

Abstract
Gramicidin A was incorporated at a peptide/lipid ratio of 1:10 mol/mol in aligned bilayers of dimyristoyl phosphatidylcholine (DMPC), phosphatidylserine (DMPS), phosphatidylglycerol (DMPG), and phosphatidylethanolamine (DMPE), from trifluoroethanol. Orientations of the peptide and lipid chains were determined by polarized attenuated total reflection infrared spectroscopy. Lipid-peptide interactions with gramicidin A in DMPC bilayers were studied with different spin-labeled lipid species by using electron spin resonance spectroscopy. In DMPC membranes, the orientation of the lipid chains is comparable to that in the absence of peptide, in both gel and fluid phases. In gel-phase DMPC, the effective tilt of the peptide exceeds that of the lipid chains, but in the fluid phase both are similar. For gramicidin A in DMPS, DMPG, and DMPE, the degree of orientation of the peptide and lipid chains is less than in DMPC. In the fluid phase of DMPS, DMPG, and DMPE, gramicidin A is also less well oriented than are the lipid chains. In DMPE especially, gramicidin A is largely disordered. In DMPC membranes, three to four lipids per monomer experience direct motional restriction on interaction with gramicidin A. This is approximately half the number of lipids expected to contact the intramembranous perimeter of the gramicidin A monomer. A selectivity for certain negatively charged lipids is found in the interaction with gramicidin A in DMPC. These results are discussed in terms of the integration of gramicidin A channels in lipid bilayers, and of the interactions of lipids with integral membrane proteins.
INTRODUCTION
Gramicidin A is a 15-residue hydrophobic peptide (Fm-VGALAVVVWLWLWLW-Etn) that is composed of alternating D- and L-amino acids, which promote the formation of β-helices. The peptide displays structural polymorphism, depending on the solvent, but a head-to-head dimer of β6.3 single helices is known to be the unit that forms ion-conducting channels in lipid-bilayer membranes. It is the latter that is the basis for the antibiotic activity of gramicidin A. A high-resolution structure of the β6.3-helix dimer has been determined in oriented lipid bilayers (Ketchem et al., 1993, 1997). This conformation is characterized by a ring of four tryptophan residues that are located at the membrane-water interface.
Depending on solvent conditions, gramicidin A may additionally form antiparallel ↑↓β7.2 double helices (Wallace and Ravikumar, 1988). In principle, a variety of both single and double helices is possible structurally. Reconstitution in dimyristoyl phosphatidylcholine (DMPC) membranes from trifluoroethanol (TFE) produces the β6.3 single-helical form, but reconstitution from chloroform-methanol causes admixture of the nonconducting ↑↓β7.2 double-helical form (Bouchard and Auger, 1993). Prolonged incubation tends to promote interconversion between these forms (Vogt et al., 1994).
The relatively small size and known structure of gramicidin A makes this a useful system for biophysical studies of membrane incorporation and lipid-protein interactions. Extensive studies have been made of the influence of gramicidin on lipid polymorphism (Killian, 1992), and of the ordering effects on the lipid chains (Rice and Oldfield, 1979; Killian, 1992). The effects on lipid dynamics have been investigated by spin-label electron spin resonance (ESR) spectroscopy (Patyal et al., 1997; Ge and Freed, 1999). Additionally, molecular dynamics (MD) simulations have been performed to characterize lipid interactions with the peptide, on a submolecular scale (Woolf and Roux, 1994, 1996).
Understanding the behavior of membranous antibiotics in bilayers of various lipids is key to the synthesis of new hydrophobic peptides with the desired membrane selectivity. Here, we study the integration of gramicidin A in bilayer membranes composed of different zwitterionic and anionic lipids, by orientation measurements with attenuated total reflection (ATR) infrared spectroscopy. There is considerable uncertainty in the literature as regards the orientations, ΘM, of the infrared transition moments of gramicidin, which are essential for determining the orientation of the peptide in the membrane (Nabedryk et al., 1982; Bouchard and Auger, 1993; Ulrich and Vogel, 1999). This problem is resolved here by spectral simulations, using the intensities and frequencies from normal mode calculations by Naik and Krimm (1986a). Further, we also investigate the stoichiometry and selectivity of lipid interactions with gramicidin A systematically by using different spin-labeled lipid species, in conjunction with ESR spectroscopy. Gramicidin A is an ideal model membrane protein with which to investigate selectivity of interactions with lipid headgroups that are not attributable to charged residues, because anchoring of the hydrophobic transmembrane peptide arises solely from interfacial tryptophan side chains (Hu and Cross, 1995). A particular goal of the ESR studies is to determine the possible role of interfacial tryptophan residues in lipid selectivity and in lipid-protein interactions in general. Samples are prepared from TFE to ensure formation of the channel structure, as characterized by infrared spectroscopy (Bouchard and Auger, 1993). It is found from spin-label spectroscopy that the lipid-peptide interactions in samples prepared from chloroform-methanol differ significantly from those prepared from TFE. Previous studies on gramicidin with spin-labeled lipids were devoted solely to the former method of preparation (Ge and Freed, 1999).
MATERIAL AND METHODS
Materials
Gramicidin A was from Sigma Chemical Co. (St. Louis, MO). Phospholipids, DMPC, DMPS, DMPG, and DMPE were from Avanti Polar Lipids (Alabaster, AL). Spin-labeled stearic acid, 14-SASL, was synthesized according to Hubbell and McConnell (1971). Spin-labeled phosphatidylcholine, 14-PCSL, was synthesized by acylation of lysophosphatidylcholine with 14-SASL, as described in Marsh and Watts (1982). Other spin-labeled phospholipids, 14-PSSL, 14-PGSL, 14-PESL, and 14-PASL, were prepared from 14-PCSL by headgroup exchange mediated by phospholipase D (Marsh and Watts, 1982).
ATR spectroscopy
Polarized ATR infrared spectra were recorded on a Bruker (Karlsruhe, Germany) IFS-25 Fourier transform spectrometer at a resolution of 2 cm−1. A horizontal ATR accessory from Specac (Orpington, U.K.), was used with a zinc selenide crystal (45° angle of incidence, six reflections). This was modified to seal the sample chamber hermetically from above, and to thermostat the cell housing with internally circulating water from a Haake (Karlsruhe, Germany) temperature-controlled bath. The surface of the ATR crystal available for coating by the sample was 8 × 45 mm. A germanium-mounted, wire-grid, linear polarizer (Specac) was used in the incident beam. Typically, 128–256 interferograms were co-added, and Fourier transformed after two levels of zero filling and apodization with a Blackman-Harris three-term function.
Samples containing 1.5–3 mg of lipid with 1:10 mol/mol gramicidin/lipid ratio were dried down on the ZnSe ATR crystal from a 10 mg/ml (DMPC), 3 mg/ml (DMPG and DMPE), or 1.5 mg/ml (DMPS) solution in TFE. Spectra of the dry sample were then recorded at the desired temperatures with radiation polarized parallel and perpendicular to the plane of incidence. The sample was then hydrated with 50 μl of D2O (DMPC and DMPE), or of 10 mM Hepes, 2 mM EDTA, 1 M NaCl, pH 7.8, D2O-buffer (DMPS and DMPG), by incubating above the chain-melting transition of the lipid bilayer, before again recording the polarized ATR spectra. Next, the sample was dried in a nitrogen gas stream for 12 h. Polarized spectra of the dry sample were then rerecorded. Finally, the sample was rehydrated with D2O, or D2O-buffer, and a second set of polarized spectra of the hydrated sample was recorded. For calculation of dichroic ratios, spectra from those dry and hydrated samples that exhibited the strongest dichroism were used. After baseline subtraction, bands in the amide I, II, and ester CO region (1520–1750 cm−1), and in the CH stretching region (2830–2975 cm−1), were fitted with Lorentzian components by using nonlinear least-squares minimization. The amide A region (3110–3640 cm−1) was fitted with matching Gaussian bandshapes. Second-derivative spectra were used to provide initial estimates of the component band positions. Dichroic ratios were calculated by using the integrated intensities of the corresponding bands.
Molecular orientations are calculated from the measured dichroic ratios, R, according to (Marsh, 1999a):
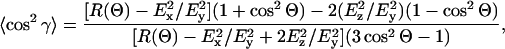 |
(1) |
where γ is the orientation of the molecular axis relative to the normal to the orienting substrate (i.e., the ATR plate), and Θ is the orientation of the transition moment relative to the molecular axis. Angular brackets in Eq. 1 indicate summation over the (normalized) orientational distribution. The strengths of the electric field components, with a zinc selenide ATR crystal, are: 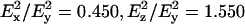 in the thick film approximation, which is appropriate to the amount of material used (Marsh, 1999a). Order parameters,
in the thick film approximation, which is appropriate to the amount of material used (Marsh, 1999a). Order parameters, 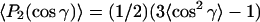 are also used to define the molecular orientations. These order parameters are a composite of the molecular orientation in the membrane and the degree of alignment of the membrane relative to the surface of the ATR crystal. Effective tilt angles, γeff, are obtained from
are also used to define the molecular orientations. These order parameters are a composite of the molecular orientation in the membrane and the degree of alignment of the membrane relative to the surface of the ATR crystal. Effective tilt angles, γeff, are obtained from 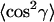 by assuming a singular value of γ. Values of γeff obtained for the lipid chains in the gel phase are used to assess the degree of macroscopic alignment of the membranes, as described in the Results section.
by assuming a singular value of γ. Values of γeff obtained for the lipid chains in the gel phase are used to assess the degree of macroscopic alignment of the membranes, as described in the Results section.
The orientation of the transition moment is taken as ΘC = 90° for the methylene and carbonyl stretch bands of the lipid. The transition moment orientations for the amide I band of gramicidin A are considered in the following main section, based on the normal mode calculations of Naik and Krimm (1986a). A value of ΘI = 32°, corresponding to the maxima in the low frequency region of the amide I band, is chosen from bandshape simulations. This approach is necessary because of the strong overlap of the modes that is predicted for the β6.3-helix (see Theoretical Background).
ESR spectroscopy
ESR spectra were recorded on a Bruker (Karlsruhe, Germany) EMX 9-GHz spectrometer with rectangular cavity and nitrogen gas-flow temperature regulation. Samples were accommodated in 1-mm diameter glass capillaries, which were placed in a standard quartz ESR tube that contained light silicone oil for thermal stability. A solution of gramicidin A plus phospholipid and 1 mol% spin label (2 mg of lipid at 7 mg/ml) was dried down under nitrogen and then incubated under vacuum overnight. The dry sample was hydrated with excess buffer (70 μl of 10 mM Hepes, 2 mM EDTA, pH 7.8) by vortex mixing above the chain-melting temperature of the lipid. This was then pelleted in the glass capillary by using a benchtop centrifuge. Excess supernatant was removed and the capillary was sealed. For samples containing spin-labeled stearic acid (14-SASL), the buffer was either 10 mM Na acetate, 2 mM EDTA pH 4.5, or 10 mM Tris, 2 mM EDTA pH 9.0.
Analysis of the two-component ESR spectra was performed by spectral addition, using least-squares optimization. The two quasi-single component spectra were taken from samples of high lipid/peptide ratio, and low lipid/peptide ratio, respectively. Spectra of the latter were recorded at low temperature, below that of chain melting.
THEORETICAL BACKGROUND
Orientation of the amide I transition moments of gramicidin A
For an isolated mode, the ratio of the intensities with radiation polarized parallel (I∥) and perpendicular (I⊥) to the helix axis, can be calculated from the vector components of the transition moment (see, e.g., Marsh et al. (2000)):
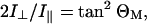 |
(2) |
where ΘM is the orientation of the transition moment, M, relative to the helix axis. Strict axial symmetry is assumed, which requires inclusion of the factor two in Eq. 2. Because the normal modes are independent, but their transition moments all transform in exactly the same way on rotation (see Eqs. 5 and 6, given in the next subsection), Eq. 2 applies equally to overlapping band components when summation is made over their contributions to the total intensities I∥ and I⊥.
Normal mode calculations by Naik and Krimm (1986a) indicate that the most intense amide I band component of gramicidin A has A-species symmetry and is predicted to have the lowest frequency and preferentially parallel polarization. This is designated by A(ν) in Table 1. However, other modes with different polarizations also contribute appreciable intensity in this region of the spectrum. The envelope of the amide I band with radiation polarized parallel or perpendicular to the helix axis that is predicted by the normal mode calculations of Naik and Krimm (1986a) is given in Fig. 1 for the β6.3 and β4.4 single-helix, and ↑↓β7.2 and ↑↓β5.6 antiparallel double-helix conformations. This figure was generated from the frequencies, intensities, and polarizations of the normal modes by assigning a Lorentzian lineshape with full-width at half-height (FWHH) of 15 cm−1 to each mode. This value of the FWHH corresponds to that found experimentally for the intense amide I component of gramicidin A in DMPC (see later). It should be noted that the vibrational frequencies correspond to calculations for fully proteated amides in gramicidin A. For the deuterated species, these frequencies are 6 cm−1 lower (Naik and Krimm, 1986b).
TABLE 1.
Parallel and perpendicular polarized relative intensities (I∥ and I⊥)
| Helix* | Mode | ν (cm−1) | I∥ | I⊥ | ΘI |
|---|---|---|---|---|---|
| β6.3 | A(ν) | 1643 | 1.000 | 0.018 | 10.8° |
| A(max) | 1643,1652 | 0.00469 | 0.00097 | 32.7° | |
| A(tot) | 1643–1654 | 1.137 | 0.233 | 32.7° | |
| β4.4 | A(ν) | 1631 | 1000 | 0.097 | 23.8° |
| A(max) | 1631,1646 | 0.00466 | 0.00083 | 30.9° | |
| A(tot) | 1631–1653 | 1.297 | 0.284 | 33.4° | |
| ↑↓β7.2 | A(ν) | 1632 | 1.000 | 0.174 | 30.6° |
| A(max) | 1632 | 0.00445 | 0.00084 | 31.6° | |
| A(tot) | 1632–1686 | 1.069 | 0.356 | 42.8° | |
| ↑↓β5.6 | A(ν) | 1636 | 1.000 | 0.754 | 50.8° |
| A(max) | 1636 | 0.00459 | 0.00341 | 50.6° | |
| A(tot) | 1636–1675 | 1.622 | 1.119 | 49.6° |
Shown are the parallel and perpendicular polarized relative intensities (I∥ and I⊥) of the strongest amide I component, A(ν), the local maximum, A(max), (see text), and the total summed intensity, A(tot), in the IR spectra of gramicidin A from normal mode calculations, and the effective orientation ΘI of the transition moment relative to the axis of the β-helix.
Primary intensity data and frequencies, ν, are taken from normal mode calculations of Naik and Krimm (1986a) for proteated gramicidin A. The effective orientation, ΘI, is obtained from Eq. 2. The local maxima are obtained from the envelope of Lorentzian components that is given in Fig. 1 (see text).
FIGURE 1.

Amide I bandshapes of proteated gramicidin A, for radiation polarized parallel (solid lines) and perpendicular (dashed lines) to the helix axis, simulated by a sum of Lorentzians of width 15 cm−1 centered at the component frequencies from normal mode calculations by Naik and Krimm (1986a). (A) β6.3 single helix and ↑↓β7.2 double-stranded helix, as indicated. (B) β4.4 single helix and ↑↓b5.6 double-stranded helix, as indicated.
The first conclusion that can be drawn from Fig. 1 is that the ↑↓β5.6-helix exhibits only small dichroism and therefore may be discarded as being incompatible with experimental observation, as will be seen later (cf. also Ulrich and Vogel (1999)). Effective values of ΘI were obtained from using Eq. 2 by using the ratios of the polarized intensities at the local maxima in the low frequency region of Fig. 1 (i.e., for parallel polarization, the highest peak in the envelope was taken and, for perpendicular polarization, the envelope peak closest in frequency to this was taken). These effective values of ΘI are designated by A(max) in Table 1. They differ appreciably from those determined from the most intense mode for the single helices (β6.3 and β4.4), but not for double-stranded helices (↑↓β7.2 and ↑↓β5.6). This is because the single helices have a neighboring mode of A-species symmetry that is less dichroic than the most intense mode. The discrepancy is greatest for the β6.3-helix and cannot be ignored because the less dichroic mode is predicted to be separated from the main (nearly parallel polarized) mode by only 9 cm−1, in this case.
It is seen from Table 1 that, with the exception of the ↑↓β5.6-helix, the effective orientations of the transition moment (Θ ∼ 31–33°) are rather similar when deduced from the local maxima in the low frequency region of the spectrum. These differ considerably, however, from values previously assumed for gramicidin A. Nabedryk et al. (1982) assumed a value of ΘI = 22° based on the geometry of a β4.4-helix and this value was adopted also by Bouchard and Auger (1993). This estimate lies close to that given for the most intense mode of the β4.4-helix in Table 1. On the other hand, Ulrich and Vogel (1999) have advocated the use of the value ΘI = 10.8° that corresponds to the most intense mode of the β6.3-helix (see Table 1). This value is inappropriate for the experimental spectra, however, because of the strong overlap with nearby unresolved modes, as represented by the band envelopes shown in Fig. 1.
Also given in Table 1 are the values of the intensities, and corresponding effective values of ΘI, that are obtained from summation over the entire amide I band. These are designated by A(tot). They may be appropriate, if the absorbance is obtained by spectral integration, without band fitting.
Simulation of dichroic spectra from gramicidin A
For a given degree of orientational order of gramicidin A, prediction of the bandshapes in the amide I region of the polarized ATR spectra, is possible also by using frequencies and intensities from the normal mode calculations of Naik and Krimm (1986a). The absorbances for parallel (A∥) and perpendicular (A⊥) polarized radiation in the ATR experiment are given by (see, e.g., Marsh (1997a)):
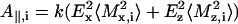 |
(3) |
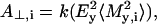 |
(4) |
where Mi = (Mx,i, My,i, Mz,i) is the transition moment of the ith amide I mode and k is a constant. Angular brackets indicate summation over all molecular orientations. The z-axis is normal to the plane of the ATR crystal and the x- and y-axes lie within and orthogonal to the incident beam, respectively. With the expressions for the components of Mi given in Marsh (1997a), and assuming axial symmetry, the absorbance for the two polarizations is given by:
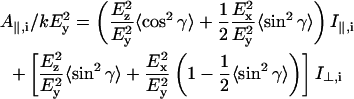 |
(5) |
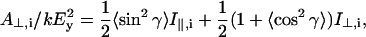 |
(6) |
where 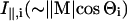 and
and 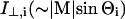 are the intensities of the amide I modes given by Naik and Krimm (1986a). The total absorbance in the amide I region is then given by summation over all modes, i. To do this, a Lorentzian bandshape of given width is associated with each mode.
are the intensities of the amide I modes given by Naik and Krimm (1986a). The total absorbance in the amide I region is then given by summation over all modes, i. To do this, a Lorentzian bandshape of given width is associated with each mode.
RESULTS
Polarized ATR spectra
FTIR spectra were recorded with radiation polarized parallel or perpendicular to the plane of incidence. Fig. 2 shows typical polarized spectra for gramicidin A in aligned DMPC membranes. Dichroism is observed both in the lipid chain methylene bands and in the amide bands of gramicidin, as seen from the differences between the solid and dashed spectra for the indicated bands. Little intensity is observed in the amide II region of the spectrum, demonstrating that all amides are accessible to H-D exchange with D2O. Table 2 lists the dichroic ratios of the amide I, amide A, lipid carbonyl, and methylene stretch bands, for both dry and hydrated samples, at temperatures in the gel and fluid phases of DMPC bilayers. These ratios, R = A∥(tot)/A⊥(tot), were determined from the integrated intensities, A(tot), of the different bands. Rehydration, after drying the hydrated samples, improved the degree of alignment in the hydrated state. Therefore, the final two stages of the repeated drying and hydration cycles in sample preparation are reported in Table 2 (see also Materials and Methods). It is seen that, in most cases, the dichroic ratios for dry and hydrated samples are rather similar, which is only possible for the thick film approximation. Table 2 includes values of the order parameters, 〈P2(cos γ)〉, that are calculated from Eq. 1 by using the electric field intensities given in the Materials and Methods section. In the dry state, all order parameters remain essentially constant with increasing temperature, whereas the lipid order parameters of the hydrated membranes decrease at the chain-melting transition, from the gel phase at 10°C to the fluid phase at 36°C. The highest order parameters in Table 2 are obtained for the lipid chains in the hydrated gel phase, at 10°C. Taking account of the fact that the lipid chains are tilted in the gel phase of DMPC (Marsh, 1990), these lipid chain order parameters show that the membrane normal is oriented almost perpendicular to the orienting surface of the ATR crystal (see Discussion).
FIGURE 2.

Polarized ATR spectra from aligned dimyristoyl phosphatidylcholine membranes containing gramicidin A at a lipid/peptide ratio of 10:1 mol/mol. Solid and dashed lines are for radiation polarized parallel and perpendicular to the plane of incidence, respectively. (Top) Dry membranes; (bottom) membranes hydrated with excess D2O. Temperature, 10°C.
TABLE 2.
Dichroic ratios, R, and order parameters, P2, of the lipid and peptide bands from DMPC + gramicidin A membranes
| Sym CH2
|
Asym CH2
|
C=O
|
Amide I
|
Amide A
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| T (°C)* | R | P2 | R | P2 | R | P2 | R | P2 | R | P2 | |
| Dried | 10 | 1.28 | 0.37 | 1.38 | 0.31 | 1.49 | 0.25 | 3.40 | 0.40 | 3.94 | 0.43 |
| 36 | 1.27 | 0.37 | 1.37 | 0.31 | 1.51 | 0.24 | 3.46 | 0.41 | 3.90 | 0.42 | |
| 40 | 1.29 | 0.36 | 1.40 | 0.3 | 1.51 | 0.24 | 3.50 | 0.42 | 4.16 | 0.46 | |
| Rehydrated | 10 | 0.99 | 0.56 | 1.04 | 0.52 | 1.22 | 0.40 | 3.05 | 0.32 | – | – |
| 36 | 1.48 | 0.25 | 1.53 | 0.22 | 1.53 | 0.22 | 3.35 | 0.39 | – | – | |
| 40 | 1.43 | 0.28 | 1.58 | 0.2 | 1.50 | 0.24 | 3.20 | 0.35 | – | – | |
Shown are the dichroic ratios, R, and order parameters, P2, of the lipid (CH2 symmetric and asymmetric stretch, and carbonyl stretch) and peptide (amide I and A) bands from DMPC + gramicidin A (10:1 mol/mol) membranes.
Order parameters, 〈P2(cos γ)〉, are calculated from Eq. 1, with orientations, Θ, of the transition moment given in Materials and Methods (see also Theoretical Background). For the amide A band, the mean effective orientation of the transition moment is calculated from the order parameters obtained from the amide I band in Tables 2–5, ΘA = 27°. The order of the entries corresponds to the chronology of the measurements.
Fig. 3 gives dichroic spectra for the amide I band of gramicidin A in four different lipid membranes: DMPC, DMPS, DMPG, and DMPE. For the two charged lipids (DMPS and DMPG), 1 M NaCl was added to the buffer to prevent infinite swelling of the aligned multilayers. The degree of dichroism differs between the various host membranes, as seen by comparing the solid and dashed lines in each panel of Fig. 3. Tables 3–5 list the dichroic ratios of the lipid and peptide bands for DMPS, DMPG, and DMPE membranes, together with the orientational order parameters, 〈P2(cos γ)〉, deduced from Eq. 1. For the amide A band, the effective orientation of the transition moment, ΘA = 27 ± 1°, was chosen as that which gives best agreement with the order parameters calculated from the amide I band, in Tables 2–5. In line with expectation (see Marsh et al. (2000)), the value of ΘA is smaller than that of ΘI. Unlike the situation for DMPC and DMPS, rehydration after drying the initially hydrated samples did not improve the degree of alignment for samples with DMPG and DMPE. Therefore, data from the first hydration stage and subsequent drying are reported in Tables 4 and 5. Trends in the temperature dependence of the lipid order parameters in Tables 3–5 are similar to those noted for DMPC samples in Table 1, when account is taken of the relative chain-melting transition temperatures of the different lipids. The order parameters of the peptide in fluid membranes (deduced from the amide I band), however, are much lower in DMPG and DMPE than in DMPC. In DMPE, they are even negative, although the lipid order parameters still remain positive. The data given in Tables 2–5 correspond to the sample for which best alignment was achieved with each of the individual lipid species, respectively. The range of dichroic ratios observed for samples differing in their degree of alignment was typically ±0.2.
FIGURE 3.

Amide I band in polarized ATR spectra from hydrated aligned phospholipid membranes containing gramicidin A at a lipid/peptide ratio of 10:1 mol/mol. Solid and dashed lines are for radiation polarized parallel and perpendicular to the plane of incidence, respectively. (A) In phosphatidylcholine (DMPC) membranes; (B) in phosphatidylserine (DMPS) membranes; (C) in phosphatidylglycerol (DMPG) membranes; and (D) in phosphatidylethanolamine (DMPE) membranes. Membranes are hydrated in D2O (DMPC and DMPE), or in D2O buffer: 10 mM Hepes, 1 M NaCl, pH 7.8 (DMPS and DMPG); temperature 10°C. Dotted lines are the components obtained from band fitting, except those overlaying the solid and dashed lines, which are the sum of the fitted components.
TABLE 3.
Dichroic ratios, R, and order parameters, P2, of the lipid and peptide bands from DMPS + gramicidin A membranes
| Sym CH2
|
Asym CH2
|
C=O
|
Amide I
|
Amide A
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| T (°C)* | R | P2 | R | P2 | R | P2 | R | P2 | R | P2 | |
| Dried | 10 | 1.11 | 0.47 | 1.12 | 0.47 | 1.17 | 0.44 | 1.94 | −0.02 | 1.92 | −0.03 |
| 60 | 1.12 | 0.47 | 1.13 | 0.46 | 1.20 | 0.42 | 1.99 | 0.0 | – | – | |
| Rehydrated | 10 | 1.09 | 0.49 | 1.10 | 0.48 | 1.21 | 0.41 | 2.50 | 0.17 | – | – |
| 24 | 1.18 | 0.43 | 1.20 | 0.42 | 1.24 | 0.39 | 2.63 | 0.21 | – | – | |
| 50 | 1.30 | 0.36 | 1.16 | 0.44 | 1.30 | 0.35 | 3.03 | 0.31 | – | – | |
| 60 | 1.26 | 0.38 | 1.30 | 0.36 | 1.33 | 0.34 | 3.23 | 0.36 | – | – | |
Shown are the dichroic ratios, R, and order parameters, P2, of the lipid (CH2 symmetric and asymmetric stretch, and carbonyl stretch) and peptide (amideI and A) bands from DMPS + gramicidin A (10:1 mol/mol) membranes.
Order parameters, 〈P2(cos γ)〉, are calculated from Eq. 1, with orientations, Θ, of the transition moment given in Materials and Methods (see also Theoretical Background). For the amide A band, the mean effective orientation of the transition moment is calculated from the order parameters obtained from the amide I band in Tables 2–5, ΘA = 27°. The order of the entries corresponds to the chronology of the measurements.
TABLE 5.
Dichroic ratios, R, and order parameters, P2, of the lipid and peptide bands from DMPE + gramicidin A membranes
| Sym CH2 | Asym CH2 | C=O | Amide I | Amide A | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| T (°C)* | R | P2 | R | P2 | R | P2 | R | P2 | R | P2 | |
| Hydrated | 10 | 1.76 | 0.11 | 1.51 | 0.23 | 1.66 | 0.16 | 1.77 | −0.09 | 0.93 | −0.43 |
| 37 | 1.54 | 0.22 | 1.68 | 0.15 | 1.86 | 0.06 | 1.70 | −0.12 | 1.63 | −0.13 | |
| 40 | 1.56 | 0.21 | 1.73 | 0.12 | 1.88 | 0.05 | 1.71 | −0.11 | 2.51 | 0.14 | |
| 63 | 1.83 | 0.08 | 1.83 | 0.08 | 2.08 | −0.03 | 1.70 | −0.12 | 2.44 | 0.13 | |
| Dried | 10 | 1.43 | 0.28 | 1.78 | 0.1 | 1.67 | 0.15 | 1.82 | −0.07 | 1.15 | −0.32 |
| 63 | 1.47 | 0.25 | 1.55 | 0.22 | 1.66 | 0.16 | 1.79 | −0.08 | 1.16 | −0.32 | |
Shown are the dichroic ratios, R, and order parameters, P2, of the lipid (CH2 symmetric and asymmetric stretch, and carbonyl stretch) and peptide (amide I and A) bands from DMPE + gramicidin A (10:1 mol/mol) membranes.
Order parameters, 〈P2(cos γ)〉, are calculated from Eq. 1, with orientations, Θ, of the transition moment given in Materials and Methods (see also Theoretical Background). For the amide A band, the mean effective orientation of the transition moment is calculated from the order parameters obtained from the amide I band in Tables 2–5, ΘA = 27°. The order of the entries corresponds to the chronology of the measurements.
TABLE 4.
Dichroic ratios, R, and order parameters, P2, of the lipid and peptide bands from DMPG + gramicidin A membranes
| Sym CH2
|
Asym CH2
|
C=O
|
Amide I
|
Amide A
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| T (°C)* | R | P2 | R | P2 | R | P2 | R | P2 | R | P2 | |
| Hydrated | 10 | 1.37 | 0.31 | 1.44 | 0.27 | 1.60 | 0.19 | 2.59 | 0.19 | – | – |
| 36 | 1.63 | 0.17 | 1.63 | 0.17 | 1.75 | 0.11 | 2.13 | 0.05 | 1.13 | −0.33 | |
| 40 | 1.66 | 0.16 | 1.60 | 0.19 | 1.76 | 0.11 | 2.12 | 0.04 | 0.75 | −0.53 | |
| Dried | 10 | 1.36 | 0.32 | 1.59 | 0.19 | 1.54 | 0.22 | 3.78 | 0.48 | 4.00 | 0.44 |
| 40 | 1.35 | 0.33 | 1.59 | 0.19 | 1.52 | 0.23 | 3.81 | 0.48 | 4.09 | 0.45 | |
Shown are the dichroic ratios, R, and order parameters, P2, of the lipid (CH2 symmetric and asymmetric stretch, and carbonyl stretch) and peptide (amide I and A) bands from DMPG + gramicidin A (10:1 mol/mol) membranes.
Order parameters, 〈P2(cos γ)〉, are calculated from Eq. 1, with orientations, Θ, of the transition moment given in Materials and Methods (see also Theoretical Background). For the amide A band, the mean effective orientation of the transition moment is calculated from the order parameters obtained from the amide I band in Tables 2–5, ΘA = 27°. The order of the entries corresponds to the chronology of the measurements.
All ATR experiments were performed with the same peptide:lipid ratio of 1:10 mol/mol. This relatively high value was chosen for direct comparison with the ESR experiments utilizing spin-labeled lipids that are to be described later. One consequence of using high-peptide contents could be the tendency to aggregation. Response of the peptide to chain melting of the hydrated lipids, perturbation of the lipid chain mobility seen by ESR and differential response to host lipid species, however, indicate that this is not a general phenomenon (see Discussion).
Simulation of dichroic spectra
The amide I band region in the polarized ATR spectra of gramicidin A in DMPC, was simulated by using the experimental order parameter, 〈P2(cos γ)〉, and the normal mode results of Naik and Krimm (1986a), as described in the section on Theoretical Background. This gives a check on consistency, because the value of 〈P2(cos γ)〉 was determined simply from the dichroic ratio and an effective value of the transition moment orientation, and not from the overall bandshape. Additionally, the bandshape simulations afford further possibility to distinguish between the channel-forming and double-helix conformations of gramicidin.
The amide I band envelopes predicted according to Eqs. 5 and 6 from the normal mode analysis for the gramicidin A β6.3-single helix and the ↑↓β7.2-double-stranded helix are given in Fig. 4, A and B, respectively. For this, a Lorentzian lineshape with full width at half-height (FWHH) of 15 cm−1 is associated with each mode. The latter corresponds to the FWHH of the intense component at ∼1630 cm−1 in the experimental spectrum of gramicidin A in DMPC (see above). Amide I bandshapes are given for radiation polarized parallel (solid lines) and perpendicular (dashed lines) to the plane of incidence, by using the thick film approximation. Corresponding calculations with the thin film approximation are given by dotted lines.
FIGURE 4.

ATR amide I bandshapes for proteated gramicidin A predicted from Eqs. 5 and 6 and the normal mode analysis of Naik and Krimm (1986a), (A) for the β6.3 single-helix conformation, and (B) for the ↑↓β7.2 double-stranded helix conformation. The thick film approximation is used with a helix tilt of γeff = 39° (〈cos2 γ〉 = 0.60) to obtain the ATR absorbance with parallel (solid line) and perpendicular (dashed line) polarized radiation. (Dotted lines are corresponding calculations with the thin film approximation, γeff = 30°; 〈cos2 γ〉 = 0.75). Each component mode has a Lorentzian lineshape with FWHH = 15 cm−1.
For comparison with the experimental spectra of gramicidin A in DMPC, it must be remembered that the uncertainty in the absolute frequencies from the normal mode calculations is ±5 cm−1 (Naik and Krimm, 1986a). Also, the predicted frequencies refer to proteated amides, and lie 6 cm−1 lower for the deuterated species (Naik and Krimm, 1986b; Ulrich and Vogel, 1999). Most reliance, therefore, can be put on the relative frequencies of the different modes and on their polarizations. The amide I ATR bandshapes predicted for the β6.3-helix with parallel and perpendicular polarized radiation (Fig. 4 A) are in reasonable agreement with the major component in the experimental spectra (Fig. 3), although absolute frequencies are considerably higher. The shoulder that extends to high frequency (up to ∼1670 cm−1) in the experimental spectra is not found in the predictions for a β6.3-helix. It has been suggested previously that this shoulder may correspond to amide carbonyls at the (blocked) N- and C-terminal regions of the β6.3-helix (Buchet et al., 1985; Axelsen et al., 1995). These are not included in the normal mode calculations, which were performed for an infinite helix (Naik and Krimm, 1986a). The amide I bandshape predicted for a ↑↓β7.2-helix (Fig. 4 B) differs from the β6.3-helix in that the major component is centered at lower frequency, and secondary components extend up to ∼40 cm−1 from the former. This splitting, which arises predominantly from transition dipole coupling between A- and E1-species modes of the antiparallel double helix, potentially might provide an alternative explanation for the high-frequency shoulder in the experimental spectrum. However, its predicted intensity relative to the major peak is greater and its dichroism differs, compared with the experimental spectra.
Quantitation by peak fitting to the parallel and perpendicular polarized spectra given in Fig. 3 (see Marsh (1999b)) reveals that the high-frequency shoulder constitutes ∼30% of the total intensity of the amide I band. This would correspond to ∼4 residues per gramicidin monomer (including the blocked N- and C-termini). This is not unreasonable for the effective number of end residues whose H-bonding does not conform strictly to the pattern for an infinite β-helix. Three residues at the membrane surface are hydrogen bonded to water and three residues at the dimer interface are involved in H-bonding to the partnering monomer (Buchet et al., 1985). For the former, the amide I band components are expected in the region of 1640–1650 cm−1. The residues at the dimer interface are arranged as antiparallel β-strands. For these a weak band is expected at 1670–1680 cm−1 and a strong band at 1630–1640 cm−1, much as for the antiparallel double helices (see Theoretical Background). This is in accord with the band fitting for the amide I region of gramicidin A in DMPC that was given above.
Lipid spin-label ESR
The ATR results of the previous section show that best orientation of gramicidin A was obtained in a DMPC host membrane. Therefore, this lipid was chosen to study the lipid-peptide interactions further by using spin-labeled lipids at probe concentrations. Fig. 5 gives the ESR spectra of phosphatidylcholine spin-labeled at the 14-position of the sn-2 chain (14-PCSL) in DMPC membrane dispersions that contain gramicidin A at different lipid/peptide molar ratios. The sample temperature is 30°C, at which DMPC membranes are in the fluid phase. For gramicidin/DMPC ratios >1:20 mol/mol, a second component with increased hyperfine splitting appears in the outer wings of the ESR spectra. This component corresponds to a lipid population with chain mobility that is more restricted than that in fluid lipid bilayers. Its intensity increases progressively, relative to that of the fluid-bilayer lipid population, as the content of gramicidin A in the membrane increases. Quantitation of the fraction, f, of motionally restricted lipids was performed as described in the Materials and Methods section. Assuming that the spin-label distribution directly reflects that of unlabeled PC (i.e., Kr ≈ 1), the motionally restricted lipid population corresponds to Nb = 3.6 ± 0.3 lipids/gramicidin, consistently over the lipid/peptide range: 5:1–15:1 mol/mol.
FIGURE 5.

ESR spectra of 14-PCSL phosphatidylcholine spin label in membranes of DMPC containing gramicidin A at the lipid/peptide molar ratios indicated on the figure. Buffer, 10 mM Hepes, 2 mM EDTA, pH 7.8; T = 30°C.
Lipid selectivity was investigated by using different lipid species, all of which have the spin label at the 14-position of the chain. Fig. 6 shows the ESR spectra of the different spin-labeled lipids in DMPC membranes that contain gramicidin at a fixed peptide/lipid ratio (1:10 mol/mol). The left-hand panel gives spectra for samples prepared from TFE, and the right-hand panel gives those for samples prepared from chloroform-methanol 2:1 v/v. (Otherwise, all samples were prepared from TFE.) For all spin-labeled lipid species, the fraction of motionally restricted lipid is greater in samples prepared from chloroform-methanol than in those prepared from TFE. Nevertheless, the pattern of lipid selectivity is consistent between the two sets of samples. The same spin-labeled lipids (e.g., 14-SASL at pH 9, or 14-PASL) display a larger motionally restricted spectral component in both cases. Table 6 lists values for the fraction, f, of motionally restricted spin label. The relative association constant, Kr, normalized to that for 14-PCSL (i.e., KrPC), is also given in Table 6, for each spin-labeled lipid:
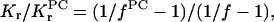 |
(7) |
where fPC is the value of f for 14-PCSL (see, e.g., Marsh and Horváth (1998)). The selectivity ranking is: SA(pH 9)>PA≥PS>PC>PG≥PE>SA(pH 4.5), for samples prepared from either solvent.
FIGURE 6.

ESR spectra of spin-labeled phospholipids: 14-PCSL, 14-PSSL, 14-PESL, 14-PASL, and 14-PGSL, in DMPC/gramicidin A (10:1 mol/mol) membranes hydrated with 10 mM Hepes, 2 mM EDTA, pH 7.8. The bottom two spectra are from spin-labeled stearic acid, 14-SASL, at pH 4.5 (10 mM Na acetate, 2 mM EDTA) and pH 9.0 (10 mM Tris, 2 mM EDTA), as indicated. (Left) Samples prepared from TFE; (right) samples prepared from chloroform-methanol (2:1 v/v). T = 30°C.
TABLE 6.
Fractions, f, of motionally restricted spin-labeled lipid in DMPC membranes containing gramicidin A
| TFE
|
CHCl3:MeOH
|
|||
|---|---|---|---|---|
| Spin label* | f | 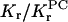 |
f | 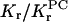 |
| 14-PCSL | 0.39 | 1.0 | 0.63 | 1.0 |
| 14-PASL | 0.44 | 1.2 | 0.65 | 1.1 |
| 14-PESL | 0.34 | 0.8 | 0.52 | 0.6 |
| 14-PGSL | 0.35 | 0.8 | 0.53 | 0.7 |
| 14-PSSL | 0.42 | 1.1 | 0.65 | 1.1 |
| 14-SASL (pH 4.5) | 0.30 | 0.7 | 0.48 | 0.5 |
| 14-SASL (pH 9.0) | 0.48 | 1.4 | 0.69 | 1.3 |
Shown are fractions, f, of motionally restricted spin-labeled lipid in DMPC membranes containing gramicidin A at 1:10 molar ratio. Kr/KrPC is the relative association constant, normalized to 14-PCSL.
Samples are prepared from TFE or from CHCl3:MeOH (2:1 v/v) and are buffered at pH 7.8, except for 14-SASL. Typical uncertainty in f from fitting the two-component spectra is ±0.01–0.02.
DISCUSSION
Orientation of gramicidin A in DMPC membranes
Data on the orientation of gramicidin obtained from polarized ATR measurements depend directly on the value taken for the orientation, ΘM, of the transition moment. Also, they depend on how well the membranes are aligned on the ATR crystal. In both of these connections, it is instructive to compare with the results of previous measurements. This is done first, before considering the implications of the present results.
Dichroic ratios for gramicidin A in DMPC in the dry state have been measured by polarized transmission spectroscopy (Nabedryk et al., 1982). The samples were prepared from chloroform. Values of ΘM were predicted from molecular coordinates for a β4.4-helix and measurements on an α-helical model compound. The maximum inclination of the amide I transition moment to the helix axis that is calculated from the experimental data of Nabedryk et al. (1982) is ΘI = 27 ± 1°, assuming that the sample is perfectly oriented (i.e., γeff = 0°). Whereas this value is within the limits set for the most intense mode of a β6.3-helix, it is less than the effective values of ΘI (≈32°) derived from the band maxima in the low-frequency region (see Table 1). One conclusion that can be drawn from this is that the samples of Nabedryk et al. (1982) are highly ordered. The other conclusion is that the effective orientations of the transition moment derived from the band maxima predicted by the normal mode analysis of Naik and Krimm (1986a) may be upper estimates.
Dichroic ratios have also been measured previously for gramicidin A in fluid hydrated DMPC bilayers by polarized ATR (Bouchard and Auger, 1993). Using the orientations of the transition moment given in the section on Theoretical Background (i.e., ΘI = 32°), rather than the older ones referred to above, yields values of 〈P2(cos γ)〉 = 0.39 ± 0.06 and γeff = 40 ∓ 2° for a sample prepared from TFE and incubated for 4 h. These values are in good agreement with those given in Table 2, which were obtained also from samples prepared from TFE but with a different ATR crystal.
The order parameter for the lipid chains in the gel phase at 10°C that is given in Table 2 corresponds to an effective tilt of γeff = 33° (CH2 symmetric stretch), when a unique orientation is assumed. For DMPC alone, a similar value of γeff = 29° was obtained (data not shown). This is comparable to the chain tilt of DMPC in the Lβ′ phase obtained from x-ray diffraction studies (≈30°, see, e.g., Marsh, 1990). From this, one can conclude that the DMPC membranes are reasonably well aligned. Nevertheless, the effective tilt of the gramicidin A molecule at 10°C somewhat exceeds this value. Possibly the peptide channel cannot incorporate perfectly in a single domain of the tilted lipid gel phase and may be associated with regions of azimuthal disorder in the chain-tilt direction. Note, however, that gramicidin A is not excluded from the lipid gel phase. Our preliminary results from differential scanning calorimetry show that the DMPC pretransition shifts progressively to lower temperature with increasing content of gramicidin A.
In the fluid phase, the effective tilt of the lipid chains increases (i.e., the order parameter decreases), relative to the gel phase. This chain disorder results from trans-gauche isomerism about the C-C bonds in the lipid chains. Under these conditions, the order parameter of gramicidin A, however, does not decrease. Instead, the order improves in the fluid phase. The hydrophobic span and effective tilt of gramicidin A match more closely those of the disordered lipid chains than for the extended chains in the gel phase (see Table 2). One, therefore, can conclude that the peptide incorporates well in the fluid Lα phase of hydrated DMPC.
Orientation of gramicidin A in different lipids
Fig. 7 gives the effective tilt angles, γeff, of gramicidin A (amide I) and the lipid chains (CH2 symmetric stretch) in the different phospholipid hosts tested. Data are given for both gel and fluid phases of the respective lipid bilayers. These values increase (i.e., the degree of orientation decreases) progressively from DMPS, via DMPG, to DMPE. With the exception of the fluid phase of DMPS, the tilt-values are also greater than for DMPC. Concomitantly, the degree of disorder of gramicidin A exceeds that of the lipid chains, in the fluid phase. This is unlike the situation for the DMPC host. The effect is particularly dramatic in the case of DMPE, where gramicidin A is almost completely disordered under all conditions. The dichroic ratios are then close to the isotropic value, RISO = 2, and γeff lies close to the magic angle. This can be interpreted as indicating that gramicidin A incorporates less well into membranes of the negatively charged lipids, DMPS and DMPG. Most likely it incorporates hardly at all into the weakly hydrated DMPE membranes, despite being entirely devoid of charged or polar residues. Internal hydrogen bonding within the DMPE surface layer (Elder et al., 1977), and its low hydration, presumably precludes hydrogen bonding of the ester carbonyls with the interfacial tryptophan residues, which is an important feature of membrane integration for gramicidin A (Woolf and Roux, 1994; Hu and Cross, 1995).
FIGURE 7.

Effective orientation, γeff, deduced from ATR order parameters of the lipid chains (CH2 symmetric stretch; left two bars for each), and of gramicidin (amide I; right two bars for each), in fully hydrated membranes of DMPC, DMPS, DMPG, and DMPE with a lipid/peptide ratio of 10:1 mol/mol. Data are given for temperatures in the gel (left bar in each pair) and fluid phases (right bar in each pair) of each lipid: DMPC, 10°C and 40°C; DMPS, 24°C and 50°C; DMPG, 10°C and 40°C; and DMPE, 37°C and 63°C.
Gramicidin A-lipid interactions in DMPC
The ESR results obtained with 14-PCSL indicate that three to four lipids per monomer are motionally restricted by interaction with the intramembranous surface of gramicidin A, when it is reconstituted with DMPC from TFE (cf., e.g., Marsh and Horváth, 1998). This is considerably less than the number of lipids that can be accommodated at the intramembranous perimeter of the peptide. In the β6.3-helical conformation, the outer diameter of the gramicidin A helix is ∼1.8 nm, and it spans a single monolayer (Ketchem et al., 1997; PDB: 1MAG). Approximately 15 lipid chains, i.e., seven to eight diacyl lipids, can be arranged to contact such a structure. In principle, the reduction in stoichiometry of lipid interaction by a factor of two could be explained by aggregation of gramicidin monomers in the plane of the membrane. Tetramer formation would be required to account for the stoichiometry observed (see, e.g., Marsh, 1997b). It is, however, perhaps significant that the number of motionally restricted lipids equals the number of interfacial tryptophans that is found in the β6.3-structure. An alternative explanation, therefore, might be offered in terms of MD simulation studies performed on lipid interactions with gramicidin A (Woolf and Roux, 1994, 1996). In a simulation for eight DMPC molecules per gramicidin A monomer, it was found that not all lipids interacted equally strongly with gramicidin, i.e., some chains are more “remote” from the intramembranous surface than are others. The four lipids that are motionally restricted therefore may correspond to more-or-less well-defined sites on the peptide surface, most probably associated with the four tryptophans. Note that this is a feature of the expanded surface of the β6.3-helix, relative to an α-helix, and possibly also of the relatively large number of interfacial tryptophans. A simple transmembrane α-helix with two interfacial tryptophans does not induce direct motional restriction of spin-labeled lipid chains (de Planque et al., 1998).
From Fig. 6, it is evident that the lipid stoichiometry in samples prepared from chloroform-methanol is greater than that in samples prepared from TFE. In the former case, the motionally restricted population, f, of 14-PCSL corresponds to six lipids per gramicidin A monomer (see Table 6). The difference is attributable to the fact that samples prepared from chloroform-methanol are composed, at least in part, of the ↑↓β7.2 double helix (Bouchard and Auger, 1993). This configuration is fully transmembrane and therefore might be more effective in restricting the motion of spin labels close to the terminal methyl group of the lipid chains than is the head-to-head dimer. Additionally, both any putative aggregation pattern and the exact disposition of the interfacial tryptophans may differ for this conformer. Indeed, the solvent history generally could affect the aggregation state of gramicidin A in the membrane.
Although gramicidin A is a hydrophobic peptide, devoid of any strongly polar residues, the results given in Table 6 demonstrate that it does express a selectivity of interaction between different lipid species. Previous ESR experiments have demonstrated that the selectivity for negatively charged lipids, which is displayed by a range of integral membrane proteins, is not solely electrostatic in origin (Marsh and Horváth, 1998). The lipid headgroup selectivity that is exhibited by gramicidin A is of the non-Coulombic type. Almost certainly it involves the strongly dipolar properties of the interfacial tryptophans that are present in both the β6.3 and ↑↓β7.2 structures.
It is of interest to compare the lipid selectivities at the peptide-lipid interface that are determined by ESR in DMPC with the results on the integration of gramicidin A in different host lipid species that was explored by ATR. Incorporation of gramicidin into lipid membranes depends not only on the specificity of lipid interactions locally at the lipid-peptide interface, as measured by different spin-labeled lipid species at probe concentrations. It depends also on the intrinsic properties of the bilayers of the host lipid, i.e., on the lipid-lipid interactions in the membrane environment into which the peptide must incorporate. For this reason, the two sets of measurements are not necessarily equivalent. Evidently, it is lipid-lipid interactions, and not direct lipid-peptide interactions, that dominate the energetics of peptide incorporation into certain lipids.
CONCLUSIONS
In addition to the role of the lipid chains, which has been investigated previously (Killian, 1992), the lipid polar groups and surface hydration also play a significant part in the successful integration of gramicidin into the lipid bilayer. Interfacial tryptophan residues in gramicidin A are important for both the stoichiometry and selectivity of lipid interactions with this peptide. This could have several implications for the interactions of lipids with integral transmembrane proteins. Tryptophan residues might quite generally nucleate the formation of crevices that could accommodate lipids at the surface of integral membrane proteins. In addition, they could be a major source of lipid selectivity that is not based directly on the net charge of the lipid headgroups—selectivity of non-Coulombic origin is a feature that is quite widespread amongst integral proteins (Marsh and Horváth, 1998).
Acknowledgments
We thank Frau B. Angerstein for synthesis of spin-labeled lipids.
This work was supported in part by the Volkswagen-Stiftung, the Hungarian National Science Fund (T029458 and T043425), and a German-Hungarian travel grant (DAAD-MOB 18/2001). D.M. and T.P. are members of the European Union COST D22 Action.
Abbreviations used: ATR, attenuated total reflection; DMPC, DMPE, DMPG, and DMPS, 1,2-dimyristoyl-sn-glycero-3-phosphocholine, -phosphoethanolamine, -phosphoglycerol, and -phosphoserine; EDTA, ethylenediaminetetraacetic acid; ESR, electron spin resonance; Hepes, N-(2-hydroxyethyl)piperazine-N′-2-ethanesulphonic acid; MD, molecular dynamics; 14-PASL, 14-PCSL, 14-PESL, 14-PGSL, and 14-PSSL, 1-acyl-2-(14-(4,4-dimethyl-oxazolidine-N-oxy))stearoyl-sn-glycero-3-phosphoric acid, -phosphocholine, -phosphoethanolamine, -phosphoglycerol, and -phosphoserine; 14-SASL, 14-(4,4-dimethyl-oxazolidine-N-oxy)stearic acid; TFE, trifluoroethanol; Tris, tris-hydroxymethylaminoethane.
References
- Axelsen, P. H., B. K. Kaufman, R. N. McElhaney, and R. N. A. H. Lewis. 1995. The infrared dichroism of transmembrane helical polypeptides. Biophys. J. 69:2770–2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard, M., and M. Auger. 1993. Solvent history dependence of gramicidin-lipid interactions: a Raman and infrared spectroscopic study. Biophys. J. 65:2484–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchet, R., C. Sandorfy, T. L. Trapene, and D. W. Urry. 1985. Infrared spectroscopic studies on gramicidin ion channels—relation to the mechanisms of anesthesia. Biochim. Biophys. Acta. 821:8–16. [DOI] [PubMed] [Google Scholar]
- de Planque, M. R. R., D. V. Greathouse, R. E. Koeppe II, H. Schäfer, D. Marsh, and J. A. Killian. 1998. Influence of lipid/peptide hydrophobic mismatch on the thickness of diacylphosphatidylcholine bilayers. A 2H NMR and ESR study using designed transmembrane α-helical peptides and gramicidin A. Biochemistry. 37:9333–9345. [DOI] [PubMed] [Google Scholar]
- Elder, M., P. Hitchcock, R. Mason, and G. G. Shipley. 1977. A refinement analysis of the crystallography of the phospholipid, 1,2-dilauroyl-DL-phosphatidylethanolamine, and some remarks on lipid-lipid and lipid-protein interactions. Proc. R. Soc. London A. 354:157–170. [Google Scholar]
- Ge, M. T., and J. H. Freed. 1999. Electron spin resonance study of aggregation of gramicidin in dipalmitoylphosphatidylcholine bilayers and hydrophobic mismatch. Biophys. J. 76:264–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, W., and T. A. Cross. 1995. Tryptophan hydrogen bonding and electric dipole moments: functional roles in the gramicidin channel and implications for membrane proteins. Biochemistry. 34:14147–14155. [DOI] [PubMed] [Google Scholar]
- Hubbell, W. L., and H. M. McConnell. 1971. Molecular motion in spin-labelled phospholipids and membranes. J. Am. Chem. Soc. 93:314–326. [DOI] [PubMed] [Google Scholar]
- Ketchem, R. R., W. Hu, and T. A. Cross. 1993. High-resolution conformation of gramicidin A in a lipid bilayer by solid-state NMR. Science. 261:1457–1460. [DOI] [PubMed] [Google Scholar]
- Ketchem, R. R., B. Roux, and T. A. Cross. 1997. High-resolution polypeptide structure in a lamellar phase lipid environment from solid state NMR derived orientational constraints. Structure. 5:1655–1669. [DOI] [PubMed] [Google Scholar]
- Killian, J. A. 1992. Gramicidin and gramicidin-lipid interactions. Biochim. Biophys. Acta. 1113:391–425. [DOI] [PubMed] [Google Scholar]
- Marsh, D. 1990. Handbook of Lipid Bilayers. CRC Press, Boca Raton FL.
- Marsh, D. 1997a. Dichroic ratios in polarized Fourier transform infrared for nonaxial symmetry of β-sheet structures. Biophys. J. 72:2710–2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh, D. 1997b. Stoichiometry of lipid-protein interaction and integral membrane protein structure. Eur. Biophys. J. 26:203–208. [Google Scholar]
- Marsh, D. 1999a. Spin label ESR spectroscopy and FTIR spectroscopy for structural/dynamic measurements on ion channels. Methods Enzymol. 294:59–92. [DOI] [PubMed] [Google Scholar]
- Marsh, D. 1999b. Quantitation of secondary structure in ATR infrared spectroscopy. Biophys. J. 77:2630–2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh, D., and L. I. Horváth. 1998. Structure, dynamics and composition of the lipid-protein interface. Perspectives from spin-labelling. Biochim. Biophys. Acta. 1376:267–296. [DOI] [PubMed] [Google Scholar]
- Marsh, D., M. Müller, and F.-J. Schmitt. 2000. Orientation of the infrared transition moments for an α-helix. Biophys. J. 78:2499–2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh, D., and A. Watts. 1982. Spin-labeling and lipid-protein interactions in membranes. In Lipid-Protein Interactions, Vol. 2. P. C. Jost and O. H. Griffith, editors. Wiley-Interscience, New York. 53–126.
- Nabedryk, E., M. P. Gingold, and J. Breton. 1982. Orientation of gramicidin A transmembrane channel. Infrared dichroism study of gramicidin in vesicles. Biophys. J. 38:243–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik, V. M., and S. Krimm. 1986a. Vibrational analysis of the structure of gramicidin A. I. Normal mode analysis. Biophys. J. 49:1131–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik, V. M., and S. Krimm. 1986b. Vibrational analysis of the structure of gramicidin A. II. Vibrational spectra. Biophys. J. 49:1147–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patyal, B. R., R. H. Crepeau, and J. H. Freed. 1997. Lipid-gramicidin interactions using two-dimensional Fourier-transform electron spin resonance. Biophys. J. 73:2201–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice, D., and E. Oldfield. 1979. Deuterium nuclear magnetic resonance studies of the interaction between dimyristoylphosphatidylcholine and gramicidin A. Biochemistry. 18:3272–3279. [DOI] [PubMed] [Google Scholar]
- Ulrich, W. P., and H. Vogel. 1999. Polarization-modulated FTIR spectroscopy of lipid/gramicidin monolayers at the water interface. Biophys. J. 76:1639–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt, T. C. B., J. A. Killian, and B. De Kruijff. 1994. Structure and dynamics of the acyl chain of a transmembrane polypeptide. Biochemistry. 33:2063–2070. [DOI] [PubMed] [Google Scholar]
- Wallace, B. A., and K. Ravikumar. 1988. The gramicidin pore crystal structure of a cesium complex. Science. 241:182–187. [DOI] [PubMed] [Google Scholar]
- Woolf, T. B., and B. Roux. 1994. Molecular dynamics simulation of the gramicidin channel in a phospholipid bilayer. Proc. Natl. Acad. Sci. USA. 91:11631–11635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf, T. B., and B. Roux. 1996. Structure, energetics and dynamics of lipid-protein interactions: a molecular dynamics study of the gramicidin A channel in a DMPC bilayer. Proteins. 24:92–114. [DOI] [PubMed] [Google Scholar]