

Abstract
The biochemical reaction networks include elementary reactions differing by many orders of magnitude in the numbers of molecules involved. The kinetics of reactions involving small numbers of molecules can be studied by exact stochastic simulation. This approach is not practical for the simulation of metabolic processes because of the computational cost of accounting for individual molecular collisions. We present the “maximal time step method,” a novel approach combining the Gibson and Bruck algorithm with the Gillespie τ-leap method. This algorithm allows stochastic simulation of systems composed of both intensive metabolic reactions and regulatory processes involving small numbers of molecules. The method is applied to the simulation of glucose, lactose, and glycerol metabolism in Escherichia coli. The gene expression, signal transduction, transport, and enzymatic activities are modeled simultaneously. We show that random fluctuations in gene expression can propagate to the level of metabolic processes. In the cells switching from glucose to a mixture of lactose and glycerol, random delays in transcription initiation determine whether lactose or glycerol operon is induced. In a small fraction of cells severe decrease in metabolic activity may also occur. Both effects are epigenetically inherited by the progeny of the cell in which the random delay in transcription initiation occurred.
INTRODUCTION
The availability of voluminous data describing the molecular components of living cells motivate mathematical and computer simulation studies aimed at understanding how the complex dynamics of cellular processes emerges as a result of the individual molecular interactions (Tyson et. al., 2001; Endy and Brent, 2001). The models of cellular processes are most commonly formulated in the framework of deterministic chemical kinetics. The elementary molecular interactions are modeled in terms of differential rate equations and the temporal changes in concentrations of molecular species or their stationary state values are studied. This approach applied to study large networks of molecular interactions has already provided valuable results, for example in studies of yeast cell cycle (Sveiczer et al., 2000) and applications in metabolic engineering (Hoefnagel et al., 2002). Whole-cell scale models of metabolic pathways are also emerging (Edwards et al., 2001, Tomita et al., 1999).
The major difficulty in applying deterministic chemical kinetics to modeling of cellular processes is that they occur in very small volumes and hence frequently involve very small numbers of molecules. For example, in gene expression processes a few molecules of transcription factor may interact with a single “molecule” of gene regulatory region (there are on average only 10 molecules of Lac repressor in E. coli cells; Levin, 1999). In these cases modeling of reactions as continuous fluxes of matter is no longer correct. Moreover, significant stochastic fluctuations that occur in reactions involving small numbers of molecules may influence biochemical processes. The presence of the stochastic effects in gene expression and signal transduction processes have been shown by both theoretical and experimental approaches (Levin et al., 1998; McAdams and Arkin, 1997; Kierzek et al., 2001; Ozbudak et al., 2002; Elowitz et al., 2002; see Rao et al. (2002) for review). Therefore, stochastic effects must be studied to understand how complex networks of molecular interactions determine the precise regulation of cellular processes, despite the inherent noise present in the system.
To study the stochastic effects in biochemical reactions, stochastic formulation of chemical kinetics and Monte Carlo computer simulation approaches have been used. The exact computer simulation methods, such as the Gillespie algorithm (Gillespie, 1977), explicitly account for the individual reactive collisions among the molecules. Using these methods the statistical samples of possible, independent time courses can be computed for systems of coupled chemical reactions. Analysis of the trajectories generated in these simulations allows for studies of the stochastic fluctuations in the numbers of molecules present in the system. The conclusions of these studies remain valid for both large and arbitrarily small numbers of molecules. However, it is not possible to use exact simulation methods to study systems containing a large number of molecules due to the computational cost of accounting for individual molecular collisions. For example, in Gillespie's direct method two random numbers must be computer generated for every elementary reaction event. It was shown that to perform a stochastic simulation of a single intensive enzymatic reaction occurring in the timescale of one cell generation, ∼109 random numbers must be generated (Kierzek, 2002). Even with the application of recent advances in the exact stochastic algorithms, the number of reaction events that may be simulated is of the order of 1010 per day on a single CPU (Endy and Brent, 2001). Taking into account that for every computational experiment many trajectories need to be simulated and that many experiments must be performed to study the system with different parameter configurations, the application of exact stochastic simulation algorithms to large metabolic networks is not practical.
As it is clear from the above considerations, there is a gap between the stochastic and deterministic regimes in the simulations of biochemical processes. Intensive metabolic reactions involving 108–109 molecules may be accurately modeled using deterministic formulation of chemical kinetics. This approach cannot be used to study cellular processes such as gene regulation that involve very small numbers of molecules. The exact stochastic computer simulation algorithms capable of modeling these processes are in turn unable to model metabolic reactions due to the computational cost. Therefore, there is a need for the consistent computer simulation algorithm allowing one to study simultaneously systems involving gene expression, signal transduction, and enzyme activity.
The problem of efficient simulation of systems involving reactions varying across multiple scales of time and molecular concentrations has been already addressed by Haseltine and Rawlings (2002) and Rao and Arkin (2003). Haseltine and Rawlings partition the system into the subsets of “slow” and “fast” reactions, and approximate the fast reactions either deterministically or as Langevin equations. In the method of Rao and Arkin, some of the reactions are explicitly simulated with the Gillespie algorithm whereas others are described by random variables distributed according to the probability density functions at quasistationary state. Both of the methods require direct intervention of the modeler to partition the system into reaction sets covering different time and concentration regimes. In this work we present the “maximal time step method,” an alternative algorithm for stochastic kinetic simulations of biochemical systems, which combines the Gibson and Bruck (2000) algorithm with Gillespie's (2001) “τ-leap” method used to simulate “slow” and “fast” reaction subsets, respectively. The new algorithm is also capable of automatic partitioning of the reaction sets. We also show the application of the algorithm to the stochastic simulation of sugar metabolism, inducer exclusion, and catabolic repression processes in E. coli. To the best of our knowledge this is the first attempt to simulate the stochastic kinetics of a system composed of reactions describing regulation of gene expression, enzyme activity, and transport and signal transduction processes simultaneously. This allows us to study the stochastic effects occurring in a system containing all the essential elements responsible for regulation of cellular metabolism. Our results indicate that stochastic fluctuations in gene expression can propagate to the level of metabolic processes and cause significant physiological effects in a particular cell. We show that random delays in the induction of glycerol and lactose operons cause population heterogeneity when the cells switch carbon source from glucose to a mixture of lactose and glycerol. In a small fraction of cells switching from glucose to lactose, random delays in the lactose operon induction result in severe decrease in metabolic activity. Both effects are epigenetically inherited by the progeny of the cell in which random delay in gene expression occurred.
In the following section we shall present formulation and justification of the maximal time step method. Theoretical background will be briefly introduced to establish notation and the algorithm will be formulated and justified by both theoretical considerations and numerical comparisons with the Gillespie algorithm. Quasistationary-state approximation will also be introduced in the context of stochastic chemical kinetics and our algorithm. In the last subsection, performance of the maximal time step method will be evaluated by comparison with the Gibson and Bruck (2000) approach. Subsequently, we shall present the model of E. coli sugar metabolism, used to test the maximal time step method, and investigate stochastic effects occurring in the complex biochemical reaction networks. In the results section we shall present computer simulations of the E. coli sugar metabolism model. The article ends with the discussion of the maximal time step method and the molecular mechanisms responsible for the stochastic effects observed in the simulation of the model system.
COMPUTER SIMULATION ALGORITHM
In the following sections we will present a novel stochastic simulation algorithm for systems of coupled chemical reactions. We shall begin by establishing our notation and briefly reviewing the details of stochastic chemical kinetics relevant to our work. Then the algorithm will be justified by both theoretical considerations and numerical tests.
Stochastic chemical kinetics and stochastic partitioning of the system
We will be concerned with the system of N chemical species (S1,…,SN) that interact through M reactions (R1,…,RM) in the specified volume V of reaction environment at constant temperature. The dynamical state of the system is specified by X(t) = (X1(t),…,XN(t)), where Xi(t) is the number of molecules of the i-th species present in the reaction environment at time t. If the system is well stirred or the number of nonreactive molecular collisions is significantly larger than the number of reactive collisions (Gillespie, 1977), each reaction Rμ can be described by its propensity function aμ such that:
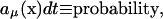 |
(1) |
given the current state of the system X(t) = x, that reaction Rμ will occur somewhere inside the volume V in the next infinitesimal time interval (t,t + dt).
Reaction Rμ is fully characterized by the propensity function aμ and the state-change vector νμ such that:
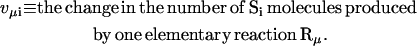 |
(2) |
For the elementary reaction mechanisms, propensity function of reaction Rμ is computed as the product of stochastic rate constant cμ and the number of distinct Rμ molecular reactant combinations available in the current state of the system x. For example, if Rμ is the reaction S1 + S2 → S3 then the aμ(x) = cμ X1 X2 and νμ = (−1,−1,+1,0,…,0). If the quasisteady-state assumption is used, aμ(x) may be computed as the other function of the numbers of reactants present in the system at state x (Rao and Arkin, 2003).
The time evolution for the system under consideration is completely described by the chemical master equation (McQuarrie, 1967), which gives the probability P(x,t|x0,t0) of the system being in the state x at time t, given the initial state of the system x0 at time t0:
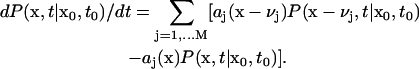 |
(3) |
Due to the intractability of the chemical master equation, the stochastic simulation algorithms were formulated, which use the reaction probability density function P(τ,μ|x,t) to generate the statistical sample of the system time courses. By definition P(τ,μ|x,t)dτ is the probability, given the state of the system X(t) = x, that the next reaction in the system will occur in the infinitesimal time interval (t + τ, t + τ + dτ) and will be an Rμ reaction, and it has the form (Gillespie 1977):
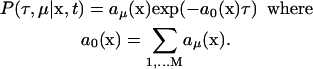 |
(4) |
It has been shown that both the chemical master equation and the reaction probability density function are rigorous consequences of Eq. 1.
In the stochastic simulation a random pair (τ,μ) is generated according to joint probability density function P(τ,μ|x,t), and the simulation variables are updated in the following way: i), the state of the system is updated by adding the state change vector νμ; ii), the time of the simulation t is increased by τ; and iii), the propensity functions of all the reactions are recomputed. Iteration of these steps, until the preset timescale is covered, results in the single time course of the system. The appropriate number of the independent simulations generates a sample of time courses that are used to compute statistical properties of the system.
There are two equivalent algorithms for generation of (τ,μ). In the direct method τ is generated as a sample of E(a0(x)), the exponential random variable with parameter a0(x), and μ is an integer number drawn from the interval [1,…,M] with the point probability aμ(x)/a0(x). In the first reaction method the tentative reaction time τμ is generated for every reaction as a sample of E(aμ(x)). Subsequently, the reaction with the least tentative time is chosen as the reaction that will occur next and therefore the random pair (τ,μ) is generated as (τmin = min(τ1,…,τM), Rmin ). The first reaction method is less effective than the direct method because in the single iteration of the algorithm the random number must be generated for every reaction whereas only two random numbers are required by the single iteration of direct method. However, this method has been modified by Gibson and Bruck (2000), which resulted in the most effective stochastic simulation algorithm referred to as the next reaction method.
In the next reaction method the tentative waiting time is computed with respect to the starting time of the simulation rather than with respect to the current simulation time. In the given step of the simulation the tentative waiting time is also generated only for the reaction that occurred in this step (for the one for which the previously generated random number was “used”). For other reactions in the system the reaction times are computed according to the formula:
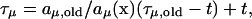 |
(5) |
where τμ is the tentative waiting time of reaction Rμ, t is simulation time, and aμ,old and τμ,old are the propensity function and waiting time of the reaction Rμ in the previous step.
Moreover, only those propensity functions, the values of which have been affected by the reaction that occurred in the current step, are computed. Therefore, single iteration of the Gibson and Bruck method requires generation of only one pseudo-random number and the number of propensity function updates is also greatly reduced. Determination of propensity functions to be updated is facilitated by the application of dependency graph and indexed priority queue. The reader is referred to the original work (Gibson and Bruck, 2000) for the details and justification of the method.
The methods described above are frequently referred to as exact simulation algorithms. These method are rigorous consequences of the fundamental hypothesis stating that every reaction may be characterized by the propensity function (Eq. 1; see Gillespie (1977) for details). This accuracy is, however, achieved at the price of a significant computational burden because for every reactive collision happening in the system at least one random number must be generated. Gillespie (2001) has formulated an approximated method that achieves significant gain in the speed of stochastic simulations with an acceptable loss in accuracy. In this approach, dubbed the τ-leap method, the number of reactive collisions kμ happening within the specified time step τ is computed for every reaction Rμ as P(aμτ), a Poisson random variable with parameter aμτ. Subsequently, the time of the simulation is increased by τ and the state of the system is updated taking into account kμ “firings” of every reaction in the system:
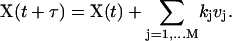 |
(6) |
Therefore, in the single iteration of the algorithm a much larger interval of simulation time is covered than in the case of exact methods because the time step τ is expected to be significantly longer than the exact waiting time for the individual reactive collision. Similar to the methods for numerical integration of deterministic kinetic equations, the accuracy of τ-leap depends on the time step. The value of τ should be such that the change of propensity functions for all the reactions in the system is “effectively infinitesimal” within the time interval (t,t + τ) where t is the current simulation time. For the systems involving large numbers of molecules, the kμ values are in the order of tens of individual reaction events and will not cause “noticeable” changes of propensity functions. In this case many elementary reactions could be “leaped over” and the time step could be large. It is also worth noting that within the limit of increasingly large numbers of molecules the τ-leap method converges to chemical Langevin equation and deterministic kinetics. It can easily be shown that when the propensity functions in the system are very large the P(aμτ) ≈ N(aμτ, √aμτ), where N(aμτ, √aμτ) is the normal random variable with a mean and variance equal to aμτ. In this case kμ = aμ(x)τ + √aμ(x)τN(0,1) and the simulation is equivalent to numerical integration of the chemical Langevin equation (Gillespie, 2001). In the thermodynamic limit aj(x) τ → ∞ and kμ = aj(x)τ. In this case, simulation becomes equivalent to the Euler formula for numerically solving deterministic reaction rate equations.
The τ-leap method and numerical solutions of chemical Langevin equation and deterministic rate equations “leap” over many reaction events on the simulation time axis, which results in a significant gain in the speed of computations, with respect to exact simulation algorithms, in systems containing large numbers of molecules. If, on the other hand, the system contains even a single reaction with very small numbers of substrate molecules the assumptions of both deterministic kinetics and the chemical Langevin equation are no longer satisfied, and these methods cannot be used. The τ-leap method can be used in such a case but it is no longer efficient because the length of the correct time step is determined by the reaction with the smallest number of reactant molecules, and it is of the order of waiting times occurring in exact simulation algorithms (Gillespie, 2001). Therefore, the approximated methods mentioned above do not provide a practical solution for systems composed of reactions with propensity functions varying by several orders of magnitude. The intuitive solution to this problem would be to apply the exact simulation algorithms to the “slow” reactions, involving small numbers of molecules, and approximate the “fast” reactions, involving large numbers of molecules by a method that does not account for individual reactive collisions. This idea has been already formally justified (Haseltine and Rawlings, 2002; Rao and Arkin, 2003) and referred to as stochastic partitioning.
In the algorithm presented below we simulate the slow reaction subset by the Gibson and Bruck method and the fast reaction subset by the τ-leap method. This approach resembles the one used by Haseltine and Rawlings (2002), with the difference that the latter authors combine exact stochastic simulation with the chemical Langevin equation and deterministic kinetics. Similar to Rao and Arkin (2003), we also use quasistationary-state approximation to model reactions for which this assumption is valid and the parameters of elementary processes are difficult to estimate. The major difference between our approach and the methods described above is the dynamic partitioning of the system. During the simulation, reactions are being moved between the subsets, according to their propensity functions, rather than being assigned to the subset at the beginning of the simulation.
Stochastic simulation algorithm
In this section, we formulate a new stochastic simulation algorithm that we name the “maximal time step method.” For the clarity of description we introduce the following three procedures. The “Partition” procedure denotes all the operations used for the stochastic partitioning of the system. The “UpdateSlow” and “UpdateFast” procedures denote all the operations necessary to update propensity functions of the slow and fast reactions, respectively, and change assignment of some of these reactions to the reaction subsets. We will first describe the algorithm using these procedures to simplify description and subsequently present them in detail. The E(a) and P(a) will denote samples from random variables distributed according to exponential and Poisson distributions, respectively. Exponentially distributed random numbers are generated using the following formula:
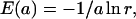 |
(7) |
where r is the random variable uniformly distributed over (0,1).
The Poisson random variable was generated as described by Atkinson (1979). The unit interval random numbers were generated by the Marsaglia et al. (1990) algorithm.
The maximal time step method
In our method the user sets the maximal time step κ (maximal τ-leap time). If, in the given iteration of the algorithm the minimal tentative time τmin of the slow reactions is within the next time step of length κ, then the slow reactions are updated according to the exact simulation method of Gibson and Bruck (2000), and the fast reactions are updated according to τ-leap method with the time step τmin−t (where t is the time of the simulation at the end of previous iteration). Otherwise, none of the slow reactions is considered to occur and the state of the system is updated according to the τ-leap method applied to the subset of fast reactions with the time step set to κ. This results in the following procedure:
Initialization
Set: i), the initial time of the simulation t to the initial value t0; ii), initial state of the system to x0 = (X1(t0),…,XN(t0)); iii), the state change vectors (ν1,…,νM) describing M reactions (R1,…,RM); iv), the maximum time step κ.
For each reaction Rμ build the list Lμ containing all reactions of which propensity functions are affected by the reaction Rμ (reactions, the substrates of which, are also either substrates or products of reaction Rμ).
For each reaction Rμ compute the propensity function aμ(x0) and the putative waiting time τμ = E(aμ(x0)) + t0.
Select reaction Rmin such that τmin = min(τ1,…,τM) set the simulation time t = τmin and compute new state of the system x(t) = νmin + x0.
Execute “Partition” procedure, which defines two reaction subsets: slow-reaction subset Sslow = (R1,…,RO) and fast-reaction subset Sfast = (RO+1,…,RM).
Iteration
If the slow reaction was executed in the previous iteration, execute “UpdateSlow” procedure.
Choose reaction Rmin ∈ Sslow such that τmin = min(τ1,…,τO).
If τmin − t = <κ, set the state of the system to νmin + x(t) and the time increment δt = (τmin−t).
If τmin − t > κ set δt = κ.
For every fast reaction Rμ ∈ Sfast, generate kμ = P(aμ δt).
Set the state of the system to x(t) + ∑j=O+1,…,M kjνj.
Increase time of the simulation t by δt.
Execute “UpdateFast” procedure.
Go to step 1.
Termination
Stop the calculations when t > Tmax.
Stochastic partitioning of the system is defined as follows. The reaction Rμ must satisfy two conditions to be assigned to the fast-reaction subset:
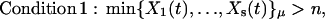 |
(8) |
where {X1(t),…,Xs(t)}μ, is the set of the numbers of molecules of the s substrates of reaction Rμ. In the case of the second order reaction involving molecules of the same species (2S1 → S2), the condition X1 > n/2 is used.
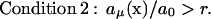 |
(9) |
The n and r are user-defined constants. It should be noted that min{X1(t),…,Xs(t)}μ corresponds to the maximal number of possible occurrences of reaction Rμ in the time interval in which it is considered to be independent of other reactions in the systems (substrates of Rμ are neither produced nor consumed by other reactions). The number of occurrences of isolated reaction Rμ cannot be larger than the amount of least numerous substrate. The ratio aμ(x)/a0 is the probability of the reaction Rμ to occur in the step of exact simulation algorithm. The “Partition” has, therefore, the following form:
Partition
Assign all reactions to the slow reaction subset Sslow.
For every reaction Rμ, move this reaction to the fast reaction subset Sfast if min{X1(t),…,Xs(t)}μ > n and aμ(x)/a0 > r.
Let τμ,old, aμ,old denote tentative waiting time and propensity function of the reaction Rμ at the last of the previous steps when aμ > 0. If aμ = 0, the τμ = +∞. Whenever, in the simulation, aμ = 0, the waiting time for the reaction Rμ is set to a very large number, exceeding Tmax. The “UpdateFast” and “UpdateSlow” procedures are defined as follows:
UpdateSlow
For the reaction Rμ ∈ Sslow that has occurred in the previous iteration execute the following steps:
Compute the propensity function aμ (x), where x is the current state of the system.
If min{X1(t),…,Xs(t)}μ > n AND aμ(x)/a0 > r move reaction Rμ to Sfast and go to step 4.
Generate τμ = E(aμ(x)) + t.
- For each reaction Rj ∈ Lμ.
- a) Compute aj(x).
- b) If min{X1(t),…,Xs(t)}j > n AND aj(x)/a0 > r move reaction Rj to Sfast.
- c) Else if Rj ∈ Sfast AND (min{X1(t),…,Xs(t)}j ≤= n OR aj(x)/a0 ≤= r), move Rj to Sslow and set: τj = E(aj(x)) + t.
- d) Else if Rj ∈ Sslow AND (min{X1(t),…,Xs(t)}j ≤= n OR aj(x)/a0 ≤= r) compute: τj = aj,old/aj(x)(τj,old − t) + t.
UpdateFast
For each reaction Rμ ∈ Sfast, execute the following steps:
Compute aμ(x).
If min{X1(t),…,Xs(t)}μ < n OR aμ(x)/a0 < r move Rμ to Sslow and set τμ = E(aμ(x)) + t.
- For every reaction Rj such that Rj ∈ Lμ AND Rj ∈ Sslow:
- a) Compute aj(x).
- b) If min{X1(t),…,Xs(t)}j > n AND aj(x)/a0 > r, move reaction Rj to Sfast and skip the next step.
- c) Compute τj = aj,old(x)/aj(x)(τj,old − t) + t.
Justification of the algorithm
Consider a particular step of the maximal time step method when the system is in the state x at the time t. We will show that this step can be considered as the following two independent simulations of length κ: i), the simulation performed with the next reaction method on the system composed of reactions belonging to Sslow; and ii), the simulation performed with the τ-leap method on the system composed of reactions belonging to Sfast. If this is the case, the algorithm presented above will be an approximated method correct within the assumptions of the τ-leap method (next reaction method is exact and brings no assumptions other than the fundamental hypothesis).
If κ will be set to the value no greater than the smallest reaction time within Sslow:
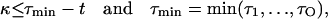 |
(10) |
then no slow reaction will occur in the time interval (t, τmin) and, as a consequence, simulation performed on Sfast will not be affected by slow reactions. If τmin − t = κ, then one reaction belonging to Sslow will occur. This single reaction event, occurring at exact time τmin, will not influence the τ-leap method simulation of Sfast within the time interval (t, τmin).
The condition for the correctness of the τ-leap simulation running on Sfast is that the changes of all propensity functions, within one time step, will be infinitesimally small (Gillespie, 2001):
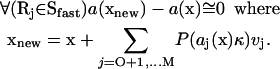 |
(11) |
If this condition is satisfied, reactions belonging to Sfast would cause infinitesimally small change in the propensity functions of reactions belonging to Sslow. Therefore, the simulation running on Sslow will be independent of the one running on Sfast within the limit of the same condition that is required for the correctness of the τ-leap method. We conclude, therefore, that the maximal time step and the τ-leap methods are correct within the limits of exactly the same approximation.
The argument presented above implies that at every step of the simulation the partitioning of the system into Sslow and Sfast and the maximal time step κ are such that in the next step the change of propensity functions of all fast reactions will be infinitesimally small. Gillespie (2001) presented various methods to estimate acceptable change of propensity functions during the simulation. In our opinion, optimization of the Sslow, Sfast, and κ at every step of the simulation would be computationally expensive. We have therefore used the two heuristic conditions, expressed by Eqs. 8 and 9 for the stochastic partitioning of the system. The maximal number of possible reaction occurrences (n) was used to assign reactions involving large numbers of molecules to Sfast, and those involving small numbers of molecules to Sslow. Additionally, the probability of the occurrence of the given reaction (r) was used to move to Sslow those reactions that have very low probability of occurrence (e.g., due to low stochastic rate constants) and avoid unnecessary generations of small kj values.
The parameters n, r, and κ can be selected empirically using the following rules. The parameter n may be arbitrarily set to 100 because in the case of lower values the change of propensity function resulting from a single “firing” of the fast reaction would be larger than 1%, which is a reasonable arbitrary threshold value to evaluate agreement with Eq. 11. The larger value of n should, however, be avoided as it would increase the number of slow reactions, for which individual reaction events are considered and decrease performance of the method. The value of maximal time step κ may be selected empirically by numerical tests. Too large maximal time step values would result in the numbers of “firings” of some fast reactions exceeding the numbers of available substrate molecules. This error is very easy to detect as it manifests itself by negative values of the numbers of molecules computed for certain molecular species. Very low maximal time step values would decrease performance of the method because the numbers of “firings” of fast reactions would become very low and the performance would become similar to that of an exact stochastic simulation. As discussed above, the parameter r is used to exclude reactions with very low rate constants and large numbers of substrate molecules from the list of fast reactions. This improves computational efficiency as the low numbers of reaction “firings” are not generated as the Poisson random numbers. Parameter r has to be set empirically by monitoring performance of the test calculations.
Fig. 1 presents a comparison of the maximal time step method and direct method for the model of LacZ and LacY gene expression described in detail elsewhere (Kierzek, 2002). This system involves slow reactions modeling gene expression processes and very intensive reactions describing enzymatic and transport activities of the LacZ and LacY products. Very good agreement with exact simulations results is achieved for the parameter values of n = 100, r = 10−4, and κ = 10−3 s. These parameters were used in other simulations described in this work.
FIGURE 1.

Comparison of the maximal time step method with exact stochastic simulation. Both methods were applied to the benchmark example of a constitutive lactose operon expression described in details elsewhere (Kierzek, 2002). The continuous lines represent the mean and ± 1 SD values computed according to 100 independent simulations with Gillespie algorithm. Dotted lines represent the mean and ± 1 SD time courses obtained with the maximal time step method. (A) The number of mRNA molecules. (B) The number of β-galactosidase reactions (denoted as product). Note that the maximal time step method is able to accurately simulate the trajectories for the chemical species, present in the same reaction environment, with amounts differing by seven orders of magnitude.
We conclude that the maximal time step method is an approximate stochastic simulation algorithm, which is correct within the limits defined by Gillespie (2001) for the τ-leap algorithm, and that the maximal time step method is able to accurately reproduce the results of exact stochastic simulations.
Quasistationary-state approximation
In the stochastic kinetic model, based on no other assumptions than the fundamental hypothesis, only elementary reaction mechanisms can be used. These are: i), conversion or decay of a single molecule; ii), reactive collision of two different molecules; and iii), reactive collision of two molecules of the same substance. The reactive collision of more than two molecules at the exact time is unlikely and can be represented as the sequence of bimolecular collisions. The stochastic rate constants of these reactions can be computed from deterministic rate constants. This rigorous approach is unfortunately not practical for modeling of large biochemical reaction networks because the kinetic constants of elementary reactions are difficult to measure and are, therefore, rarely available. On the contrary, in many cases the parameters of the deterministic rate equations describing complex reaction mechanisms (e.g., Michelis-Menten, Monod-Wyman-Changeux, etc.) are available. The complex reaction mechanisms are valid within the assumption that the instantaneous rates of change of some transitory intermediate species (e.g., enzyme-substrate complexes) are equal to zero on the timescale of interest. Following Rao and Arkin (2003) we will refer to this assumption as the quasisteady-state assumption (QSSA). The QSSA allows elimination of intermediate species from the model. Their presence is implicitly accounted for in the equations describing complex reaction mechanisms.
Rao and Arkin (2003) have shown that QSSA can be applied in the context of stochastic kinetics and Gillespie algorithm simulations. They have derived chemical master equations describing Michaelis-Menten and competitive inhibition reaction mechanisms. In both cases the enzymatic activities could be approximated, in the stochastic framework, by the reactions describing overall enzymatic activity with the propensity functions set to familiar expressions of deterministic kinetics. For example, the enzyme with a Michaelis-Menten mechanism could be modeled as a single reaction:
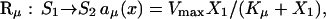 |
(12) |
where S1, S2 denote substrate and product, respectively. Rao and Arkin (2003) concluded that similar derivations could probably be provided also for other complex reaction mechanisms known from deterministic kinetics. They have also shown that the computation of propensity functions according to complex reaction mechanisms is one of the ways to include QSSA into Gillespie algorithm simulation. The same idea has been also numerically tested by van Gend and Kummel (2001). On the other hand, recent results of Bundschuh et al. (2003) show that if the Hill equation is used to model the propensity of transcription initiation in autoregulated genes, the magnitude of stochastic fluctuations is significantly overestimated. This example shows that application of certain complex reaction mechanisms in the context of particular network topologies may result in errors in noise levels.
The primary goal of our modeling studies is to show the applicability of the maximal time step method to simulation of biochemical reaction networks, including simultaneously gene expression signal transduction and enzymatic activities. Building of a suitable test case model without assuming complex reaction mechanisms would not be possible. It is also clear that in the near future, complex reaction mechanisms, properly describing both means and variances will be formulated and used in the stochastic simulations. For example Bundschuh et al. (2003) formulated an effective reaction mechanism, alternative to the Hill equation, that allowed correct variance estimation in the case of autoregulated genes. Thus, it is useful to test the applicability of the maximal time step method on a biochemical reaction network involving all the essential components, even if some of the propensity functions are modeled by equations that may introduce errors in noise estimation. We have, therefore, tested the applicability of the method on the model involving various complex reaction mechanisms, and discussed the influence of this approximation on the simulation results.
Performance of maximal time step method
We have used the model of LacZ and LacY gene expression (Kierzek, 2002) as a benchmark example to evaluate performance of the maximal time step method in comparison with the Gibson and Bruck method, the most effective algorithm of exact stochastic simulations. Simulation of the single trajectory for this system by the maximal time step method required an average of 3.5 × 107 random number generations whereas simulations with the Gibson and Bruck method required 1.5 × 109 random numbers. Thus, the maximal time step method required ∼42 times fewer executions of the random number generator than the Gibson and Bruck method. In terms of computational times (compared on Athlon 1700XP+ processor under Linux operating system) the simulation of the single trajectory for the benchmark system with the maximal time step method was on average 36 times faster than the simulation with Gibson and Bruck algorithm as implemented in our program. The discrepancy between the performance gains expressed in terms of random number generations and execution times results from a high cost of the Poisson random number generation. The computational efficiency could be further increased, without a noticeable loss of accuracy, by the application of the Michaelis-Menten reaction mechanism to model β-galactosidase activity. When the complex reaction mechanism was applied, the average number of random number generations per trajectory was 155 times smaller than in the case of the Gibson and Bruck method (9.6 × 106) and execution time was 107 times shorter.
STOCHASTIC KINETIC MODEL OF GLUCOSE, LACTOSE, AND GLYCEROL METABOLISM IN E. COLI
We have constructed the stochastic kinetic model of glucose, lactose, and glycerol metabolism in E. coli to test the applicability of the maximal time step method and to study the stochastic effects in a system composed of all the essential elements of biochemical reaction networks. The model included the following biochemical processes: i), all enzymatic activities taking part in the conversion of glucose, lactose, and glycerol to pyruvate; ii), metabolic regulation of these enzymes; iii), PTS-dependent transport of glucose and PTS-independent transport of lactose and glycerol; iv), inducer exclusion by the PTS-dependent kinase cascade; v), cAMP synthesis by adenylate cyclase; vi), expression of all the genes that products take part in the metabolic and regulatory processes present in the model; vii), gene regulation by CRP, LacR, and GlpR transcription factors. The model contains 94 substances interacting through 120 reaction channels. It contains both reactions involving very small numbers of molecules, such as regulation of the lactose operon (10 LacR molecules binding to 1 promoter “molecule”) and very intensive reactions with the rates reaching 107 reaction events per second (activity of the transporters). The difference between the smallest and largest values of the propensity functions in our model may be as large as nine orders of magnitude. One should also note that the model includes representative examples of many essential cellular processes, i.e., gene regulation, activity, and regulation of metabolic enzymes, transport, and signal-transduction cascades. Therefore, the model provides a difficult and realistic example on which the maximal time step method can be tested. It also offers opportunity to study the stochastic effects in the large biochemical reaction network.
Another reason for selecting glucose, lactose, and glycerol metabolism in E. coli as our model system is the detailed knowledge of the biochemical reactions involved and the availability of quantitative parameters. In the series of recent works (Wang et al., 2001; Kremling et al., 2001; Kremling and Gilles, 2001) quantitative data concerning the metabolism of glucose, lactose, saccharose, and glycerol were collected from the literature and the dynamics of biochemical reaction networks was studied using deterministic kinetics. Authors built models of bacterial strains growing on either glucose/lactose (Kremling et al., 2001) or saccharose/glycerol (Wang et al., 2001). To verify the model and identify unknown quantitative parameters, measurements of gene-induction kinetics and substrate composition have been performed. Thus, the models of Kremling et al. (2001) and Wang et. al. (2001) constitute a unique collection of experimentally verified kinetic parameters for a large biochemical reaction network.
We have used the results of the works described above to construct our model. Most of the reactions in these publications were modeled by the complex reaction mechanisms, and it was not possible to estimate the parameters of the elementary reactions. Therefore, the propensity functions of enzymatic, transport, and most of the signal-transduction reactions in our model were computed using complex reaction mechanisms parameterized by Kremling et al. (2001) and Wang et al. (2001). We have, however, used a more detailed model of gene expression. In the works of Kremling et al. and Wang et al., mRNA was treated as a transient intermediate and was not present in the model. We have treated mRNA explicitly and used a two-step model of gene expression in which transcription, translation, and mRNA degradation were modeled as separate reactions. On the other hand we did not use more detailed models of gene expression, involving isomerization of a closed complex and treating explicitly RNA polymerase and ribosome binding (e.g., Kierzek et al. (2001)). Recently, Swain et al. (2002) have shown that a simplified two-step model of gene expression is capable of reproducing the properties of the stochastic kinetics of a more complex model. Our numerical comparisons of the detailed model (Kierzek et al., 2001) and the simplified, two-step one (data not shown) are in agreement with these conclusions. We believe, therefore, that simplified models of gene expression, in which transcription and translation initiation reactions are “lumped” together and modeled as pseudo-first-order reactions or complex reaction mechanisms, can be used for the modeling of complex biochemical reaction networks. Transcriptional regulation by CRP, LacR, and GlpR proteins was modeled by computing propensity functions of transcription initiation reactions according to complex reaction mechanisms parameterized by Kremling et al. (2001) and Wang et al. (2001).
A simple model of cell division was used to perform simulations in the timescale of several cellular generations. In all the simulations the generation time of 2100 s was used. The dependence of the generation time on the state of the system was not studied in the current version of the model. During the simulation of the particular bacterial generation, the volume of the reaction environment was linearly doubled and after the simulation reached the generation time, the numbers of all molecules except DNA elements were divided by two and the volume was reset to its initial value. The growth of the volume was simulated by linear decrease of all the volume-dependent rate and equilibrium constants in the model (see Kierzek et al. (2001); Kierzek (2002) for details).
In the deterministic simulations, the concentrations of molecules in the total volume of all the cells growing in the medium are studied. To account for the stochastic effects, the numbers of molecules contained in the volume of a single cell must be used. Therefore, the initial amounts of extracellular sugar molecules must be calculated as the numbers of molecules per single cell rather than as the concentrations in the medium. We have assumed that the volume of the environment in which the cells grow is much larger than the total volume of growing cells and, therefore, does not change during the experiment. Thus, the parameters of reactions describing transport of substances from the extracellular environment into the cell were not changed during the simulation.
A full list of reactions, complex reaction mechanism formulas and parameters of our model are given in the supplementary material. Fig. 2 shows the schema of a biochemical reaction network defined by the model.
FIGURE 2.

Schema representing the model of E. coli glucose, lactose, and glycerol metabolism used in this work. Nodes represent substances and arrows represent reactions. The node shapes have the following meanings: ellipsoid, DNA and mRNA species; tetragonal, metabolites; hexagonal, enzymes and transporters; trapezoid, external pools of glucose, lactose, and glycerol. The names of the molecular species are explained (see online supplementary material). Different lines denote different reaction classes: metabolic reactions (solid line); protein synthesis (dotted line); transcription regulation (dashed line); and metabolic regulation (dash-dotted line). Color version of the schema is included (see online supplementary material).
RESULTS
We have performed the maximal time step method simulation of the system described above in which initial conditions were set to the following values. The numbers of all DNA elements were set to 1, the number of external glucose molecules was set to 1012, the number of ATP molecules was set to 106, and the numbers of all other molecules were set to 0. Within the timescale of the simulation, covering 10 bacterial generations, the system reached stationary state and the average number of molecules, present at the end of simulation, was computed for every molecular species according to 100 independent time courses. These values (see online supplementary material) served as the initial conditions for seven subsequent maximal time step method simulations in which the initial numbers of external glucose, lactose, and glycerol molecules were varied (Table 1). In every experiment 100 individual time courses of the system, representing individual cells, were simulated. In simulations 1–4 a large number of external glucose molecules was accompanied by a large number of lactose and/or glycerol molecules. The amounts of sugar molecules were set in such a way that the numbers of carbon units were equal in the different carbon sources; the number of lactose molecules was twice lower than the number of glucose molecules and the number of glycerol molecules was twice higher. The number of glucose molecules corresponded to experimental conditions used by Kremling et al. (2001). According to the results of simulations 1–4, presented in Table 1, lactose and glycerol operons remain repressed if glucose is available in the medium. As a consequence, only glucose is used as a carbon source, and glycerol and lactose are not consumed under these conditions. In the three subsequent simulations the initial number of glucose molecules was decreased 1000 times. Under these conditions the external glucose was depleted after ∼5000 s, the glycerol or lactose operons were induced, and the cells used other carbon sources. When both glycerol and lactose were present in the medium, the lactose operon was expressed at a higher level than the glycerol operon and lactose was the preferred carbon source (simulation 7, Table 1).
TABLE 1.
Initial conditions of the computer simulations performed in this work and resulting induction of Lac and Glp operons
| Number of external glucose molecules/cell* | Number of external lactose molecules/cell* | Number of external glycerol molecules/cell* | Number of LacZ molecules† | Number of GlpF molecules† | |
|---|---|---|---|---|---|
| 1 | 1012 | 0 | 0 | 0.00 ± 0.00 | 0.26 ± 1.21 |
| 2 | 1012 | 5 × 1011 | 0 | 0.02 ± 0.20 | 0.70 ± 3.83 |
| 3 | 1012 | 0 | 2 × 1012 | 0.00 ± 0.00 | 0.26 ± 1.21 |
| 4 | 1012 | 5 × 1011 | 2 × 1012 | 1.67 ± 16.51 | 0.64 ± 2.59 |
| 5 | 109 | 0 | 2 × 1012 | 10.22 ± 14.77 | 10,992 ± 685.6 |
| 6 | 109 | 5 × 1011 | 0 | 7694 ± 2051 | 79.55 ± 41.04 |
| 7 | 109 | 5 × 1011 | 2 × 1012 | 7968 ± 1572 | 621.9 ± 1864 |
The total number of sugar molecules present in the medium in the experimental setup of Kremling et al. (2001) was divided by the total number of cells. The volume of the cell and the cell density were set to 10−15 L and 280 g/L, respectively.
The mean and standard deviation of the number of protein molecules at the beginning of the 10th bacterial generation.
We conclude that our model qualitatively reproduces the well-known phenomena of inducer exclusion, catabolic repression, and diauxic shift growth. The cells grown on a mixture of sugars use exclusively glucose as a carbon source, and the genes encoding proteins used for metabolism of other sugars are expressed at very low levels. If the glucose is depleted in the medium, the cells switch to one of the other available carbon sources. To assess quantitative accuracy of the model, we have compared the numbers of β-galactosidase molecules present in the cells grown on lactose (simulation 6) with the quantitative data of Kremling et al. (2001). According to our simulations, 7524 ± 2356 β-galactosidase molecules were present in the cell at the beginning of the 10th generation. The stationary number of LacZ protein molecules calculated according to the data of Kremling et al. (2001), for the cell volume equal to 10−15 L was 5500 (experimental error was not given). We considered this level of quantitative accuracy sufficient for testing the maximal time step method on a realistic example and to justify conclusions concerning stochastic effects on the model. We have not, therefore, fit model parameters further to achieve better agreement with β-galactosidase induction data.
The results of simulations 5, 6, and 7 indicate that the fate of single cells may be significantly different than the average population behavior after the switch of carbon source. Fig. 3 shows the time courses for the glycolytic pathway outflow obtained in the simulations. During the change of carbon source the glycolytic pathway outflow decreases as a result of glucose depletion and delay time in the synthesis of proteins necessary for the consumption of other sugars. On average, the activity of glycolytic pathway returns to its previous values after lactose or glycerol operons are activated and their products synthesized. However, in the case of several individual time courses the delay time of the return to the previous metabolic activity is much longer than the average, and there is one particular trajectory in which the outflow of glycolytic pathway drops close to zero. This trajectory represents the cell that switches to the stationary phase or dies before it is able to use a carbon source other than glucose.
FIGURE 3.

The glycolytic pathway outflow under different conditions. The time courses for 10 bacterial generations are shown. After every generation time of 2100 s the number of all molecules in the system except DNA elements is divided by two and the volume is reset to its initial value. The glycolytic pathway outflow is computed as the number of pyruvate molecules processed by the first order reaction representing the total pyruvate consumption by all metabolic processes in the cell. The rate constant of this reaction was set to 10 1/s following Wang et al. (2001). Four plots represent glycolytic pathway outflow under different conditions. (A) Large number of glucose molecules. The time course shown was obtained in simulation 1 where glucose alone was present. Results for simulations 2, 3, and 4 were nearly identical. (B) Small number of glucose and large number of glycerol molecules. (C) Small number of glucose and large number of lactose molecules. (D) Small number of glucose and large numbers of glycerol and lactose molecules.
Fig. 4 shows the time courses for cAMP, EIIAP, and β-galactosidase in simulation 6 in which the single trajectory with significantly decreased metabolic activity occurred. The cAMP level is low when the cell grows on glucose, reaches its maximal value immediately after glucose depletion, and drops to the new stationary state. Very similar cAMP time courses were also obtained in simulations 5 and 7. According to the simulation results in the majority of individual cells catabolic operons are activated when the number of cAMP molecules is maximal. In the case of eight trajectories, activation of the lactose operon was significantly delayed but the operon was eventually activated despite the fact that cAMP level was no longer at its maximal value. In one of the cells, activation of the lactose operon was delayed so much that the activity of the glycolytic pathway became very low and the number of PEP molecules was not sufficient for phosphorylation of the EIIA protein. This resulted in the dilution of the remaining EIIAP pool in this particular cell, lack of adenylate cyclase activation by EIIAP, and a decrease in the cAMP level. In the absence of cAMP the lactose operon could not be activated, the cell could not use lactose as a carbon source, and the glycolytic pathway outflow was decreased close to zero.
FIGURE 4.

Induction of lactose operon. The time courses shown on the plots were obtained in simulation 6 (small number of glucose and large number of lactose molecules). Glucose is completely depleted during the first 5000 s of the simulation. Plots A, B, and C show trajectories for cAMP, EIIAP, and LacZ, respectively.
Significant differences between individual cells and the average outcome were also observed in simulation 7, modeling the growth on a mixture of lactose and glycerol. Fig. 3 D shows that all of the cells returned to the same stationary state of glycolytic pathway after the change of carbon source. One cell was significantly delayed but managed to recover. However, results of simulation 7 (Fig. 5) indicate that the population is heterogeneous. Most of the cells use lactose as a carbon source, but some of them grow on glycerol. The burst of cAMP level occurring after glucose depletion results in the activation of both glycerol and lactose operons in most of the cells. Subsequently, in the majority of individual time courses the glycerol operon is switched off. In ∼10% of cells, activation of the lactose operon is delayed and the glycerol operon becomes fully activated. In some of these cells the lactose operon becomes activated after a long delay time and the activity of the glycerol operon is repressed. In none of the individual cells both operons reach their stationary states simultaneously. Activation of the lactose operon results in repression of the glycerol operon. The mechanism that is responsible for this effect involves inhibition of glycerol kinase by fructose-1,6-bisphosphate, which is an intermediate product of hexose metabolism. Inhibition of glycerol phosphorylation results in a decrease in glycerol-3P, which activates the expression of the glycerol operon by binding to GlpR repressor.
FIGURE 5.

Induction of glycerol and lactose operons in the cells switching from glucose to the mixture of lactose and glycerol. Results of simulation 7 (small number of glucose and large numbers of glycerol and lactose molecules) are shown. Plots A, B, C, and D show the time courses for LacZ, GlpF, cAMP, and external glycerol, respectively. Plots B and D show the small subpopulation of cells that express proteins encoded by the glycerol operon and consume glycerol as a carbon source.
DISCUSSION
In this work we formulate the maximal time step method, a novel algorithm for the stochastic kinetic simulation of biochemical reaction networks. The method is an approximated stochastic simulation algorithm correct within the limit of the large values of propensity functions within the fast reaction subset. Numerical tests, performed on the example of constitutive lactose operon expression (Kierzek, 2002), show that the maximal time step method accurately reproduces the results of exact stochastic simulations in the case of systems in which the propensity functions of individual reactions differ by many orders of magnitude. The maximal time step method simulation is >30-fold faster than the exact stochastic simulation performed with the Gibson and Bruck algorithm. The simulation using both the maximal time step method and quasistationary-state approximation is ∼100-fold more efficient than the simulation performed with the Gibson and Bruck algorithm. Our method is an alternative to the related approach of Haseltine and Rawlings (2002). The two major differences are: i), our method uses Gibson and Bruck for the slow reaction subset and the Gillespie τ-leap method for the fast reaction subset instead of combining Gillespie's direct method with the integration of the chemical Langevin equation; ii), our method dynamically partitions the reactions to slow and fast subsets, according to the state of the system, whereas in the Haseltine and Rawlings method the partition of the system is specified during the initialization stage.
Despite the effort that is required to empirically determine parameters n, r, and κ, the user would benefit from the automatic partitioning of the reaction sets. In the course of simulations described above, many of the reactions change their propensity functions by many orders of magnitude. Therefore, permanent assignment of these reactions to the reaction subsets may result in the simulation of the individual reaction events for the fast reaction, which would decrease method performance, or in the τ-leap step applied to slow reaction, which could lead to an error. Moreover, according to our experience, both accuracy and performance of the maximal time step method is robust with respect to parameter choice. For example, in the simulations described above, the parameter κ rarely influenced calculations. In most cases one of the slow reactions occurred fast enough to determine the time step of the τ-leap step applied to fast reaction subset. The robustness of parameter choice also may be illustrated by the fact that parameters selected for the simulation of a simple LacZ, LacY gene expression model were applicable to the simulation of the complex system, including most of the classes of interactions observed in the biochemical reaction networks. This shows that the parameters presented in this work will most probably be a reasonable choice for the simulation of a variety of systems or at least provide a good starting point for further fine tuning. The choice of κ = 10−3 s is additionally supported by the observation that on average, the small number of propensity functions change within the time interval of 10−3 s. In the simulation of E. coli sugar metabolism model, containing 120 reactions, the average number of propensity functions that are affected within the time interval of 10−3 s is 22.
We apply the maximal time step method to the simulation of glucose, lactose, and glycerol metabolism in E. coli cells. To the best of our knowledge this is the first stochastic simulation of the kinetics of a system involving simultaneously the reactions of gene expression, signal transduction, transport, and enzymatic activity. We use the quasistationary-state approximation, recently reformulated in terms of stochastic chemical kinetics by Rao and Arkin (2003), to describe reactions by the complex mechanisms. This lets us use kinetic parameters experimentally verified by Kremling et al. (2001) and Wang et al. (2001). The simulations reproduce the catabolic repression and inducer exclusion phenomena of the E. coli cells growing on a mixture of carbon sources. Despite the reduction of the model, by the application of complex reaction mechanisms, there is still a difference of many orders of magnitude between the propensity functions of the reactions modeling gene expression and enzymatic activity. The maximal timestep method allows stochastic simulations of such a system that would not be feasible if the Gillespie algorithm alone was used.
Our simulations show that stochastic fluctuations in the reactions involving small numbers of molecules may propagate through the reaction network and influence the time courses of other processes in the system, including metabolic pathways in which large numbers of molecules are processed. In particular, the random delay in transcription initiation of the lactose operon in an individual cell, switching the carbon source from glucose to glycerol may result in almost complete shutdown of the glycolytic pathway. Another effect of the random delay in the activation of the lactose operon is the heterogeneity within the cellular population switching from glucose to a mixture of lactose and glycerol. Moreover, in both cases the effect of the random fluctuation occurring in a particular cell is inherited by its progeny. Delay in the activation of the lactose operon in the single cell may lead to a continuous decrease in the glycolytic pathway activity, spanning six generations of progeny cells (Fig. 3 C). It may also cause the progeny cells to use glycerol as the carbon source in the course of several generations (Fig. 5 B). Our results could be subjected to experimental verification by creating bacterial strains in which fluorescent proteins are expressed under control of the lactose and glycerol operons and measuring fluorescence intensity in single cells. This approach has been already applied to verify other hypotheses concerning stochastic effects in cellular processes (Blake et al., 2003, Elowitz et al., 2002; Ozbudak et al., 2002; Rosenfeld and Alon, 2003; Setty et al., 2003).
Both glycerol and lactose operons are regulated by a positive feedback loop. Increase in transcription activity of the operon results in an increased amount of the lactose/glucose-3-P, due to an increase in transport activities, which further derepresses the operon. It has been shown in the classical papers of Novick and Weiner (1957) and Cohn and Horibata (1959a,b) that the positive feedback loop present in bacterial operon is able to maintain an induced state, in the progeny of the induced cells, even if the inducer concentration is significantly decreased. The multistationary behavior of the positive feedback loop can also be presented by theoretical analysis (see Thomas (1998) and Savageau (2001) for reviews). A similar phenomenon occurs in our simulations, where both lactose and glycerol operons are induced by high amounts of cAMP and in most of the individual time courses are capable of maintaining the induced level when the cAMP concentration is lowered to the new stationary state.
A fast increase in cAMP amount, above the final stationary level, is an example of the overshooting phenomenon recently described as the property of the regulatory motif in which the negatively autoregulated unit is activated by an external signal (Rosenfeld and Alon, 2003). The cAMP negatively autoregulates its synthesis because the cAMP-CRP complex represses the expression of adenylate cyclase. This negatively autoregulated unit is activated by the increase in EIIAP, resulting from depletion of glucose (Fig. 2).
In a recent work, Thattai and Shraiman (2003) performed stationary state analysis of the differential equation model describing key processes in the PTS-dependent metabolic switching. Although the deterministic modeling techniques did not allow the authors to study stochastic effects directly, they have noted that biochemical noise may result in the heterogeneity within cellular populations and simultaneous occupation of all stationary states available for the biochemical reaction network under given conditions. The existence of the subpopulations in which either lactose or glycerol are used as a carbon source, predicted by our detailed kinetic simulations, are in agreement with this hypothesis. The effect of random fluctuations in gene expression in a particular cell on its progeny and the role of overshooting phenomenon in the activation of a catabolic operon have not been studied by Thattai and Shraiman (2003).
As discussed before, our model may include equations describing complex reaction mechanisms that introduce unrealistic fluctuations in particular reactions. It is therefore possible that our simulation exaggerates the variance of the lactose operon transcription initiation, and that under experimental conditions the population heterogeneity is smaller than in the case of our simulation results. Even if this is the case, our simulation still shows that the random event occurring at gene expression level may propagate, through the positive and negative feedback loops present in the biochemical reaction networks, and result in an epigenetically inheritable effect on cellular metabolism. This mechanism is an important factor in the natural selection of architectures and kinetic constants of reaction networks controlling gene expression. In some cases “noisy solutions” would be eliminated in the course of evolution because propagation of noise through the network would cause excessive perturbations in cellular physiology. In other cases, the phenomena described above may be explored as the way to increase variability of isogenic microbial population.
It is also important to note that the maximal time step method does not require any particular approach to modeling of the complex reaction mechanisms, because the propensity functions computed from elementary reaction mechanisms and other functions of molecular populations in the system can be used. Therefore, both the detailed parameters obtained by novel experimental approaches (Setty et al., 2003) and the effective reaction mechanisms correctly accounting for the noise may be easily incorporated into maximal time step method simulations. Moreover, simulation results presented in this paper show that stochastic effects are of interest in studies of the dynamics of complicated systems involving all essential components of bacterial physiological processes. We therefore believe, that the maximal time step method, will be useful in future studies of the complex dynamics of cellular processes. The software used for simulations presented in this work is available on request.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Supplementary Material
Acknowledgments
We are grateful to Professor Julio Collado-Vides for valuable comments and discussions.
A.K. acknowledges financial support from the European Union Center of Excellence MAMBA (QLRI-CT-2002-90383).
References
- Atkinson, A. C. 1979. The computer generation of poisson random variables. Appl. Statist. 28:29–35. [Google Scholar]
- Blake, W. J., M. Kærn, C. R. Cantor, and J. J. Collins. 2003. Noise in eukaryotic gene expression. Nature. 422:633–637. [DOI] [PubMed] [Google Scholar]
- Bundschuh, R., F. Hayot, and C. Jayaprakash. 2003. Fluctuations and slow variables in genetic networks. Biophys. J. 84:1606–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn, M., and K. Horibata. 1959a. Inhibition by glucose of the induced synthesis of the beta-galactoside-enzyme system of Escherichia coli. Analysis of maintenance. J. Bacteriol. 78:601–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn, M., and K. Horibata. 1959b. Analysis of the differentiation and of the heterogeneity within a population of Escherichia coli undergoing induced beta-galactosidase synthesis. J. Bacteriol. 78:613–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elowitz, M. B., E. D. Levine, E. D. Siggia, and P. S. Swain. 2002. Stochastic gene expression in a single cell. Science. 297:1183–1186. [DOI] [PubMed] [Google Scholar]
- Endy, D., and R. Brent. 2001. Modelling cellular behaviour. Nature. 409:391–395. [DOI] [PubMed] [Google Scholar]
- Edwards, J. S., R. U. Ibarra, and P. O. Palsson. 2001. In silico predictions of Escherichia coli metabolic capabilities are consistent with experimental data. Nat. Biotechnol. 19:125–130. [DOI] [PubMed] [Google Scholar]
- Gibson, M. A., and J. Bruck. 2000. Efficient exact stochastic simulation of chemical systems with many species and many channels. J. Phys. Chem. 104:1876–1889. [Google Scholar]
- Gillespie, D. T. 1977. Exact stochastic simulation of coupled chemical reactions. J. Phys. Chem. 81:2340–2361. [Google Scholar]
- Gillespie, D. T. 2001. Approximate accelerated stochastic simulation of chemically reacting systems. J. Chem. Phys. 115:1716–1733. [Google Scholar]
- Haseltine, E. L., and J. B. Rawlings. 2002. Approximate simulation of coupled fast and slow reactions for stochastic chemical kinetics. J. Chem. Phys. 117:6959–6969. [Google Scholar]
- Hoefnagel, M. H. N., M. J. C. Starrenburg, D. E. Martens, J. Hugenholtz, M. Kleerebezem, I. I. van Swam, R. Bongers, H. V. Westerhoff, and J. L. Snoep. 2002. Metabolic engineering of lactic acid bacteria, the combined approach: kinetic modelling, metabolic control and experimental analysis. Microbiology. 148:1001–1013. [DOI] [PubMed] [Google Scholar]
- Kierzek, A. M., J. Zaim, and P. Zielenkiewicz. 2001. The effect of transcription and translation initiation frequencies on the stochastic fluctuations in procaryotic gene expression. J. Biol. Chem. 276:8165–8172. [DOI] [PubMed] [Google Scholar]
- Kierzek, A. M. 2002. STOCKS: STOChastic Kinetic Simulations of biochemical systems with Gillespie algorithm. Bioinformatics. 18:470–481. [DOI] [PubMed] [Google Scholar]
- Kremling, A., and E. D. Gilles. 2001. The organization of metabolic reaction networks II. Signal processing in hierarchical structured functional units. Metab. Eng. 3:138–150. [DOI] [PubMed] [Google Scholar]
- Kremling, A., K. Bettenbrock, B. Laube, K. Jahreis, J. W. Lengeler, and E. D. Gilles. 2001. The organization of metabolic reaction networks III. Application for diauxic growth on glucose and lactose. Metab. Eng. 3:362–379. [DOI] [PubMed] [Google Scholar]
- Levin, B. 1999. Genes VII. Oxford University Press, Oxford, UK.
- Levin, M. D., C. J. Morton-Firth, W. N. Abouhamad, R. B. Bourett, and D. Bray. 1998. Origins of individual swimming behavior in bacteria. Biophys. J. 74:175–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsaglia, G., A. Zaman, and W. Tseng. 1990. Toward a universal random number generator. Stat. Prob. Letter. 8:35–39. [Google Scholar]
- McAdams, H. H., and A. Arkin. 1997. Stochastic mechanisms in gene expression. Proc. Natl. Acad. Sci. USA. 94:814–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuarrie, D. A. 1967. Stochastic approach to chemical kinetics. J. Appl. Prob. 4:413. [Google Scholar]
- Novick, A., and M. Weiner. 1957. Enzyme induction as an all-or-none phenomenon. Proc. Natl. Acad. Sci. USA. 45:553–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozbudak, E. O., M. Thattai, I. Kurtser, A. D. Grossman, and A. van Oudenaarden. 2002. Regulation of noise in the expression of single gene. Nat. Genet. 31:69–73. [DOI] [PubMed] [Google Scholar]
- Rao, C. V., D. M. Wolf, and A. P. Arkin. 2002. Control, exploitation and tolerance of intracellular noise. Nature. 420:231–237. [DOI] [PubMed] [Google Scholar]
- Rao, C. V., and A. P. Arkin. 2003. Stochastic chemical kinetics and the quasi-steady-state assumption: application to the Gillespie algorithm. J. Chem. Phys. 118:4999–5010. [Google Scholar]
- Rosenfeld, N., and U. Alon. 2003. Response delays and the structure of transcription networks. J. Mol. Biol. 329:645–654. [DOI] [PubMed] [Google Scholar]
- Savageau, A. M. 2001. Design principles for elementary gene circuits: elements, methods, and examples. Chaos. 11:142–159. [DOI] [PubMed] [Google Scholar]
- Setty, Y., A. E. Mayo, M. G. Surette, and U. Alon. 2003. Detailed map of a cis-regulatory input function. Proc. Natl. Acad. Sci. USA. 100:7702–7707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain, P. S., M. B. Elowitz, and E. D. Siggia. 2002. Intrinsic and extrinsic contributions to stochasticity in gene expression. Proc. Natl. Acad. Sci. USA. 99:12795–12800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sveiczer, A., A. Csikasz-Nagy, B. Gyorffy, and J. J. Tyson. 2000. Modeling the fision yeast cell cycle: quantized cycle times in wee1-Cdc25Δ mutant cells. Proc. Natl. Acad. Sci. USA. 97:7865–7870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thattai, M., and B. Shraiman. 2003. Metabolic switching in the sugar phosphotransferase system of Escherichia coli. Biophys. J. 85:744–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, R. 1998. Laws for the dynamics of regulatory networks. Int. J. Dev. Biol. 42:479–485. [PubMed] [Google Scholar]
- Tomita, M., K. Hashimoto, K. Takahashi, T. S. Shimizu, Y. Matsuzaki, F. Miyoshi, K. Saito, S. Tanida, K. Yugi, J. C. Venter, and C. A. Hutchinson 3rd. 1999. E-CELL: software environment for whole-cell simulation. Bioinformatics. 15:72–84. [DOI] [PubMed] [Google Scholar]
- Tyson, J. J., K. Chen, and B. Novak. 2001. Network dynamics and cell physiology. Nat. Rev. Mol. Cell Biol. 2:908–915. [DOI] [PubMed] [Google Scholar]
- Wang, J., E. D. Gilles, J. W. Lengeler, and K. Jahreis. 2001. Modeling of inducer exclusion and catabolite repression based on a PTS-dependent sucrose and non-PTS-dependent glycerol transport systems in Escherichia coli K-12 and its experimental verification. J. Biotechnol. 92:133–158. [DOI] [PubMed] [Google Scholar]
- van Gend, K. and U. Kummer. 2001. STODE—automatic stochastic simulation of systems described by differential equations. In Proceedings of the International Conference on Systems Biology. T. M. Yi, M. Hucka, M. Morohashi., and H. Kitano, editors. Omnipress, Madison, WI. 326–332.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.