taking the difference between the Ca2+ level at the beginning of the depolarizing step to the peak value and dividing by 2. Fitting the data to Eq. 2 gives an estimate of Bmax and KD. Note that d[BCa]/d[Ca2+] is the chord BP at any given cytosolic Ca2+ level. Fig. 5, A–C, show nonlinear fits of the data for the three experimental ages. The KD was similar at all three developmental ages, whereas the Bmax increased by 38% between ED5 and 15 with most of the increase occurring after ED11. These results can be compared to data from adult rat myocytes (Berlin et al., 1994), where estimated KD was 0.96 μM and Bmax was 123 μmol/liter cell H2O.
FIGURE 5.

Cytosolic Ca buffering. [Ca2+]m and d[BCa]/d[Ca] were calculated and fitted to Eq. 2 as described in the text. The KD and Bmax (mean ± SE) for each age are shown in A–C, and were determined by fitting the data set for each myocyte individually. The cumulative data for each age is shown in the graphs. Note that d[BCa]/d[Ca] is the chord buffer power for any given level of cytosolic-free Ca2+. N = 5, 8, and 8, for ED5, 11, and 15, respectively. The data are from four experiments for each age and 1–3 cells per experiment. Units of Bmax are in μM/liter cell water and KD values are in μM.
Efficiency of CICR increases with development
The efficiency for SR CICR was expected to increase with development, with increasing SR Ca2+ content and development of mature peripheral junctions. CICR efficiency was approximated by computing a simple gain factor for EC coupling. For this study, gain was approximated as
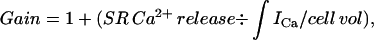 |
where ∫ICa/cell vol is the concentration of Ca2+ entering the cell via Ca2+ channels, referred to as the total accessible cell volume (55% of total cell volume; see Methods). Under this definition, gain is 1 if there is no contribution from the SR. Gain can be computed from the data with and without ryanodine shown in Fig. 1 B, since
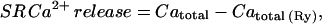 |
where Catotal is the change in total Ca in the cytosol and is equal to the total Ca at the peak of the Ca2+ transient minus total Ca2+ at the base level. Catotal (Ry) is the total change in Ca in the cytosol in the presence of ryanodine and is equal to the total Ca in the presence of ryanodine at the peak minus that at the base in the presence of this drug. Total Ca at any free Ca2+ level is computed from Eq. 1 and the experimentally determined Bmax and KD at each age. The results for gain at the three experimental ages are shown in Fig. 6. As expected, gain increases with development as SR proliferates, and as peripheral couplings mature, with most of the increase occurring between ED5 and 11 (gain = 1.05, 1.71, and 1.92, for ED5, 11, and 15, respectively). Another analogous way of expressing the efficiency of CICR is to compute the percent contribution of the SR to the total Ca2+ transient using the equation
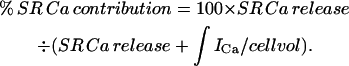 |
Since SR Ca release is defined as gain × ∫ICa/cell vol,
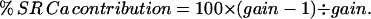 |
These data are also shown in Fig. 6.
FIGURE 6.

Computed gain factor for CICR and percent contribution to the Ca2+ transient from the SR are illustrated together for comparison. The values were calculated using the Ca2+ buffer power and the data from the field-stimulated Ca2+ transients illustrated in Fig. 1 as detailed in the text. The increase in gain and SR contribution parallels the structural development of SR junctions as demonstrated in Protasi et al. (1996; compare this Fig. 6 with their Fig. 4).
Theoretical estimation of cytosolic BP
Cytosolic BP was calculated using KD and Bmax values of the major intracellular Ca2+ binding moieties given in Table 10 of Bers (2001) for adult rat cardiac myocytes, scaled appropriately for embryonic chick myocytes (Table 1). The major calcium binding moieties considered are TnC, SR Ca2+ pump, and low- and high-affinity binding to the sarcolemma (SL), as well as calmodulin, ATP, and PCr (creatine phosphate). The KD and Bmax values for these moieties are given in Table 1. Using these values, we generated the relationship between total bound Ca and free cytosolic Ca2+ over the range of 0.1 to 1 μM, which gave values of Bmax and KD for total cytosolic Ca buffering at the three embryonic ages. These values are displayed in Fig. 7 and are in reasonable agreement with experiment, especially given the uncertainties in the Bmax and KD of chick proteins.
FIGURE 7.

Measured data in comparison with theoretically calculated Bmax and KD values from estimates of the major fast Ca binding moieties in the cytoplasm. See text and Table 1.
Simulation of Ca2+ binding to these moieties using the on- and off-rates of binding demonstrate that peak binding occurred within a few ms of the peak of the Ca2+ transient (data not shown). The distribution of Ca among the major binding moieties at the peak of the Ca2+ transient at the three ages (from Fig. 1) is shown in Fig. 8. Note that, at all three ages, TnC is the major Ca binding moiety in the cytosol, and that the values for SR binding increase, as would be expected with SR proliferation with age. The relative increase in SR Ca binding is largely offset by declines in Ca bound by calmodulin and ATP.
FIGURE 8.

Graphical representation of the proportion of Ca2+ bound by the major cytoplasmic fast Ca2+ binding moieties at the peak of the intracellular Ca2+ transient at the three developmental ages. Note that TnC binds ∼57% of the Ca2+ at each age. The proportion of Ca2+ bound by the SR increases, and this is offset primarily by corresponding decreases in Ca2+ bound by calmodulin and ATP. Ca2+ binding to total sarcolemmal (SL) sites and to creatine phosphate (PCr) is small and has been combined in the figure.
DISCUSSION
This is the first report providing a detailed analysis of variations in the Ca2+ handling properties of ventricular myocytes over an extended period of development. The findings indicate that the magnitude of electrically stimulated Ca2+ transients in embryonic ventricular myocytes declines by ∼35% between ED5 and ED15 of development. The decline is due largely to a decline in the magnitude of voltage-gated Ca2+ current although the observed increase in the cytosolic buffering capacity would be expected to account for some of this decline. In contrast, the SR Ca2+ contribution to the Ca2+ transient increases markedly by nearly 12.5-fold over the same period of development, which corresponds to a 13.5-fold increase in the amount of Ca2+ stored by the SR. Important determinants of the free Ca2+ detected by fura-2 are the fast-acting Ca2+ buffers in the cytoplasm, which are expected to vary with development. The Bmax for the fast Ca buffers increases by ∼40% between ED5 and 15. These data indicate that the cytoplasmic chord buffer power for any given value of [Ca2+]i increases with developmental age. Theoretical calculations indicate that this is due to increases in the Bmax for both TnC and SR. Calculations utilizing buffer power measurements and the integral of the calcium current show that the gain factor for CICR increases ∼20-fold between ED5 and 15.
Developmental decline in the magnitude of the Ca2+ transient
Most of the decline of electrically stimulated Ca2+ transients occurred between ED5 and 11, with a slight further decline by ED15. In the chick embryo the shapes of the action potentials differ only slightly between ED5 and 15, and probably have minimal, if any, measurable effect on the magnitude of electrically stimulated transients (see Sperelakis, 1982). Our results indicate that the decline appears to be mostly due to a parallel decline in both L-and T-type Ca2+ currents. The decrease in the L-type Ca2+ current is not due to a decline in the number of L-type Ca2+ channels, as the number of DHP receptor binding sites is not significantly different throughout in-ovo development in the chick embryo (Marsh and Allen, 1989). One possible explanation is that the open channel probability for L-type channels decreases with development (Tohse et al., 1992) which may be related to the phosphorylation state of the channel. As of yet, the role of protein phosphorylation on Ca2+ channel activity in development has not been investigated. Another possibility is that there may be an embryonic form of the L-type channel that is not expressed in later development and that possesses different functional properties. This possibility is supported by a recent report of Ca2+ currents in embryonic mouse heart in a targeted knockout of α1C by Seisenberger et al. (2000). The Seisenberger study provides convincing evidence for anas-yet unidentified L-type-like Ca2+ channel in the early developing heart that is later replaced by the cardiac α1C isoform characteristic of adult myocardium.
Another observation of note is that the L-type Ca2+ current shows significantly faster inactivation at ED5 than at the later ages. The most likely explanation is that there is greater Ca2+-dependent inactivation because of the relatively large Ca2+ current at this early age. A contributing factor may be the lower fast buffering capacity of the cytoplasm from less Ca2+ binding by SR and TnC. Recent evidence indicates that calmodulin, which appears to be tethered to the L-type channel, is thought to be a critical component of Ca2+-dependent inactivation (reviewed in Bers, 2001). Although we assumed that the concentration of calmodulin remained constant during heart development, our calculations indicate that a higher proportion of entering Ca2+ is bound by calmodulin at ED5 than at ED11 or 15 (see Table 1 and Fig. 8). Taken together, these factors likely contribute to the fast inactivation even in the relative absence of local SR Ca2+ release, which is thought to contribute significantly to Ca2+-dependent inactivation in adult heart (Puglisi et al., 1999).
Cytoplasmic Ca2+ buffering during development
The summed Bmax for fast Ca2+ buffers increases with development. This is to be expected, inasmuch as the rate of cell division declines with development and the myocytes become more packed with myofibrils and the content of SR increases. The experimentally determined KD and Bmax values were in reasonable agreement with our theoretical predictions (see Fig. 7). We used these values in a unique approach to estimate the gain for CICR and the percent contribution from the SR in the global field-stimulated Ca2+ transients which increase with development.
Our theoretical estimations of Ca binding moieties indicates that TnC binds ∼57% of entering Ca2+ during a transient at each of the three ages investigated. As expected, the proportion of Ca2+ bound by SR Ca2+-ATPase increases with development, largely at the expense of that which is bound by ATP and calmodulin. The summed Bmax for the fast Ca2+ buffers measured in the present study is approximately one-third that reported by Berlin et al. (1994). This appears to be largely due to ∼50% lower Bmax for TnC and ∼70% lower value for SR binding compared to adult mammalian ventricle (compare our Table 1 with Table 10 in Bers, 2001).
In our approach to estimating cytosolic Ca2+ buffering, we used the global Ca2+ transient, which assumes that the transient was uniform throughout the myocyte. This is true for adult ventricular myocytes with an extensive t-tubular system regardless of whether SR function is blocked. A report by Haddock et al. (1999) on neonatal rabbit myocytes which lack t-tubules demonstrated with confocal line scans that the magnitude of the transient is greatest near the periphery and least toward the center of the myocyte. This nonuniform distribution of Ca2+ during a transient is likely to be similar in embryonic chick myocytes which have not-tubules and no extended junctional SR. In this scenario, Ca2+ measurements restricted to the cell periphery are likely to lead to an underestimation of the cytosolic buffering capacity whereas central measurements will lead to an overestimation. Embryonic myocytes are spherical after enzymatic dissociation and this geometry should lead to an even distribution of the transient from periphery to center. Therefore, we assumed that regional differences in [Ca2+]i are averaged-out in the global Ca2+ transient and, along with our detailed measurements of the assessable cell volume, we have obtained a good estimate of the total cytosolic Ca2+ buffering capacity.
Functional development of the SR
As expected, SR function as determined by the efficacy of ryanodine blockade of field-stimulated transients markedly increases between ED5 and 11 with a small increase thereafter (see Fig. 1). These data would suggest that most of the development of SR occurs between the ages of ED5 and 11. This hypothesis is supported by the report from Protasi, Franzini-Armstrong, and colleagues (Protasi et al., 1996), which shows a dramatic increase in the relative frequency of complete junctions that occur over the same period of development, and is consistent with our determinations of the gain factor for CICR and the proportion of transient due to SR Ca release (see Fig. 4 in Protasi et al., 1996, and compare with our Fig. 6). Complete junctions, defined on the basis of electron microscopic observations, are SR-plasma membrane junctional complexes in which the junctional gap is fully zippered by the closely spaced feet of the ryanodine receptor/Ca2+ release channel. There are virtually no complete junctions at ED5 but by ED11, nearly 80% of junctions are complete. Other measured parameters indicate that the length of junctional profiles with zippered feet and the number of peripheral couplings per unit length of plasma membrane are similar to adult by ED11. Moreover, the area of plasmalemmal junctional domains increases markedly between ED5 and 11, while not changing significantly between ED11 and 15. Together with the Protasi study, our data shows that the development of structure and function of SR junctions are well correlated.
The study by Protasi et al. (1996) suggests that the relatively low gain factor for SR CICR in ED5 myocytes is likely due to several factors including less SR, reduced SR Ca2+ stores from a lack of calsequestrin, and incomplete SR junctions lacking fully zippered arrays of RyRs. This conclusion is further supported by a recent study of EC coupling development in mouse embryonic stem cells by Sauer et al. (2001). These investigators found that the elementary SR Ca2+ release events (Ca2+ sparks) seen at an earlier stage of in vitro development were of lower frequency and magnitude than at a later stage, and attributed these observations to fewer RyRs and less Ca2+ load in the SR. These conclusions from the study of Sauer and co-workers are consistent with the SR morphology reported by Protasi and co-workers, and with our measurements of SR Ca2+ content and CICR gain factor in developing chick heart.
Species differences and EC coupling in embryonic versus adult heart
In mammals, it is usually considered that SR content is sparse at birth and undergoes most of its development postnatally (Nakanishi and Jarmakani, 1984). However, the level of maturity and function of the SR is species-dependent. For example, as mentioned in the Introduction, SR is sparse in rabbit and rat neonatal hearts (Seguchi et al., 1986b; Nakanishi et al., 1992) but not in guinea pigs, which have a fully developed SR at birth (Goldstein and Traeger, 1985). Moreover, during a Ca2+ transient, Ca2+ removal from the cytoplasm is dependent more on extrusion into the extracellular space via the Na+/Ca2+ exchanger in neonatal rabbit, whereas the SR Ca2+-ATPase plays a larger role in neonatal rat (Bassani and Bassani, 2002; Balaguru et al., 1997). By contrast, SR morphology and function in the chick is well developed by hatching (Protasi et al., 1996). In agreement, our results indicate that CICR from the SR parallels the morphological development, in that CICR is well developed by ED15 in the chick (see Discussion above).
Regardless of the species, in early embryonic heart development the SR is likely to be very sparse or entirely absent. This implies a simplified EC coupling, in which all of the Ca2+ contributing to the Ca2+ transient is derived from the extracellular space via voltage-gated Ca2+ channels and possible entry from reverse-mode Na+/Ca2+ exchange and extrusion of Ca2+ via the exchanger working in its normal forward mode. However, EC coupling in the early heart tube has not been investigated, and there may be other contributing factors unique to early development. In working with a targeted knockout of the cardiac Na+/Ca2+ exchanger (NCX1), we observed relatively normal Ca2+ transients in heart tubes from 9.5 days postcoitum mouse embryos (Koushik et al., 2001). This intriguing finding suggests the possibility of other potent mechanisms for the extrusion of Ca2+ in early heart development.
A common feature of embryonic and early neonatal or posthatching ventricular myocytes is the absence of t-tubules, and SR junctional complexes are all at the periphery of the cell, producing spatial-temporal characteristics of the global Ca2+ transient different from that of adult (discussed above; Haddock et al., 1999). In this feature and along with smaller size, these myocytes resemble atrial and nodal pacemaking cells. Within a week of postnatal development mammalian ventricular myocytes begin to develop an extensive t-tubular system, although in avian species there are no t-tubules, but an extensive system of extrajunctional SR that develops instead (Sommer, 1995). A major advantage in the use of the avian embryo is that because of its early maturity, SR CICR can be studied without the complicating influences of t-tubules and extrajunctional or corbular SR.
A second but less studied common feature of immature ventricle is the presence of significant T-type Ca2+ current as demonstrated in the chick embryo in this report and also in embryonic mouse (Cribbs et al., 2001). T-type currents are absent in adult ventricle and are confined largely to atrial and nodal cells and Purkinje fibers, and are thought to be important for cardiac autorhythmicity. We recently reported that Ca2+ entry via T-type channels contributes significantly to the global Ca2+ transient and can stimulate CICR after nifedipine block of L-type channels (Kitchens et al., 2003). More work is needed to clearly establish the function of this important ion channel in the developing myocardium.
SUMMARY AND CONCLUSIONS
To our knowledge, this is the first report to quantify in detail the maturation of cardiac EC coupling and Ca2+ handling over an extended period of development. We made direct measurements of the Ca buffering capacity of the cytoplasm and used these data in a unique approach to estimate the gain factor for CICR and the contribution of the SR to the global Ca2+ transient. Not surprisingly, these factors increase markedly with development and moreover, the increases occur in a manner consistent with detailed published data on the structural development of SR peripheral junctions. We have provided estimates of the changes in the contributions of the major fast Ca binding moieties over an extended period of development. Finally, at all ages examined, TnC binds at least half of the entering Ca2+ during a transient and as expected, the proportion bound by the SR increases.
From the data presented here and the above discussion it is apparent that all of the primary structural components necessary for a relatively mature level of EC coupling are present by ED11. Does this mean that the efficiency or, in other words, the gain factor for CICR, develops in synchrony with the appearance of the primary components for cardiac EC coupling? The results from this report indicate that this is the case, and thus it appears that the development of structure and function of SR junctions are well correlated.
Acknowledgments
We thank Ms. Jane Chu for her careful determinations of the accessible cell volume, and for SDS PAGE of ventricular strips.
This work was supported by National Institutes of Health grants HL58861, HL36059, and HL71015.
References
- Balaguru, D., P. S. Haddock, J. L. Puglisi, D. M. Bers, W. A. Coetzee, and M. Artman. 1997. Role of the sarcoplasmic reticulum in contraction and relaxation of immature rabbit ventricular myocytes. J. Mol. Cell. Cardiol. 29:2747–2757. [DOI] [PubMed] [Google Scholar]
- Bassani, R. A., and J. W. M. Bassani. 2002. Contribution of Ca2+ transporters to relaxation in intact ventricular myocytes from developing rats. Am. J. Physiol. Heart Circ. Physiol. 282:H2406–H2413. [DOI] [PubMed] [Google Scholar]
- Berlin, J. R., J. W. M. Bassani, and D. M. Bers. 1994. Intrinsic cytosolic calcium buffering properties of single rat cardiac myocytes. Biophys. J. 67:1775–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers, D. M. 2001. Excitation-Contraction Coupling and Cardiac Contractile Force, 2nd ed. Kluwer Academic Publishers, Boston, MA.
- Brotto, M. A. D., and T. L. Creazzo. 1996. Ca2+ transients in embryonic chick heart: contributions from Ca2+ channels and the sarcoplasmic reticulum. Am. J. Physiol. Heart Circ. Physiol. 270:H518–H525. [DOI] [PubMed] [Google Scholar]
- Brotto, M. A. P., R. T. H. Fogaça, T. L. Creazzo, R. E. Godt, and T. M. Nosek. 1995. The effect of 2,3-butanedione 2-monoxime (BDM) on ventricular trabeculae from the avian heart. J. Muscle Res. Cell Motil. 16:1–10. [DOI] [PubMed] [Google Scholar]
- Creazzo, T. L., M. A. P. Brotto, and J. Burch. 1995. Reduced Ca2+ transients and Ca2+ currents during early and late cardiac dysmorphogenesis. FASEB J. 9:A557. [Google Scholar]
- Creazzo, T. L., R. E. Godt, L. Leatherbury, S. J. Conway, and M. L. Kirby. 1998. Role of cardiac neural crest cells in cardiovascular development. Annu. Rev. Physiol. 60:267–286. [DOI] [PubMed] [Google Scholar]
- Creazzo, T. L., Q. Wang, and R. E. Godt. 2001. Colocalization of dihydropyridine and ryanodine receptors in developing heart with a neural crest-associated defect. Exp. Cardiol. 6:11–16. [PMC free article] [PubMed] [Google Scholar]
- Cribbs, L. L., B. L. Martin, E. A. Schroder, B. B. Keller, B. P. Delisle, and J. Satin. 2001. Identification of the T-type calcium channel (CaV3.1d) in developing mouse heart. Circ. Res. 88:403–407. [DOI] [PubMed] [Google Scholar]
- Diaz, M. E., S. J. Cook, J. P. Chamunorwa, A. W. Trafford, M. K. Lancaster, S. C. O'Neill, and D. A. Eisner. 1996. Variability of spontaneous Ca2+ release between different rat ventricular myocytes is correlated with Na+-Ca2+ exchange and [Na+]i. Circ. Res. 78:857–862. [DOI] [PubMed] [Google Scholar]
- Dutro, S. M., J. A. Airey, C. F. Beck, J. L. Sutko, and W. R. Trumble. 1993. Ryanodine receptor expression in embryonic avian cardiac muscle. Dev. Biol. 155:431–441. [DOI] [PubMed] [Google Scholar]
- Fujii, S., R. K. Ayer, Jr., and R. L. DeHaan. 1988. Development of the fast sodium current in early embryonic chick heart cells. J. Membr. Biol. 101:209–223. [DOI] [PubMed] [Google Scholar]
- Fabiato, A. 1983. Calcium-induced release from the cardiac sarcoplasmic reticulum. Am. J. Physiol. 245:C1–14. [DOI] [PubMed] [Google Scholar]
- Godt, R. E., R. T. H. Fogaça, and T. M. Nosek. 1991. Changes in force and calcium sensitivity in the developing avian heart. Can. J. Physiol. Pharmacol. 69:1692–1697. [DOI] [PubMed] [Google Scholar]
- Godt, R. E., and D. W. Maughan. 1981. Influence of osmotic compression on calcium activation and tension in skinned muscle fibers of the rabbit. Pflüegers Arch. 391:334–337. [DOI] [PubMed] [Google Scholar]
- Goldstein, M. A., and L. Traeger. 1985. Ultrastructural changes in postnatal development of the cardiac myocyte. In The Developing Heart. M. L. Legato, editor. Martinus Nijhoff, Boston, MA. 1–20.
- Grynkiewicz, G., M. Poenie, and R. Y. Tsien. 1985. A new generation of calcium indicators with greatly improved fluorescence properties. J. Biol. Chem. 260:3440–3450. [PubMed] [Google Scholar]
- Haddock, P. S., W. A. Coetzee, E. Cho, L. Porter, H. Katoh, D. M. Bers, M. S. Jafri, and M. Artman. 1999. Subcellular [Ca2+]i gradients during excitation-contraction coupling in newborn rabbit ventricular myocytes. Circ. Res. 85:415–427. [DOI] [PubMed] [Google Scholar]
- Jewett, P. H., S. D. Leonard, and J. R. Sommer. 1973. Chicken cardiac muscle. J. Cell Biol. 56:595–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junker, J., J. R. Sommer, M. Sar, and G. Meissner. 1994. Extended junctional sarcoplasmic reticulum of avian cardiac muscle contains functional ryanodine receptors. J. Biol. Chem. 269:1627–1634. [PubMed] [Google Scholar]
- Kawano, S., and R. L. DeHaan. 1991. Developmental changes in the calcium currents in embryonic chick ventricular myocytes. J. Membr. Biol. 120:17–28. [DOI] [PubMed] [Google Scholar]
- Kitchens, S. A., J. Burch, and T. L. Creazzo. 2003. T-type Ca2+ current contribution to Ca2+-induced Ca2+ release in developing myocardium. J. Mol. Cell. Cardiol. 35:515–523. [DOI] [PubMed] [Google Scholar]
- Koushik, S. V., J. Wang, R. Rogers, D. Moskophidis, N. A. Lambert, T. L. Creazzo, and S. J. Conway. 2001. Targeted inactivation of the sodium-calcium exchanger (Ncx1) results in the lack of a heartbeat and abnormal myofibrillar organization. FASEB J. 15:1209–1211. [DOI] [PubMed] [Google Scholar]
- Manasek, F. J. 1970. Histogenesis of the embryonic myocardium. Am. J. Cardiol. 25:149–168. [DOI] [PubMed] [Google Scholar]
- Marsh, J. D., and P. D. Allen. 1989. Developmental regulation of cardiac calcium channels and contractile sensitivity to [Ca]o. Am. J. Physiol. Heart Circ. Physiol. 256:H179–H185. [DOI] [PubMed] [Google Scholar]
- Moore, E. D. W., P. L. Becker, K. E. Fogarty, D. A. Williams, and F. S. Fay. 1990. Ca2+ imaging in single living cells: theoretical and practical issues. Cell Calcium. 11:157–179. [DOI] [PubMed] [Google Scholar]
- Nakanishi, T., and J. M. Jarmakani. 1984. Developmental changes in myocardial mechanical function and subcellular organelles. Am. J. Physiol. Heart Circ. Physiol. 246:H615–H625. [DOI] [PubMed] [Google Scholar]
- Nakanishi, T., M. Seguchi, and A. Takao. 1992. Developmental changes in myocardial mechanical function and subcellular organelles. Experientia. 44:936–944. [DOI] [PubMed] [Google Scholar]
- Nosek, T. M., R. T. Fogaca, C. J. Hatcher, M. A. Brotto, and R. E. Godt. 1997. Effect of cardiac neural crest ablation on contractile force and calcium uptake and release in chick heart. Am. J. Physiol. Heart Circ. Physiol. 273:H1464–H1471. [DOI] [PubMed] [Google Scholar]
- Protasi, F., X. H. Sun, and C. Franzini-Armstrong. 1996. Formation and maturation of the calcium release apparatus in developing and adult avian myocardium. Dev. Biol. 173:265–278.8575628 [Google Scholar]
- Puglisi, J. L., W. L. Yuan, J. W. M. Bassani, and D. M. Bers. 1999. Ca2+ influx through Ca2+ channels in rabbit ventricular myocytes during action potential clamp: influence of temperature. Circ. Res. 85:E7–E16. [DOI] [PubMed] [Google Scholar]
- Satoh, H., L. M. D. Delbridge, L. A. Blatter, and D. M. Bers. 1996. Surface:volume relationship in cardiac myocytes studied with confocal microscopy and membrane capacitence measurements: species dependence and developmental effects. Biophys. J. 70:1494–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer, H., T. Theben, J. R. Hescheler, M. Lindner, M. C. Brandt, and M. Wartenberg. 2001. Characteristics of calcium sparks in cardiomyocytes derived from embryonic stem cells. Am. J. Physiol. Heart Circ. Physiol. 281:H411–H421. [DOI] [PubMed] [Google Scholar]
- Seguchi, M., J. Harding, and J. Jamrmakani. 1986a. Developmental change in the function of sarcoplasmic reticulum. J. Mol. Cell. Cardiol. 18:189–195. [DOI] [PubMed] [Google Scholar]
- Seguchi, M., J. M. Jarmakani, B. L. George, and J. Harding. 1986b. Effect of Ca2+ antagonists on mechanical function in the neonatal heart. Pediatr. Res. 20:838–842. [DOI] [PubMed] [Google Scholar]
- Seisenberger, C., V. Specht, A. Welling, J. Platzer, A. Pfeifer, S. Kühbandner, J. Striessnig, N. Klugbauer, R. Feil, and F. Hofmann. 2000. Functional embryonic cardiomyocytes after disruption of theL-type α1C (Cav1.2) calcium channel gene in the mouse. J. Biol. Chem. 275:39193–39199. [DOI] [PubMed] [Google Scholar]
- Sommer, J. R. 1995. Comparative anatomy: in praise of a powerful approach to elucidate mechanisms translating cardiac excitation into purposeful contraction. J. Mol. Cell. Cardiol. 27:19–35. [DOI] [PubMed] [Google Scholar]
- Sperelakis, N. 1982. Pacemaker mechanisms in myocardial cells during development of embryonic chick heart. In Cardiac Rate and Rhythm. L. N. Bouman and H. J. Jongsna, editors. Martinus Nijhoff, Dordrecht, The Netherlands. 129–65.
- Sun, X.-H., F. Protasi, M. Takahashi, H. Takeshima, D. G. Ferguson, and C. Franzini-Armstrong. 1995. Molecular architecture of membranes involved in excitation-contraction coupling of cardiac muscle. J. Cell Biol. 129:659–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohse, N., J. Mészáros, and N. Sperelakis. 1992. Developmental changes in long-opening behavior of L-type Ca2+ channels in embryonic chick heart cells. Circ. Res. 71:376–384. [DOI] [PubMed] [Google Scholar]
- Vetter, R., H. Will, I. Kuttner, C. Kemsies, and L. Will-Shahab. 1986. Developmental changes of Ca transport systems in chick heart. Biomed. Biochim. Acta. 45:219–222. [PubMed] [Google Scholar]
- Wiggins, G. R., J. Reiser, D. F. Fitzpatrick, and J. L. Bergey. 1980. Inotropic actions of diacetyl monoxime in cat ventricle muscle. J. Pharmacol. Exp. Ther. 212:217–224. [PubMed] [Google Scholar]
- Witcome, M., Y. M. Khan, J. M. East, and A. G. Lee. 1995. Binding of sesquiterpene lactone inhibitors to the Ca2+-ATPase. Biochem. J. 310:859–868. [DOI] [PMC free article] [PubMed] [Google Scholar]